
A Bioinformatic Approach for Molecular Characterization and Functional Annotation of an Uncharacterized Protein from Vibrio cholerae †




Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Sequence Retrieval
2.2. Physicochemical Characterization
2.3. Functional Annotation Prediction
2.4. Subcellular Location Identification
2.5. Secondary Structural Assessment
2.6. Tertiary Structure Modeling and Validation
2.7. Active Site Determination
3. Results and Discussion
3.1. Protein Sequence Retrieval
3.2. Physicochemical Characterization
3.3. Functional Annotation Prediction
3.4. Subcellular Location Determination
3.5. Secondary Structure Inquiry
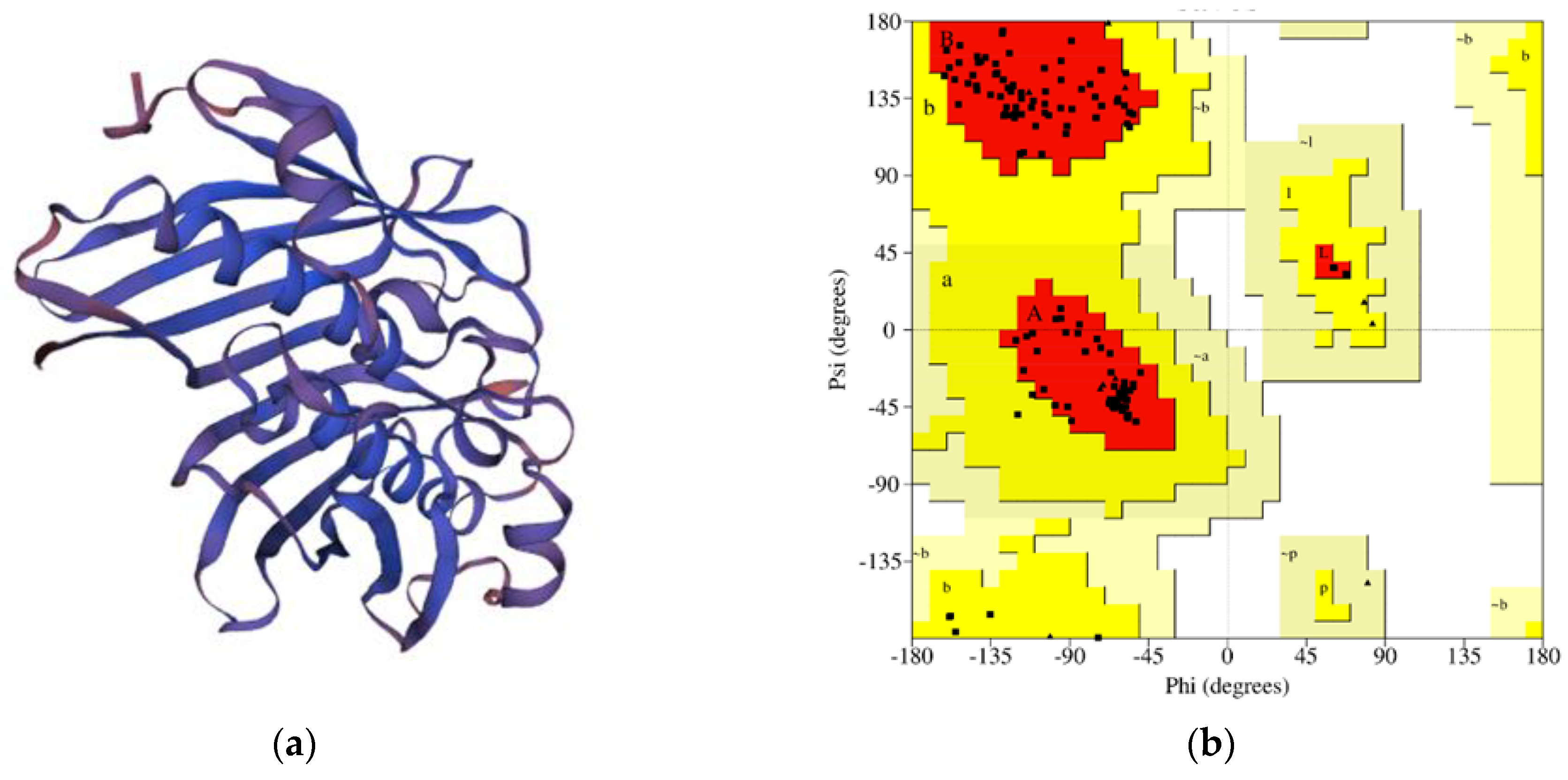
3.6. Tertiary-Structure Modeling and Validation
3.7. Active Site Determination
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weil, A.A.; Becker, R.L.; Harris, J.B. Vibrio cholerae at the Intersection of Immunity and the Microbiome. mSphere 2019, 4, e00597-19. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y.; Liu, R.; Macbeth, J.C.; Hsiao, A. The Interface of Vibrio cholerae and the Gut Microbiome. Gut Microbes 2021, 13, 1937015. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.G.; Teschler, J.K.; Jones, C.J.; Yildiz, F.H. Staying Alive: Vibrio cholerae’s Cycle of Environmental Survival, Transmission, and Dissemination. Microbiol. Spectr. 2016, 4, 593–633. [Google Scholar] [CrossRef] [PubMed]
- Baker-Austin, C.; Oliver, J.D.; Alam, M.; Ali, A.; Waldor, M.K.; Qadri, F.; Martinez-Urtaza, J. Vibrio spp. infections. Nature reviews. Dis. Prim. 2018, 4, 8. [Google Scholar] [CrossRef]
- Deen, J.; Mengel, M.A.; Clemens, J.D. Epidemiology of cholera. Vaccine 2020, 38 (Suppl. S1), A31–A40. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Saikat, A.S.M.; Paul, A.K.; Dey, D.; Das, R.C.; Das, M.C. In-Silico Approaches for Molecular Characterization and Structure-Based Functional Annotation of the Matrix Protein from Nipah henipavirus. Chem. Proc. 2022, 12, 21. [Google Scholar] [CrossRef]
- de Castro, E.; Sigrist, C.J.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Saikat, A.S.M.; Islam, R.; Mahmud, S.; Imran, A.S.; Alam, M.S.; Masud, M.H.; Uddin, E. Structural and Functional Annotation of Uncharacterized Protein NCGM946K2_146 of Mycobacterium Tuberculosis: An In-Silico Approach. Proceedings 2020, 66, 13. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Tusnády, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M.; Uddin, M.E.; Ahmad, T.; Mahmud, S.; Imran, M.A.S.; Ahmed, S.; Alyami, S.A.; Moni, M.A. Structural and Functional Elucidation of IF-3 Protein of Chloroflexus aurantiacus Involved in Protein Biosynthesis: An In Silico Approach. BioMed Res. Int. 2021, 2021, 9050026. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M. Computational approaches for molecular characterization and structure-based functional elucidation of a hypothetical protein from Mycobacterium tuberculosis. Genom. Inform. 2023, 21, e25. [Google Scholar] [CrossRef] [PubMed]
- Jisna, V.A.; Jayaraj, P.B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein J. 2021, 40, 522–544. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M.; Ripon, A. Structure Prediction, Characterization, and Functional Annotation of Uncharacterized Protein BCRIVMBC126_02492 of Bacillus cereus: An In Silico Approach. Am. J. Pure Appl. Biosci. 2020, 2, 104–111. [Google Scholar] [CrossRef]
- Eisenhaber, F.; Persson, B.; Argos, P. Protein structure prediction: Recognition of primary, secondary, and tertiary structural features from amino acid sequence. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 1–94. [Google Scholar] [CrossRef]
- Saikat, A.S.M.; Das, R.C.; Das, M.C. Computational Approaches for Structure-Based Molecular Characterization and Functional Annotation of the Fusion Protein of Nipah henipavirus. Chem. Proc. 2022, 12, 32. [Google Scholar] [CrossRef]
- Dubey, S.P.N.; Kini, N.G.; Balaji, S.; Kumar, M.S. A Review of Protein Structure Prediction Using Lattice Model. Crit. Rev. Biomed. Eng. 2018, 46, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chem. Proc. 2022, 8, 5. [Google Scholar] [CrossRef]

{kind=link}
| Analysis | Results |
|---|---|
| PSORTb (v.3.0.2) | Cytoplasmic |
| SOSUIGramN | Cytoplasmic |
| PSLpred | Cytoplasmic |
| HMMTOP (v.2.0) | No transmembrane helices present |
| TMHMM (v.2.0) | No transmembrane helices present |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousuf, M.; Saikat, A.S.M.; Uddin, M.E. A Bioinformatic Approach for Molecular Characterization and Functional Annotation of an Uncharacterized Protein from Vibrio cholerae. Eng. Proc. 2023, 37, 62. https://doi.org/10.3390/ECP2023-14644
Yousuf M, Saikat ASM, Uddin ME. A Bioinformatic Approach for Molecular Characterization and Functional Annotation of an Uncharacterized Protein from Vibrio cholerae. Engineering Proceedings. 2023; 37(1):62. https://doi.org/10.3390/ECP2023-14644
Chicago/Turabian StyleYousuf, Md., Abu Saim Mohammad Saikat, and Md. Ekhlas Uddin. 2023. "A Bioinformatic Approach for Molecular Characterization and Functional Annotation of an Uncharacterized Protein from Vibrio cholerae" Engineering Proceedings 37, no. 1: 62. https://doi.org/10.3390/ECP2023-14644
APA StyleYousuf, M., Saikat, A. S. M., & Uddin, M. E. (2023). A Bioinformatic Approach for Molecular Characterization and Functional Annotation of an Uncharacterized Protein from Vibrio cholerae. Engineering Proceedings, 37(1), 62. https://doi.org/10.3390/ECP2023-14644