
Electrochemical Detection of Cocaine in Authentic Oral Fluid †



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
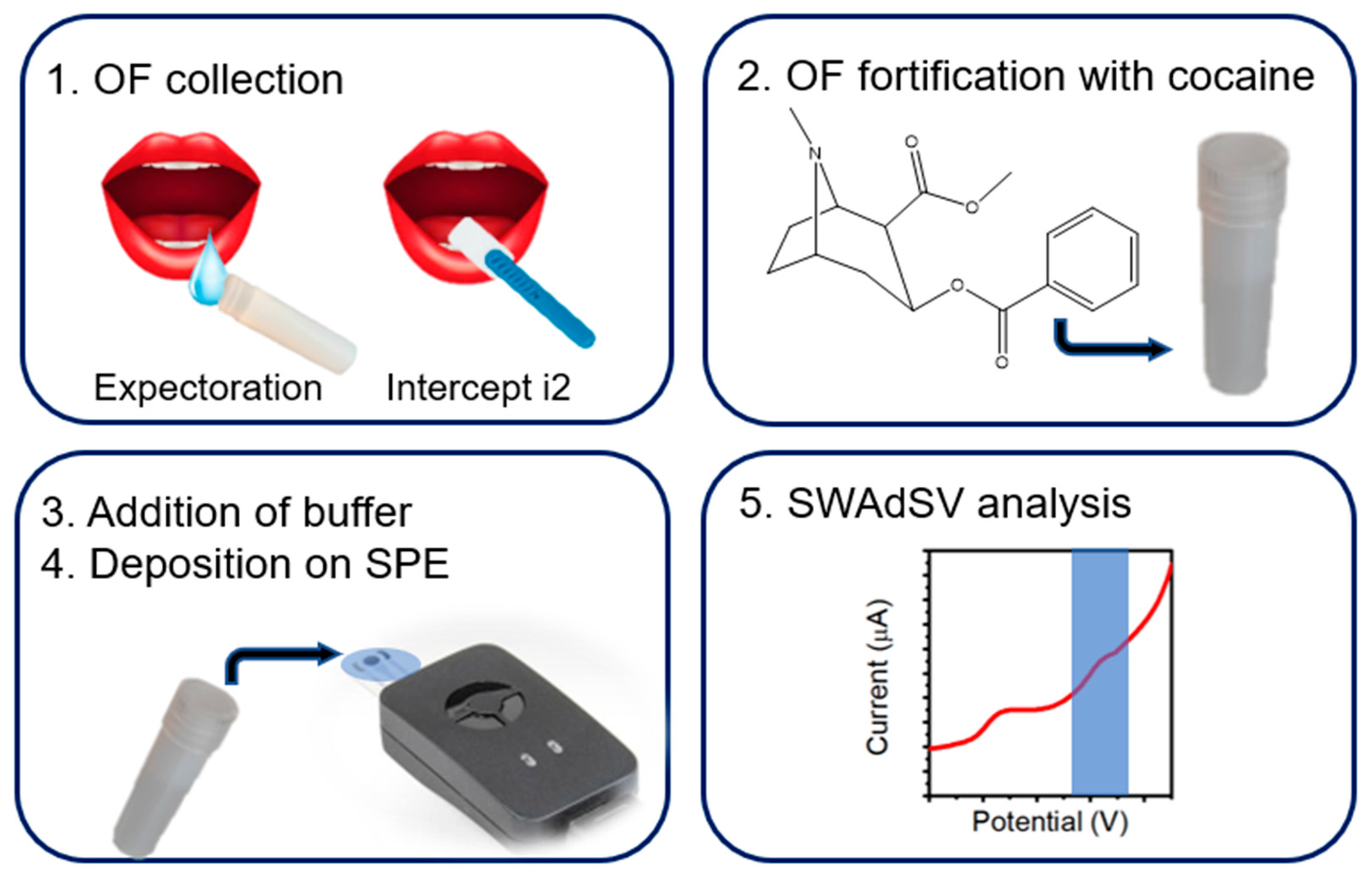
2.3. Cocaine Detection in Oral Fluid
3. Results and Discussion
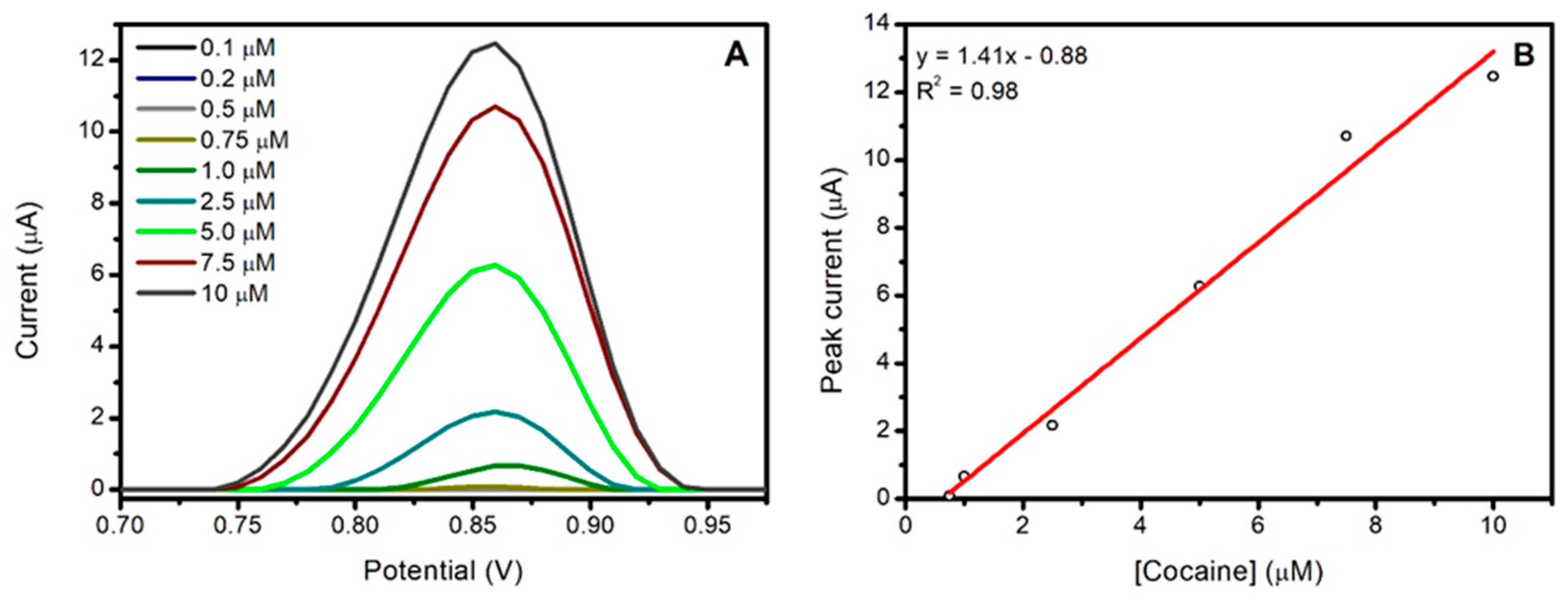
3.1. Analytical Characterization of Cocaine in Buffer Solution
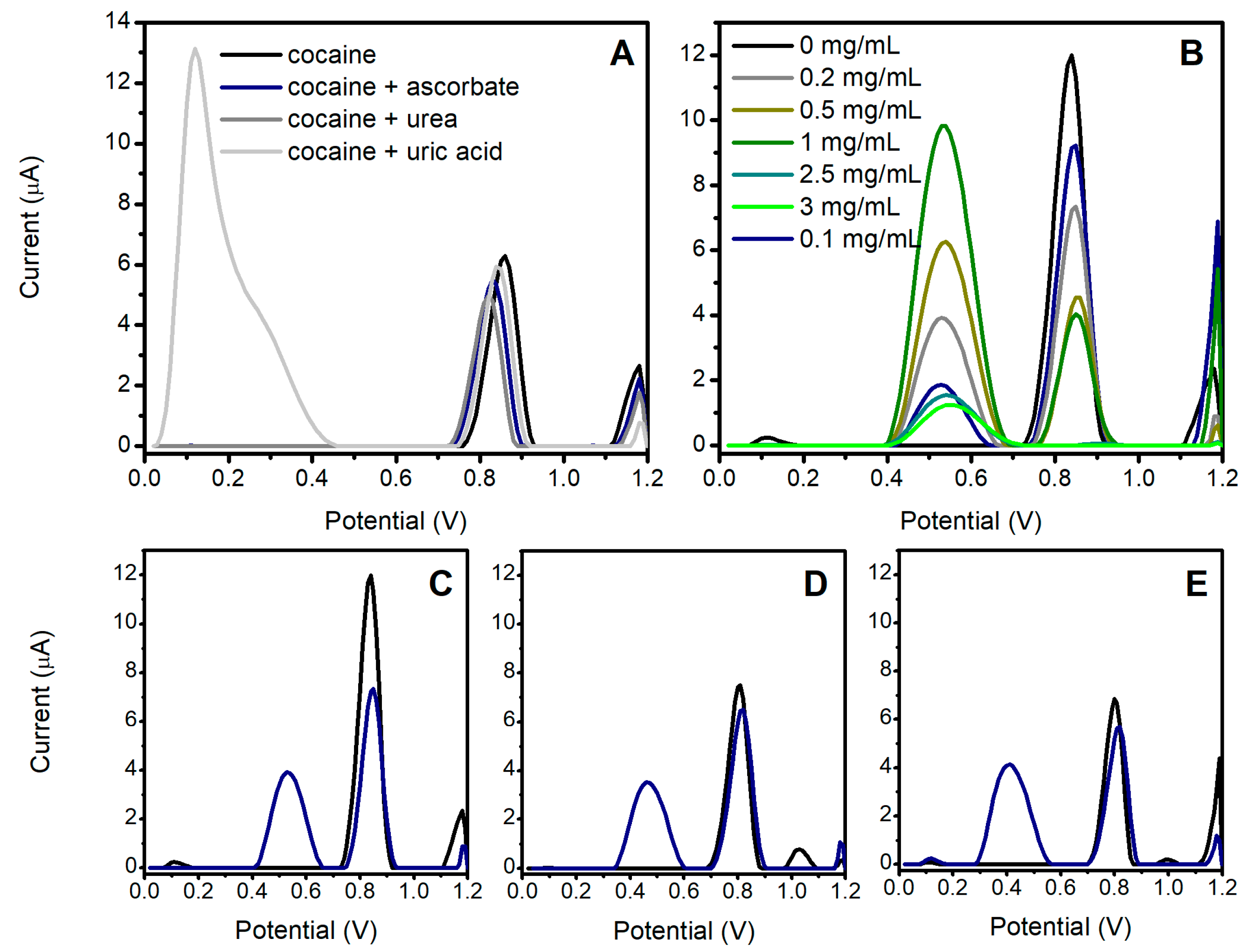
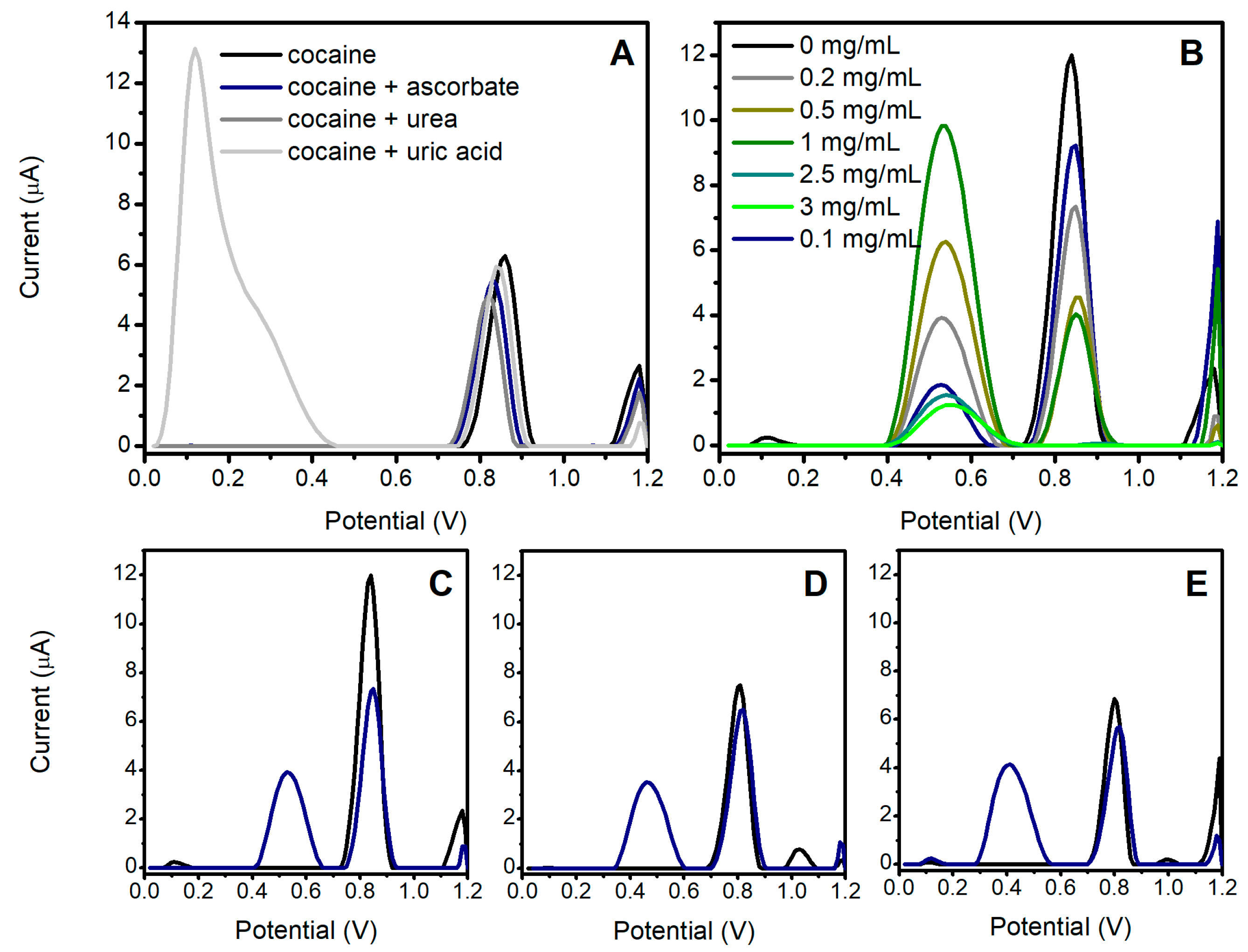
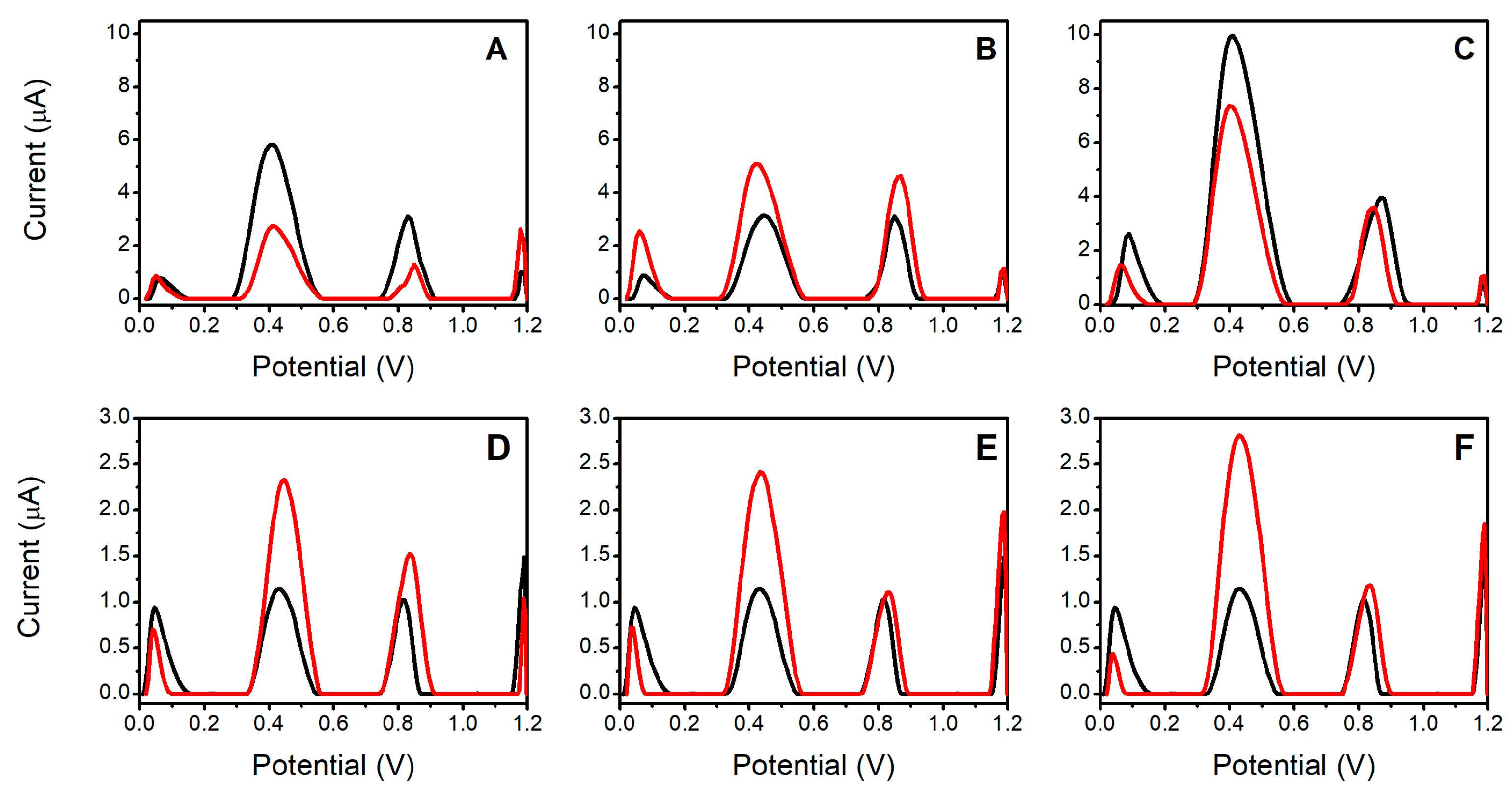
3.2. Study of the OF Matrix Effects
3.3. Investigation of Two OF Collection Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Drug Report 2021; United Nations Office on Drugs and Crime (UNODC): Vienna, Austria, 2021.
- World Drug Report 2020; United Nations Office on Drugs and Crime (UNODC): Vienna, Austria, 2020.
- European Drug Report 2020: Trends and Developments; European Monitoring Centre for Drugs and Drug Addiction (EMCDDA): Lisbon, Portugal, 2020.
- Drug Use and Road Safety; World Health Organization (WHO): Geneva, Switzerland, 2016.
- Schulze, H.; Schumacher, M.; Urmeew, R.; Auerbach, K.; Alvarez, J.; Bernhoft, I.M.; de Gier, H.; Hagenzieker, M.; Houwing, S.; Knoche, A.; et al. Driving Under the Influence of Drugs, Alcohol and Medicines in Europe—Findings from the DRUID Project; Publications Office of the European Union: Luxembourg, 2012. [Google Scholar]
- Scientific Working Group for the Analysis of Seized Drugs (SWGDRUG). Scientific Working Group for the Analysis of Seized Drugs (SWGDRUG) Recommendations. Available online: https://www.swgdrug.org/Documents/SWGDRUG%20Recommendations%20Version%208_FINAL_ForPosting_092919.pdf (accessed on 24 June 2021).
- Ahmed, S.R.; Chand, R.; Kumar, S.; Mittal, N.; Srinivasan, S.; Rajabzadeh, A.R. Recent Biosensing Advances in the Rapid Detection of Illicit Drugs. TrAC Trends Anal. Chem. 2020, 131, 116006. [Google Scholar] [CrossRef]
- Posthuma-Trumpie, G.A.; Korf, J.; Van Amerongen, A. Lateral Flow (Immuno)Assay: Its Strengths, Weaknesses, Opportunities and Threats. A Literature Survey. Anal. Bioanal. Chem. 2009, 393, 569–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrilla, M.; Joosten, F.; De Wael, K. Enhanced Electrochemical Detection of Illicit Drugs in Oral Fluid by the Use of Surfactant-Mediated Solution. Sens. Actuators B Chem. 2021, 348, 130659. [Google Scholar] [CrossRef]
- De Jong, M.; Sleegers, N.; Kim, J.; Van Durme, F.; Samyn, N.; Wang, J.; De Wael, K. Electrochemical Fingerprint of Street Samples for Fast On-Site Screening of Cocaine in Seized Drug Powders. Chem. Sci. 2016, 7, 2364–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, R.G.; Stefano, J.S.; Arantes, I.V.S.; Ribeiro, M.M.A.C.; Santana, M.H.P.; Richter, E.M.; Munoz, R.A.A. Simple Strategy for Selective Determination of Levamisole in Seized Cocaine and Pharmaceutical Samples Using Disposable Screen-Printed Electrodes. Electroanalysis 2019, 31, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Del Vigna de Almeida, P.; Trindade Grégio, A.M.; Naval Machado, M.Â.; Adilson Soares de Lima, A.; Reis Azevedo, L. Saliva Composition and Functions: A Comprehensive Review. J. Contemp. Dent. Pract. 2008, 9, 72–80. [Google Scholar]
- Battino, M.; Ferreiro, M.S.; Gallardo, I.; Newman, H.N.; Bullon, P. The Antioxidant Capacity of Saliva. J. Clin. Periodontol. 2002, 29, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehak, N.N.; Cecco, S.A.; Csako, G. Biochemical Composition and Electrolyte Balance of “unstimulated” Whole Human Saliva. Clin. Chem. Lab. Med. 2000, 38, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.J.; Han, M.; Desroches, P.E.; Manasa, C.S.; Dennaoui, J.; Quigley, A.F.; Kapsa, R.M.I.; Moulton, S.E.; Guijt, R.M.; Greene, G.W.; et al. Antifouling Strategies for Electrochemical Biosensing: Mechanisms and Performance toward Point of Care Based Diagnostic Applications. ACS Sens. 2021, 6, 1482–1507. [Google Scholar] [CrossRef] [PubMed]
- Inzitari, R.; Cabras, T.; Rossetti, D.V.; Fanali, C.; Vitali, A.; Pellegrini, M.; Paludetti, G.; Manni, A.; Giardina, B.; Messana, I.; et al. Detection in Human Saliva of Different Statherin and P-B Fragments and Derivatives. Proteomics 2006, 6, 6370–6379. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, O.; Fleissig, Y.; Zaks, B.; Krief, G.; Aframian, D.J.; Palmon, A. An Approach to Remove Alpha Amylase for Proteomic Analysis of Low Abundance Biomarkers in Human Saliva. Electrophoresis 2008, 29, 4150–4157. [Google Scholar] [CrossRef] [PubMed]
- Shaila, M.; Pai, G.P.; Shetty, P. Salivary Protein Concentration, Flow Rate, Buffer Capacity and PH Estimation: A Comparative Study among Young and Elderly Subjects, Both Normal and with Gingivitis and Periodontitis. J. Indian Soc. Periodontol. 2013, 17, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt University. Amino Acids. Available online: https://www.vanderbilt.edu/AnS/Chemistry/Rizzo/stuff/AA/AminoAcids.html (accessed on 25 June 2021).
- Aframian, D.; Davidowitz, T.; Benoliel, R. The Distribution of Oral Mucosal PH Values in Healthy Saliva Secretors. Oral Dis. 2006, 12, 420–423. [Google Scholar] [CrossRef] [PubMed]
- OraSure Technologies. Oral Fluid Drug Testing. Available online: https://www.orasure.com/products-substance-abuse/i2.html (accessed on 25 June 2021).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joosten, F.; Parrilla, M.; De Wael, K. Electrochemical Detection of Cocaine in Authentic Oral Fluid. Eng. Proc. 2022, 16, 13. https://doi.org/10.3390/IECB2022-12284
Joosten F, Parrilla M, De Wael K. Electrochemical Detection of Cocaine in Authentic Oral Fluid. Engineering Proceedings. 2022; 16(1):13. https://doi.org/10.3390/IECB2022-12284
Chicago/Turabian StyleJoosten, Florine, Marc Parrilla, and Karolien De Wael. 2022. "Electrochemical Detection of Cocaine in Authentic Oral Fluid" Engineering Proceedings 16, no. 1: 13. https://doi.org/10.3390/IECB2022-12284
APA StyleJoosten, F., Parrilla, M., & De Wael, K. (2022). Electrochemical Detection of Cocaine in Authentic Oral Fluid. Engineering Proceedings, 16(1), 13. https://doi.org/10.3390/IECB2022-12284