Abstract
The increasing rates of multidrug-resistant (MDR) and extremely drug-resistant (XDR) cases of tuberculosis (TB) strains are alarming, and eventually hampered the effective control of the pathogenic disease. Epigallocatechin gallate (EGCG) is a major polyphenolic constituent of green tea, which previously demonstrated in vitro potency against TB strains. However, efforts to elucidate the exact mechanism of the interaction are still ongoing. Aiming to elucidate the probable mechanism of its anti-TB action as decaprenylphosphoryl-beta-D-ribose 2′-epimerase (DprE) inhibition, we conducted a molecular modeling analysis. Our molecular docking analysis for a set of 65 bioactive compounds of tea realized that EGCG has the highest binding affinity (docking score: −142.98 kcal/mol) against DPRE (pdb id: 4p8c) from Mycobacterium tuberculosis. Further, a molecular dynamics analysis of 100 ns resulted in extreme stability of the ligand–protein complex. We further assessed the in silico pharmacokinetics and toxicities of the top three green tea polyphenols, based on the docking scores. Our results provide critical insights into the mechanism of action of EGCG and other green tea polyphenols, and their use as potential therapeutic agents (DprE1) against TB.
1. Introduction
Tuberculosis (TB) is considered to be a public health crisis, which is hampering healthcare systems, especially in low-economic countries [1,2,3,4,5]. As per the latest WHO statistics, 1.4 million people died from TB in 2019 (10 million people fell ill) [2]. Furthermore, considering the increasing numbers of multidrug-resistant TB (MDR-TB), or multidrug- or rifampicin-resistant TB (MDR/RR-TB) cases, there is an urgent need to develop newer anti-TB agents with unique mechanisms of action. The mycobacterial cell wall is made up of mycolic acids, which are long fatty acids. The synthesis of mycolic acid is regulated via enzymes of the fatty acid synthase (FAS) complex. Enoyl reductase, pantothenate synthetase, and decaprenylphosphoryl-β-d-ribose 2′-epimerase (DprE1) are key attractive targets for the discovery of newer anti-TB agents. From our literature analysis, it is clear that polyphenols (Figure 1) from the leaves of green tea (GTPs) and black tea (Camellia sinensis) have significant pharmacological potentials against a variety of biological targets [3]. Recently, Anand et al. (2006) showed the potential of GTP, epigallocatechin-3-gallate, to inhibit Mycobacterium tuberculosis survival within human macrophages [5]. Their study suggested that epigallocatechin-3-gallate has the ability to down-regulate tryptophan aspartate-containing coat protein gene transcription [5]. Sun et al. (2015) proposed a probable mechanism for the effects of epigallocatechin gallate (EGCG) on the growth of Mycobacterial smegmatis mc(2)155 [4]. Moreover, their analysis revealed that EGCG had an impact on the integrity of the mycobacterial cell wall. The probable targets of EGCG for the inhibition of Mycobacterium tuberculosis have not been studied in detail, although a few reports have mentioned some antimycobacterial activity based on enzyme inhibition studies utilizing compounds such as triclosan. Considering these facts, we particularly aimed to explore the probable anti-TB drug target for epigallocatechin gallate (EGCG), a major bioactive compound from green tea extracts [6]. Furthermore, we have also calculated docking affinity scores for other GTPs. The top three GTPs were subjected to in silico absorption, distribution, metabolism, excretion (ADME) and toxicity (T) analyses. Finally, the best docked hit, epigallocatechin gallate (EGCG), the 4P8C complex, was simulated and analyzed using 100 ns molecular dynamics analysis.
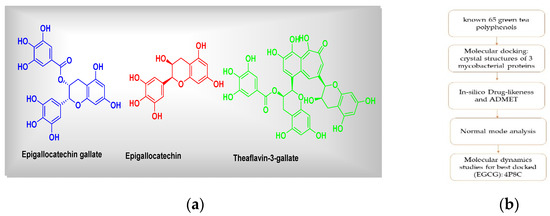
Figure 1.
(a) Chemical structures of green tea polyphenols (representatives) and (b) the graphical summary of workflow followed.
Lastly, we have signified a probable lead that could be developed as a drug candidate against mycobacterial targets.
2. Materials and Methods
2.1. Molecular Docking Simulations
For the current study, we have taken a set of 65 known green tea polyphenols or compounds [7]. Overall, molecular docking comprises the following 5 main steps: (1) protein preparation, (2) ligand preparation, (3) receptor grid generation, (4) ligand docking procedure and then (5) viewing the docking results. All necessary structures have been drawn using ‘ChemSketch’. All the 3D crystal structures of 3 mycobacterial proteins (the enoyl reductase receptor protein (PDB IDs: 2 × 22), decaprenylphosphoryl-β-d-ribose 2′-epimerase (DprE1), 4p8c, and the pantothenate synthetase, 3IVX) were downloaded from the protein database bank (PDB database, www.rcsb.org, accessed on 26 February 2021). The data set including the inbound (co-crystallized) ligand was docked into the binding pocket of pantothenate synthetase and DprE1 (PDB-ID: 3IVX, 4p8c) target enzymes. For the reliability of the docking protocol, we evaluated it through the RMSD value. The grid was centered around active binding site residues Gly158, Met195, Pro38, etc. (for case of 3ivx). We have performed our docking analysis using ‘iGemDock’ as per standard procedures by making 15 Å radius from binding pocket, followed by visualization using Discovery Studio 2020 Visualizer [8,9].
2.2. In Silico Drug Likeness and ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) Analysis
Top 3 higher-affinity GTPs were subjected to an in silico ADME analysis using SWISS tools (http://www.swissadme.ch, accessed on 21 April 2021). For the toxicity assessments, we used ‘admetSAR’ (http://lmmd.ecust.edu.cn:8000/, accessed on 21 April 2021).
2.3. Normal Mode Analysis
To gain more insights into the conformational flexibilities [8] of proteins with their best docked hits, we have performed normal mode analysis (NMA) with internal coordinates (IC) using a fast and easy server, iMODS (http://imods.chaconlab.org/, accessed on 21 April 2021).
2.4. Molecular Dynamics Studies
The docking studies do not take into account the flexible nature of the protein. For confirming the exact binding mode and stability, we have to study MD simulations with the Desmond program. The stability of epigallocatechin gallate (EGCG), 4P8C complex, was evaluated through 100 ns molecular dynamics simulations. The simulations provided the exact binding interaction of the docking complex with system embedded with water molecules, temperature and pressure. The standard NTP conditions were followed for MD simulation setup. The complex was originated in all proper binding poses with an acceptable RMSD value (<3 Å). Molecular dynamics (MD) simulation for a period of 100 ns was carried out for best docked hit with epigallocatechin gallate (EGCG), 4P8C complex, and this was achieved with the GROMACS simulation package, 2020, as per known literature protocols [9].
3. Results and Discussion
3.1. Molecular Docking Simulations
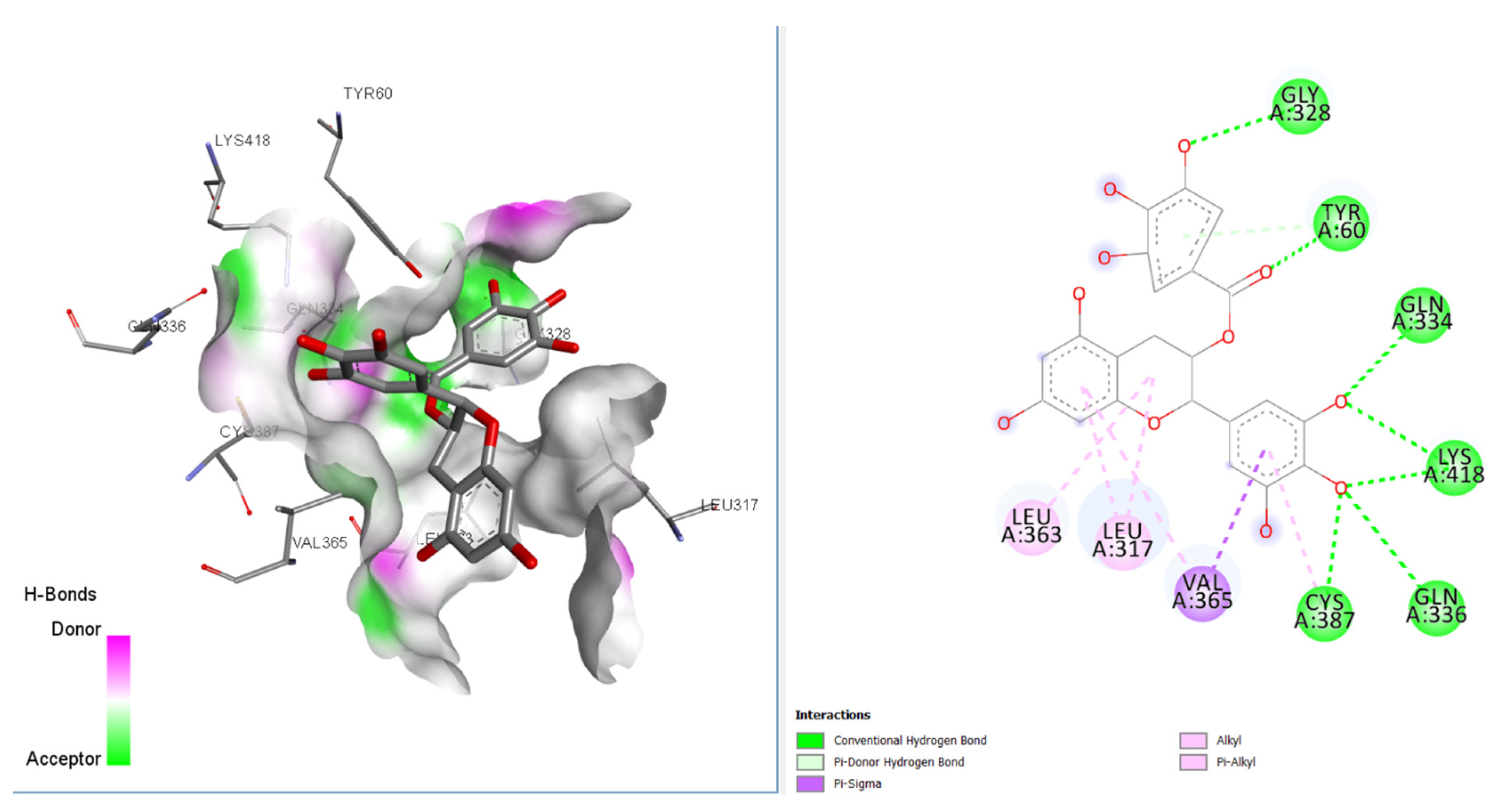
Our molecular docking analysis of 65 GTPs on 3 anti-TB targets suggested that epigallocatechin gallate (EGCG) had the highest affinity towards the DprE1 target, rather than 2X22 and 3IVX. The docking interactions depicted that this compound had interactions with GLN A:328, TYR A:60, GLY A:334, LYS A:418, VAL A:365, and LEU A:317 amino acids, with seven conventional hydrogen bonds present at the receptor site of the target 4P8C (Figure 2) [9,10]. VAL A:365 formed a pi–sigma interaction with the aromatic portion. Alkyl and pi–alkyl interactions were also observed for LEU A:363 and LEU A:317 amino acids. In Figure 2, it is also revealed that the binding mode of EGCG has favorable H-bond donors (purple colored) and acceptable (green colored) regions in the binding cavity. Table 1 and Table 2 give better insights into the interaction profiles of the studied green tea/black tea molecules. From our docking analysis of 65 bioactive compounds of tea on 4p8c, we found the top three best docked hits to be theaflavin-3-gallate (docking score: −124.06 kcal/mol), epigallocatechin gallate (docking score: −142.98 kcal/mol), and epigallocatechin (docking score: −127.73 kcal/mol). The docking affinities for these three bioactive compounds were found to be greater than the standard drug ciprofloxacin* (docking score: −109.20 kcal/mol). The standard drug isoniazid interacted with LYS A:367, VAL A:365, CYS A:387, and ASN A:385 amino acid residues (docking score: −121.21 kcal/mol). The binding cavity residues for the inbound ligands were found to be VAL A: 365, LYS A: 418, ASN A: 385, and LYS A: 418 (salt bridge). Moreover, our re-docking validation protocol also resulted in an RMSD value below 2 Å, suggesting that the docking results are valid.
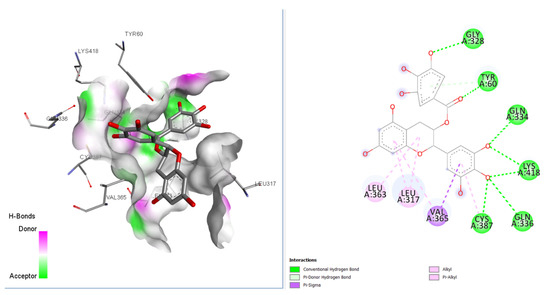
Figure 2.
2D and 3D interaction profiles for best docked epigallocatechin gallate with 4p8c.

Table 1.
Docking interaction energies * of selected 65 bioactive molecules and 3 FDA-approved drugs for target protein 4P8C.
Table 2.
Energy contribution of the key residues computed by docking methodology.
3.2. Molecular Dynamics Simulation and Normal Mode Analysis
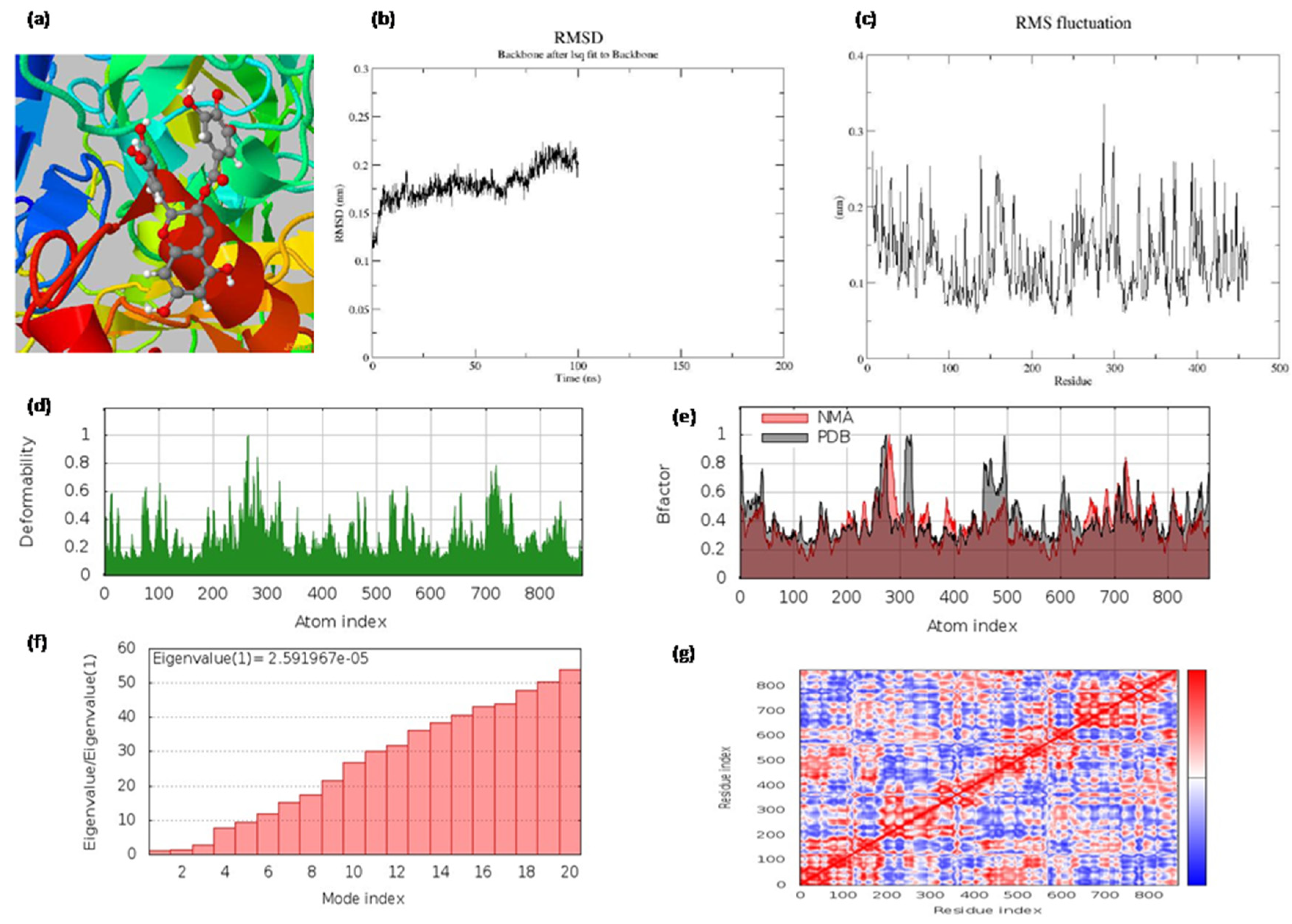
The best docked hit, epigallocatechin gallate (EGCG), with the target 4P8C, was simulated for 100 ns molecular dynamics and normal mode analysis. The MD simulations showed that the obtained root mean square fluctuation (RMSF) values were within tolerable ranges (0.4 nm). The obtained root mean square deviation (RMSD) value was below 0.25 nm, suggesting that the complex was stable (Figure 3). From our NMA results, we observed that epigallocatechin gallate, with the protein 4P8C complex, was retained with good deformability and eigenvalue profiles (Figure 3). The radius of the gyration value was also below 2.15 nm. The accessible surface areas of the solvent were found to be around 180 nm2.
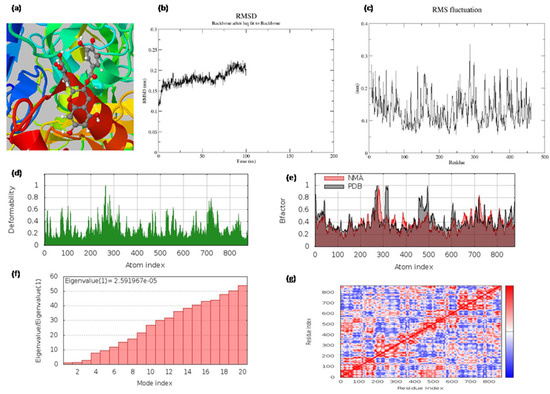
Figure 3.
(a) The binding pocket; (b) the root mean square deviations (RMSD) of backbone atoms relative to the starting complexes during 100 ns MD; (c) protein RMSF plot (on this plot, peaks indicate areas of the protein that fluctuate the most during the simulation, and protein residues that interact with the ligand are marked with green-colored vertical bars); (d) deformability and (e) B-factor profiles; (f) eigenvalue and (g) covariance matrix of the complex for epigallocatechin gallate with 4p8c, respectively.
3.3. In Silico ADMET Studies
Cytochrome P450 (CYPs) enzymes are key enzymes responsible for various factors of metabolism. Our in silico calculated ADMET (absorption, distribution, metabolism, excretion, and toxicity) values, in the body properties evaluated, for the top three best docked hits are represented in Table 3. Three GTPs, theaflavin-3-gallate, epigallocatechin, and epigallocatechin gallate (EGCG), exhibited positive human intestinal absorption profiles, negative blood–brain barrier passage, and non-carcinogenic, non-AMES toxicity, and class IV acute oral toxicity profiles.
Table 3.
In silico ADMET profiling for top 3 best docked hits against target 4P8C.
4. Conclusions
In our current study, we noticed that epigallocatechin gallate (EGCG) has strong interactions with the 4p8c enzyme (docking score: −142.98 kcal/mol), and the following amino acid residues: GLN A:328; TYR A:60; GLY A:334; LYS A:418; VAL A:365; LEU A:317. The binding energy obtained for EGCG was higher when compared with the standard drug ciprofloxacin (docking score: −109.20 kcal/mol). Moreover, in silico ADMET analysis revealed that this compound has low human ether-a-go-go-related gene inhibition, no AMES toxicity, and no carcinogens. Considering a previous literature report on EGCG as an antimycobacterial, we investigated the same against the DprE1 enzyme via molecular docking analysis. Considering the benefits of GTPs, this study may provide further directions to develop more potent anti-TB compounds. Moreover, we also believe that synthetic analogues of EGCG could be tested for in vitro anti-TB potentials. The tea extract containing EGCG could also be tested in vitro for anti-TB enzymatic assessments.
Author Contributions
Conceptualization, S.N.M. and A.P.; methodology, S.N.M.; software, S.N.M.; writing—review and editing, S.N.M. and A.P.; visualization, S.N.M. and A.P.; supervision, A.P. All authors have read and agreed to the published version of the manuscript.
Funding
We wish to thank the Dept. of Pharmaceutical Sciences, Birla Institute of Technology, Mesra, India for financial assistance. Authors are thankful to the team ‘Molegro virtual docker’ and BIOVIA Discovery studio for their free trial licenses. S.M. is also thankful for the provision of IRF (PHD/PH/10006/20) (Ref. No. GO/Estb/Ph.D./IRF/2020-21/2484A) provided by BIT, Mesra, India.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the Head, Department of Pharmaceutical Sciences and Technology, BIT, Mesra for providing the research facilities for performing the current study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Poro, K.E.; Hoekou, Y.; Pissang, P.; Kpabi, I.; Novidzro, K.M.; Dagnra, A.Y.; Tchacondo, T.; Batawila, K. In vitro Antimycobacterial Activity of Selected Medicinal Plants against Mycobacterium tuberculosis. Int. J. Curr. Microbiol. App. Sci. 2021, 10, 3201–3208. [Google Scholar]
- WHO TB Factsheet. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 17 July 2021).
- Paiva, L.; Rego, C.; Lima, E.; Marcone, M.; Baptista, J. Comparative analysis of the polyphenols, caffeine, and antioxidant activities of green tea, white tea, and flowers from Azorean Camellia sinensis varieties affected by different harvested and processing conditions. Antioxidants 2021, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Qin, B.; Gao, M.; Yin, Y.; Wang, C.; Zang, S.; Li, X.; Zhang, C.; Xin, Y.; Jiang, T. Effects of epigallocatechin gallate on the cell-wall structure of Mycobacterial smegmatis mc2155. Nat. Prod. Res. 2015, 29, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.K.; Kaul, D.; Sharma, M. Green tea polyphenol inhibits Mycobacterium tuberculosis survival within human macrophages. Int. J. Biochem. Cell Biol. 2006, 38, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Ramesh, K.V. Epigallocatechin gallate, a green tea polyphenol inhibits Mycobacterium smegmatis: In silico and in vitro studies. Indian J. Pharm. Sci. 2017, 79, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020, 39, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.; PChacón, R. Abagyan. Predictions of Protein Flexibility: First Order Measures. PROTEINS: Structure, Function, and Bioinformatics. Proteins 2004, 56, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Mali, S.N.; Pandey, A. Balanced QSAR and Molecular Modelling to Identify Structural Requirements of Imidazopyridine Analogues as Anti-infective Agents against Trypanosomiases. J. Comput. Biophys. Chem. 2021, 21, 83–114. [Google Scholar] [CrossRef]
- Hsu, K.C.; Chen, Y.F.; Lin, S.R.; Yang, J.M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, S33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).