
A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking †



Abstract
:1. Introduction
2. Methodology
2.1. Dataset
2.2. Protein Preparation
2.3. D-Similarity
2.4. Molecular Docking
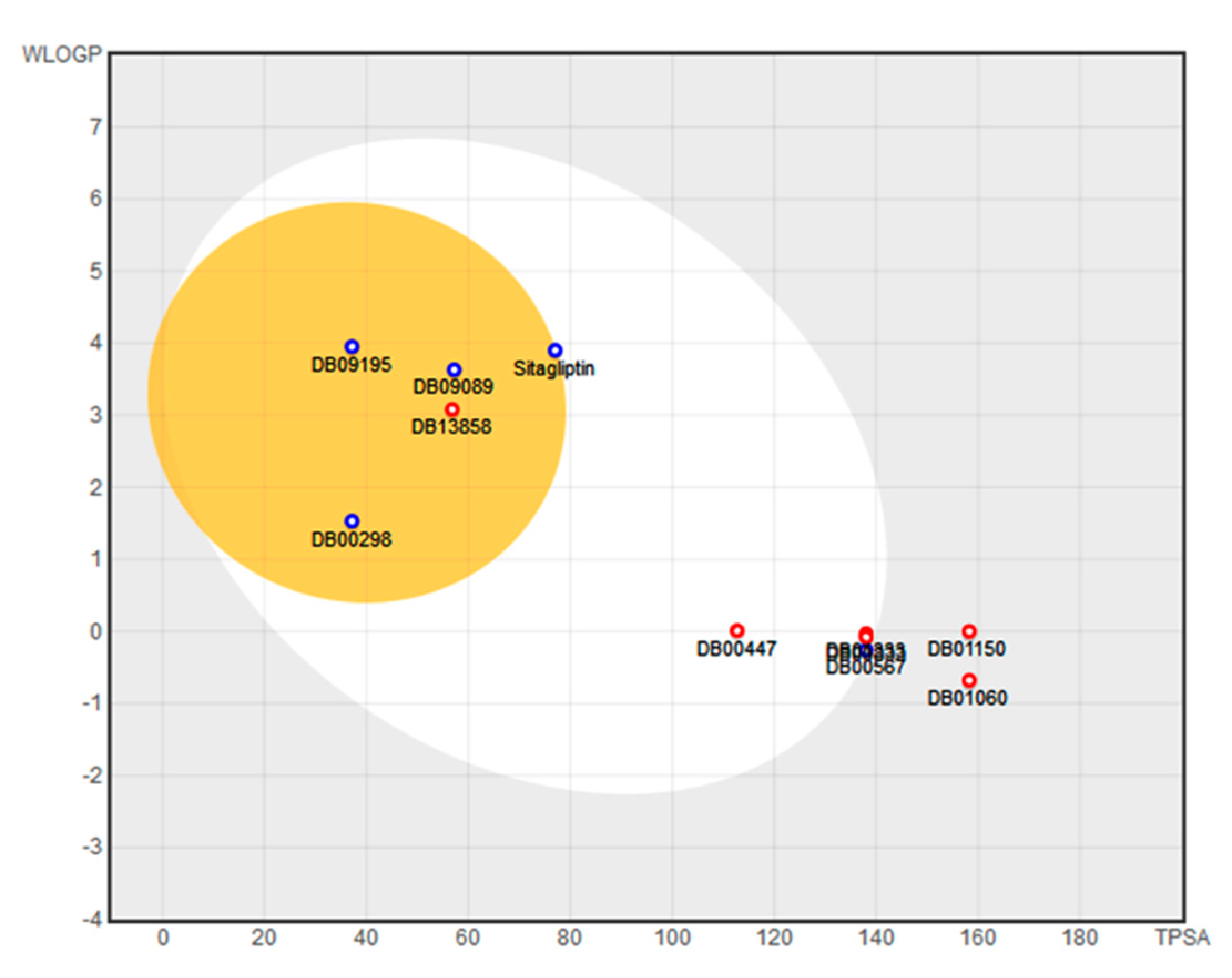
2.5. Pharmacokinetic Profile
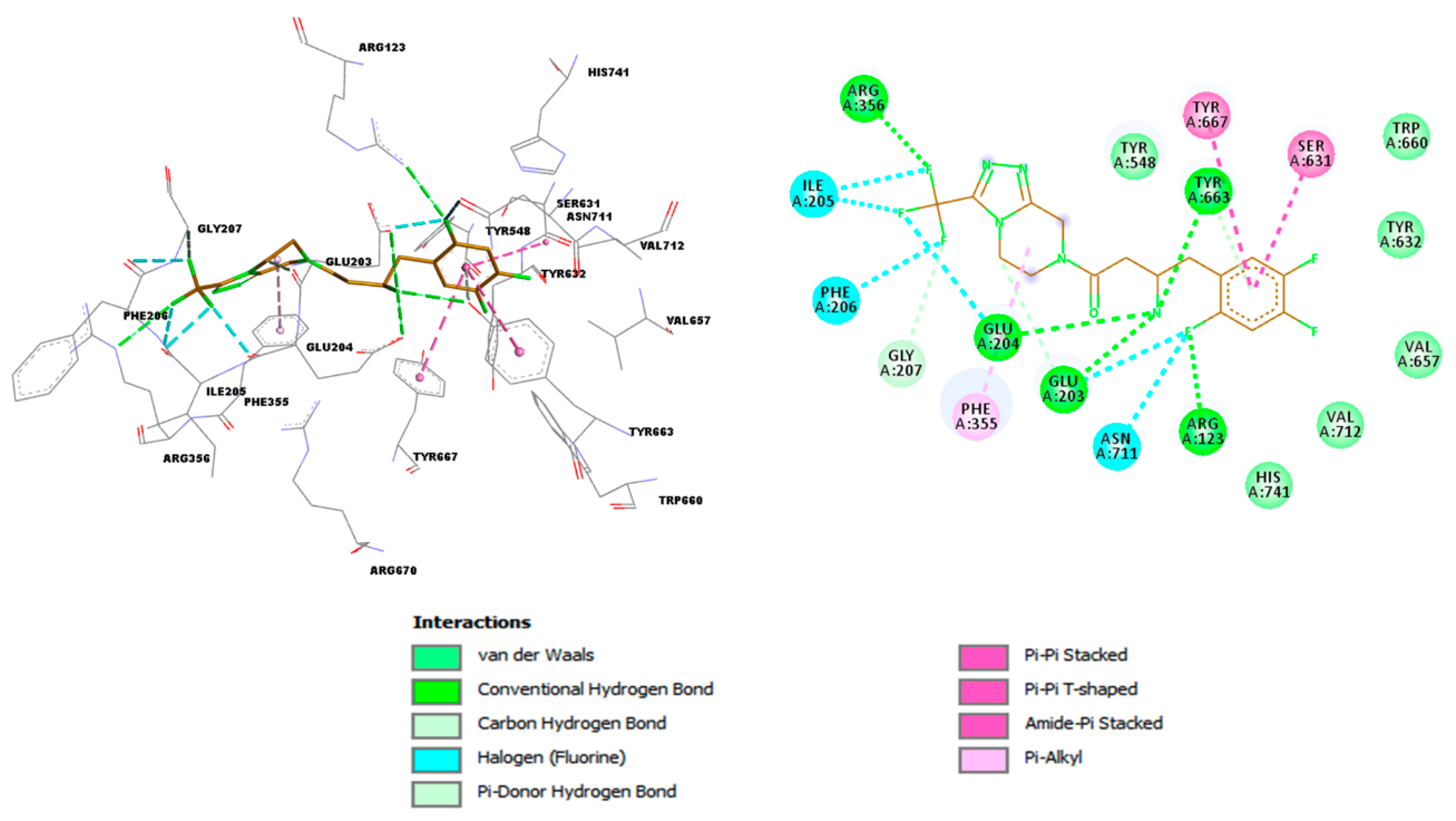
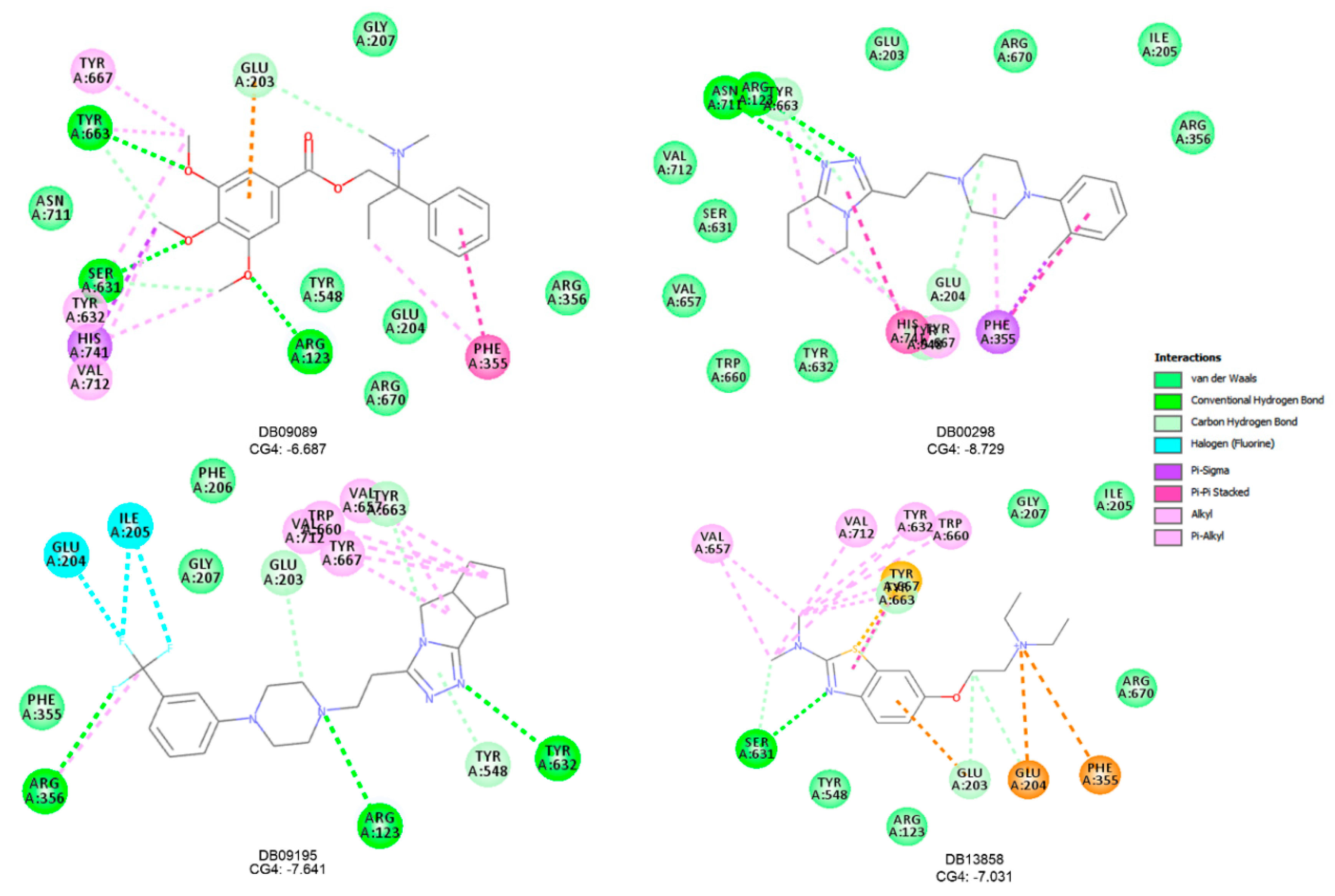
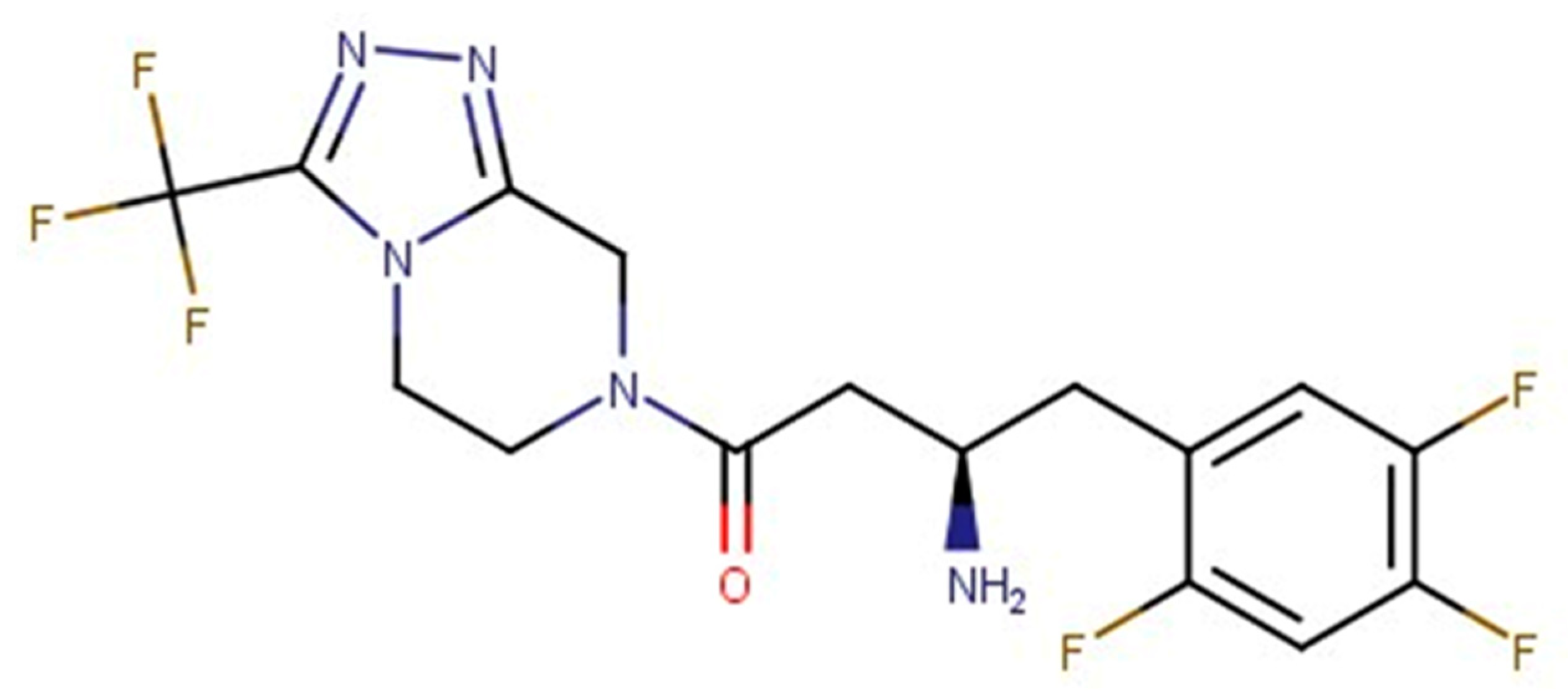
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goyal, R.; Jialal, I. Diabetes Mellitus Type 2. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus-present and future perspectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef]
- Zhu, Y.; Meng, X.; Cai, Z.; Hao, Q.; Zhou, W. Synthesis of phenylpyridine derivatives and their biological evaluation to-ward dipeptidyl peptidase-4. Chem. Heterocycl. Compd. 2017, 53, 350–356. [Google Scholar] [CrossRef]
- Lai, Z.-W.; Li, C.; Liu, J.; Kong, L.; Wen, X.; Sun, H. Discovery of highly potent DPP-4 inhibitors by hybrid compound design based on linagliptin and alogliptin. Eur. J. Med. Chem. 2014, 83, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Mulakayala, N.; Reddy, U.C.H.; Iqbal, J.; Pal, M. Synthesis of dipeptidylpeptidase-4-inhibitors: A brief overview. Tetrahedron 2010, 66, 4919–4938. [Google Scholar] [CrossRef]
- Augustyns, K.; Van der Veken, P.; Senten, K.; Haerners, A. Dipeptidyl peptidase IV inhibitors as new therapeutic agents for the treatment of Type 2 diabetes. Expert Opin. Ther. Patents 2003, 13, 499–510. [Google Scholar] [CrossRef]
- Rosenstock, J.; Brazg, R.; Andryuk, P.J.; Lu, K.; Stein, P. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: A 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin. Ther. 2006, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B.; Simonsson, E.; Larsson, H.; Landin-Olsson, M.; Torgeirsson, H.; Jansson, P.-A.; Sandqvist, M.; Båvenholm, P.; Efendic, S.; Eriksson, J.W.; et al. Inhibition of Dipeptidyl Peptidase IV Improves Metabolic Control Over a 4-Week Study Period in Type 2 Diabetes. Diabetes Care 2002, 25, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Padhy, B.M.; Gupta, Y.K. Drug repositioning: Re-investigating existing drugs for new therapeutic indications. J. Postgrad. Med. 2011, 57, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledge-base for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2016-1: LigPrep; v.3.1; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- OpenEye Scientific Software. OMEGA; v.2.5.1.4; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2013. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Nicholls, A.N. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- RCS Protein Data Bank. Available online: https://www.rcsb.org/structure/4FFW (accessed on 16 March 2021).
- OpenEye Scientific Software. MakeReceptor; v.3.2.0.2; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2015. [Google Scholar]
- OpenEye Scientific Software. ROCS; v. 3.2.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Venhorst, J.; Núñez, S.; Terpstra, J.W.; Kruse, C.G. Assessment of Scaffold Hopping Efficiency by Use of Molecular Interaction Fingerprints. J. Med. Chem. 2008, 51, 3222–3229. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific Software. FRED; v.3.2.0.2; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2015. [Google Scholar]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput.-Aid Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.-F.; Cornell, W.D. Comparison of Topological, Shape, and Docking Methods in Virtual Screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer; v20.1.0, vol19295; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
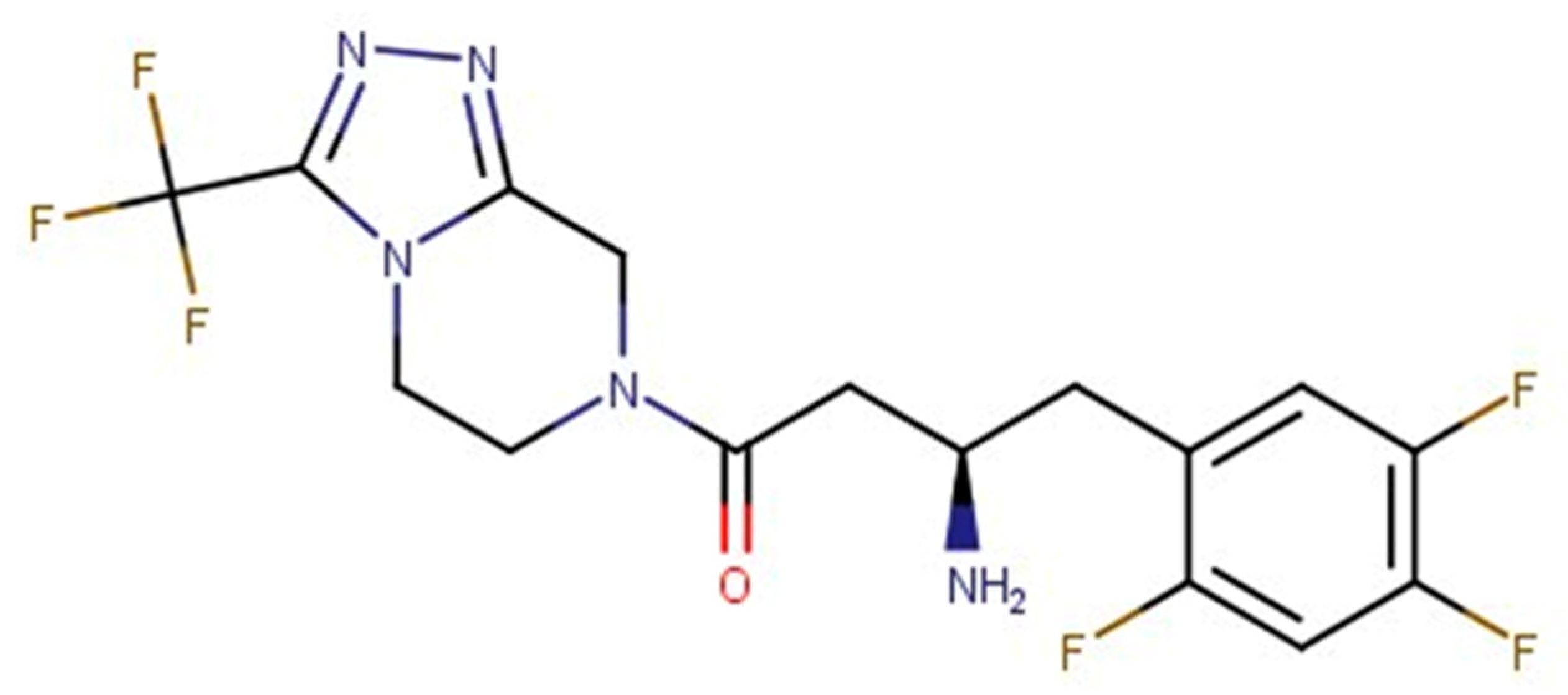
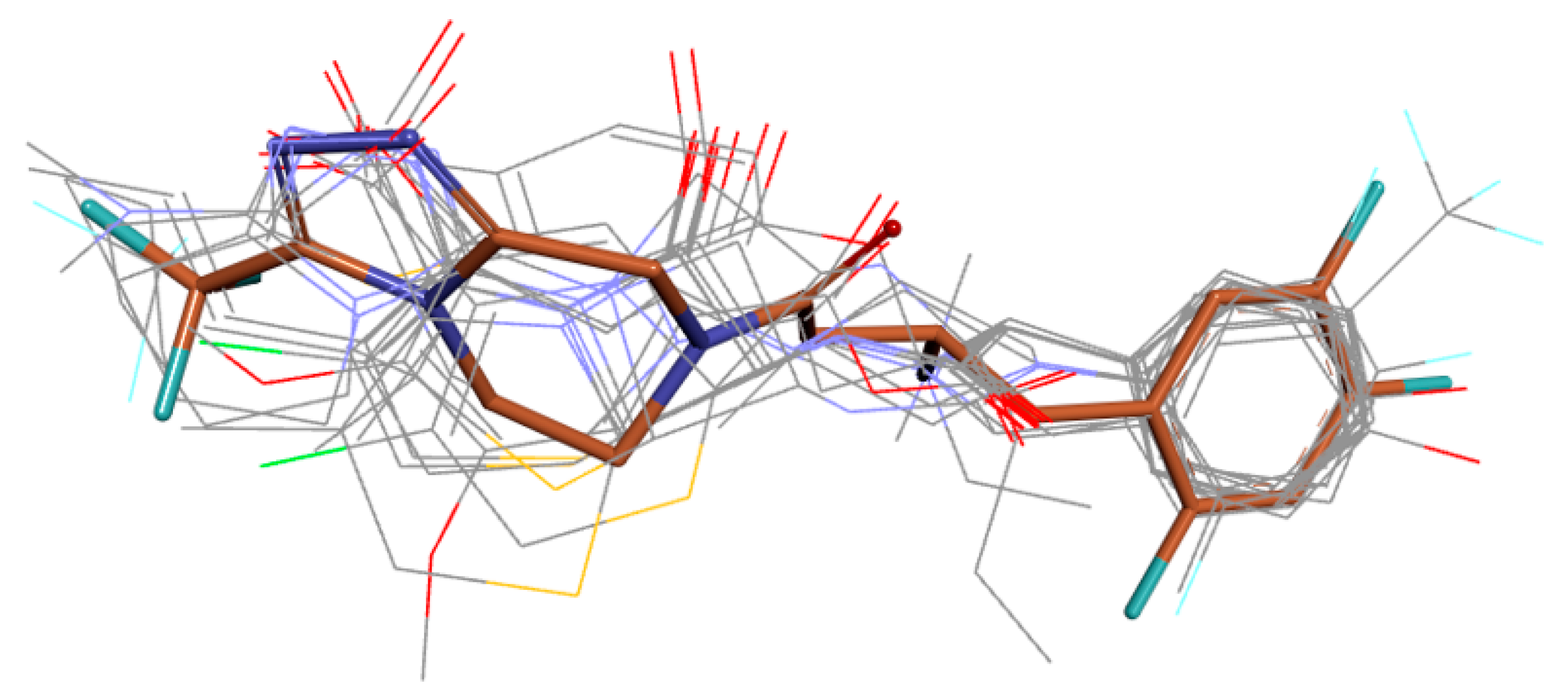
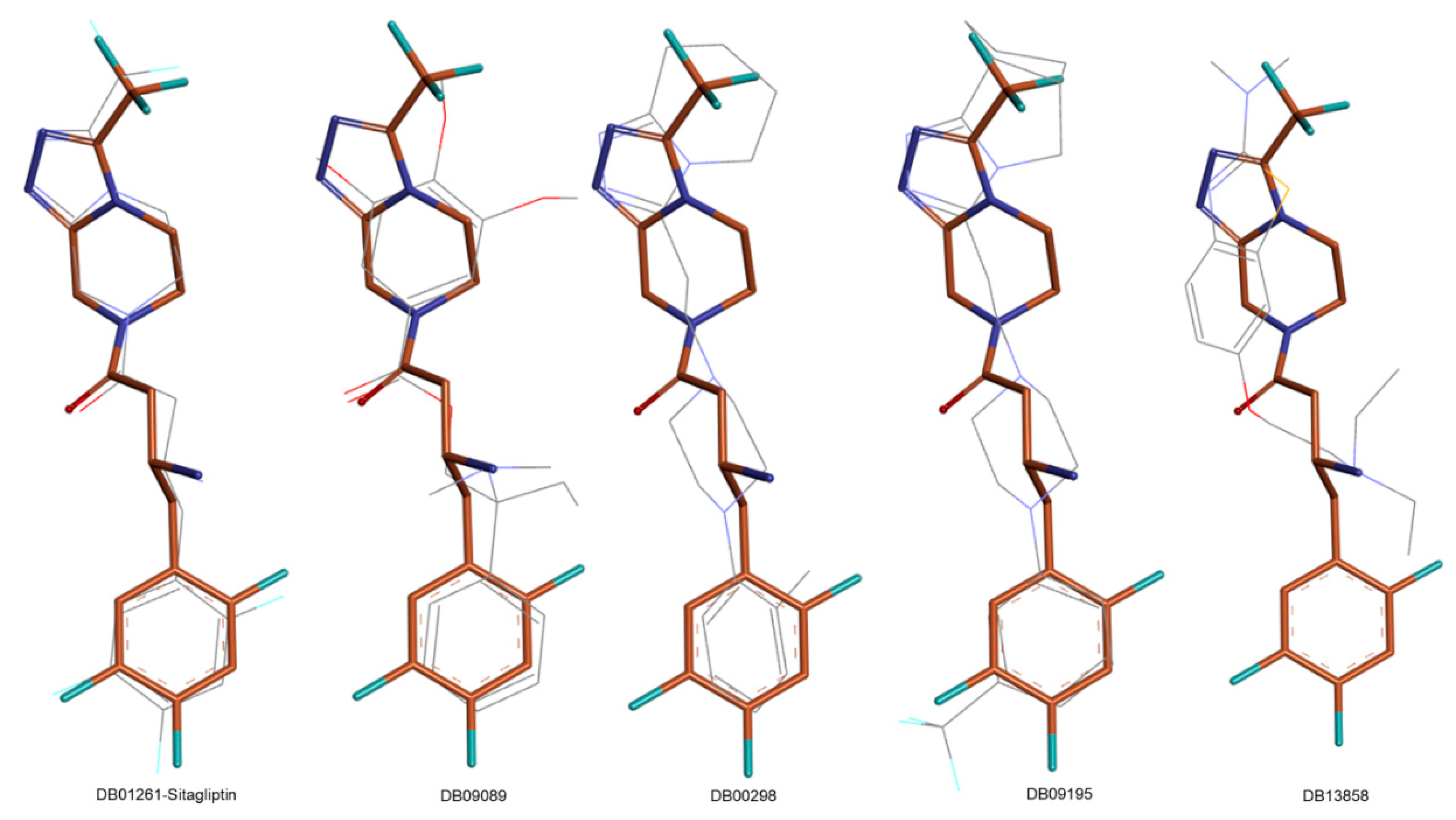
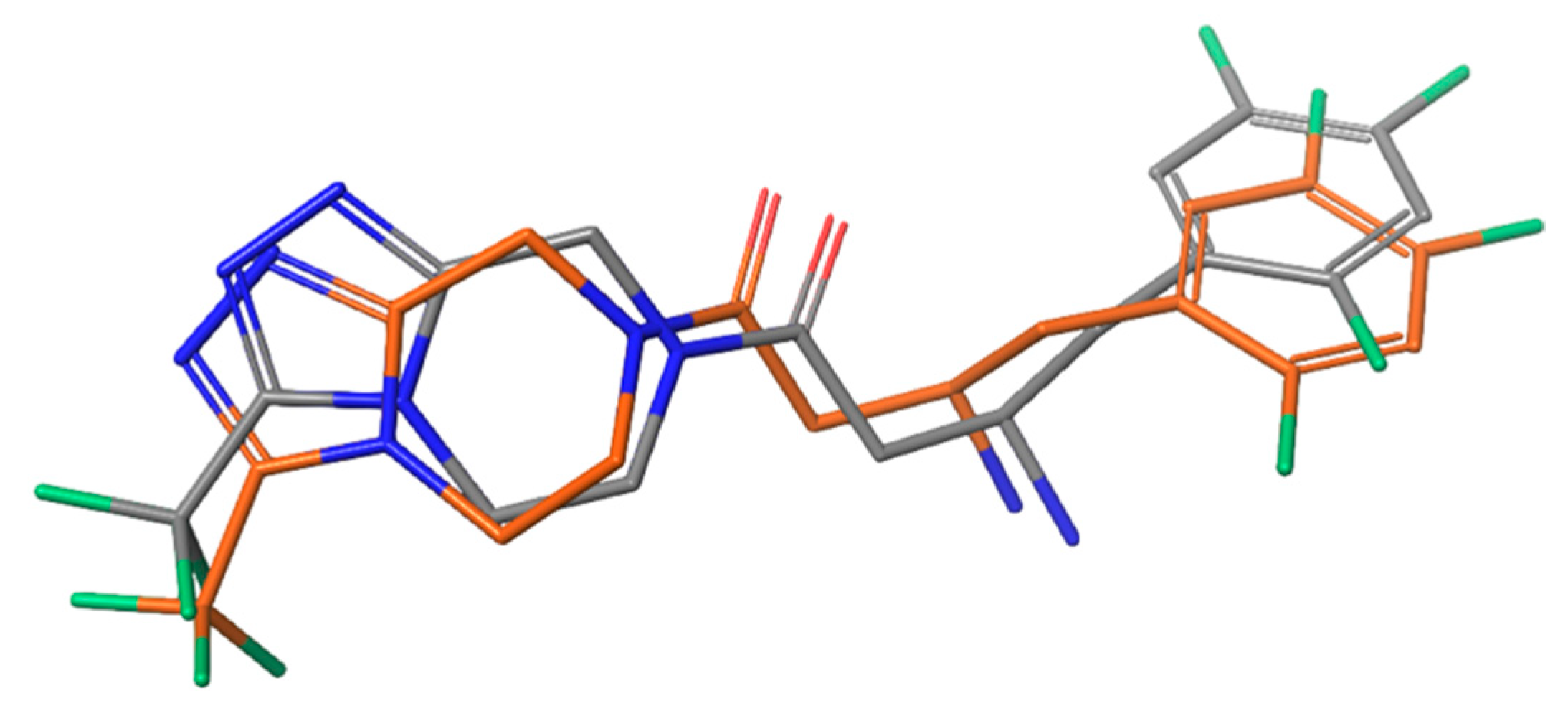

{kind=link}
{kind=link}
{kind=link}
{kind=link}
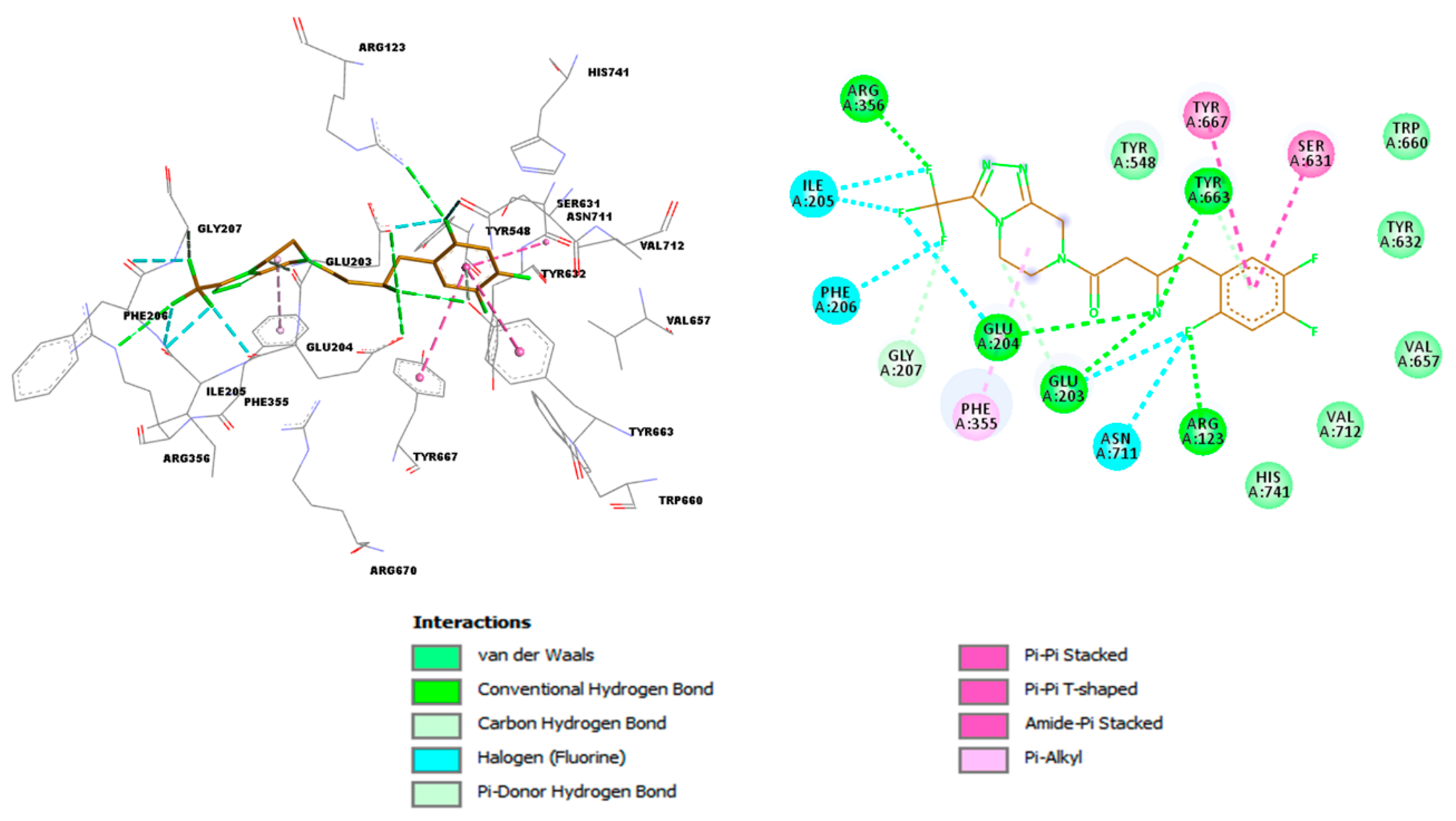{kind=link}
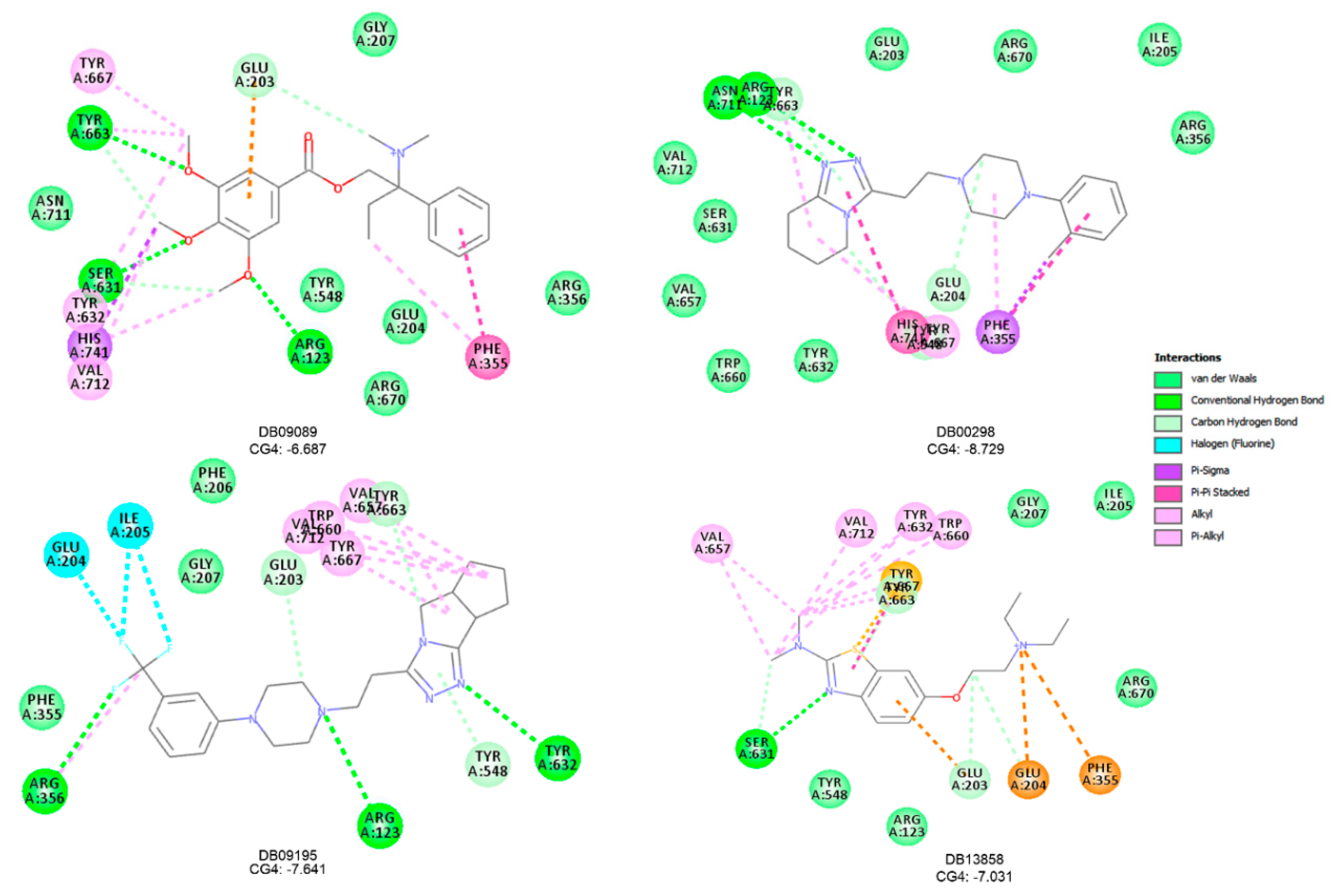{kind=link}
{kind=link}
| Name | TC | ShT | CoT | ScCo | CS | CoS |
|---|---|---|---|---|---|---|
| DB09089 | 1.203 | 0.690 | 0.513 | 0.721 | 1.411 | −5.765 |
| DB00298 | 1.098 | 0.770 | 0.328 | 0.463 | 1.233 | −3.705 |
| DB09195 | 1.068 | 0.765 | 0.303 | 0.466 | 1.231 | −3.725 |
| DB01333 | 1.020 | 0.707 | 0.312 | 0.565 | 1.273 | −4.523 |
| DB00447 | 1.017 | 0.704 | 0.313 | 0.566 | 1.270 | −4.528 |
| DB00567 | 1.008 | 0.670 | 0.339 | 0.601 | 1.270 | −4.805 |
| DB13858 | 1.004 | 0.450 | 0.554 | 0.624 | 1.074 | −4.993 |
| DB00833 | 1.000 | 0.677 | 0.322 | 0.579 | 1.257 | −4.633 |
| DB01150 | 1.000 | 0.748 | 0.252 | 0.553 | 1.301 | −4.427 |
| DB01060 | 0.995 | 0.718 | 0.277 | 0.623 | 1.342 | −4.988 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Istrate, D.; Bora, A.; Crisan, L. A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking. Chem. Proc. 2021, 3, 7. https://doi.org/10.3390/ecsoc-24-08368
Istrate D, Bora A, Crisan L. A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking. Chemistry Proceedings. 2021; 3(1):7. https://doi.org/10.3390/ecsoc-24-08368
Chicago/Turabian StyleIstrate, Daniela, Alina Bora, and Luminita Crisan. 2021. "A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking" Chemistry Proceedings 3, no. 1: 7. https://doi.org/10.3390/ecsoc-24-08368
APA StyleIstrate, D., Bora, A., & Crisan, L. (2021). A First Attempt to Identify Repurposable Drugs for Type 2 Diabetes: 3D-Similarity Search and Molecular Docking. Chemistry Proceedings, 3(1), 7. https://doi.org/10.3390/ecsoc-24-08368