
Rational Design, Synthesis, and In-Silico Evaluation of Homologous Local Anesthetic Compounds as TASK-1 Channel Blockers †

 , and
, and
Abstract
:1. Introduction
2. Methods
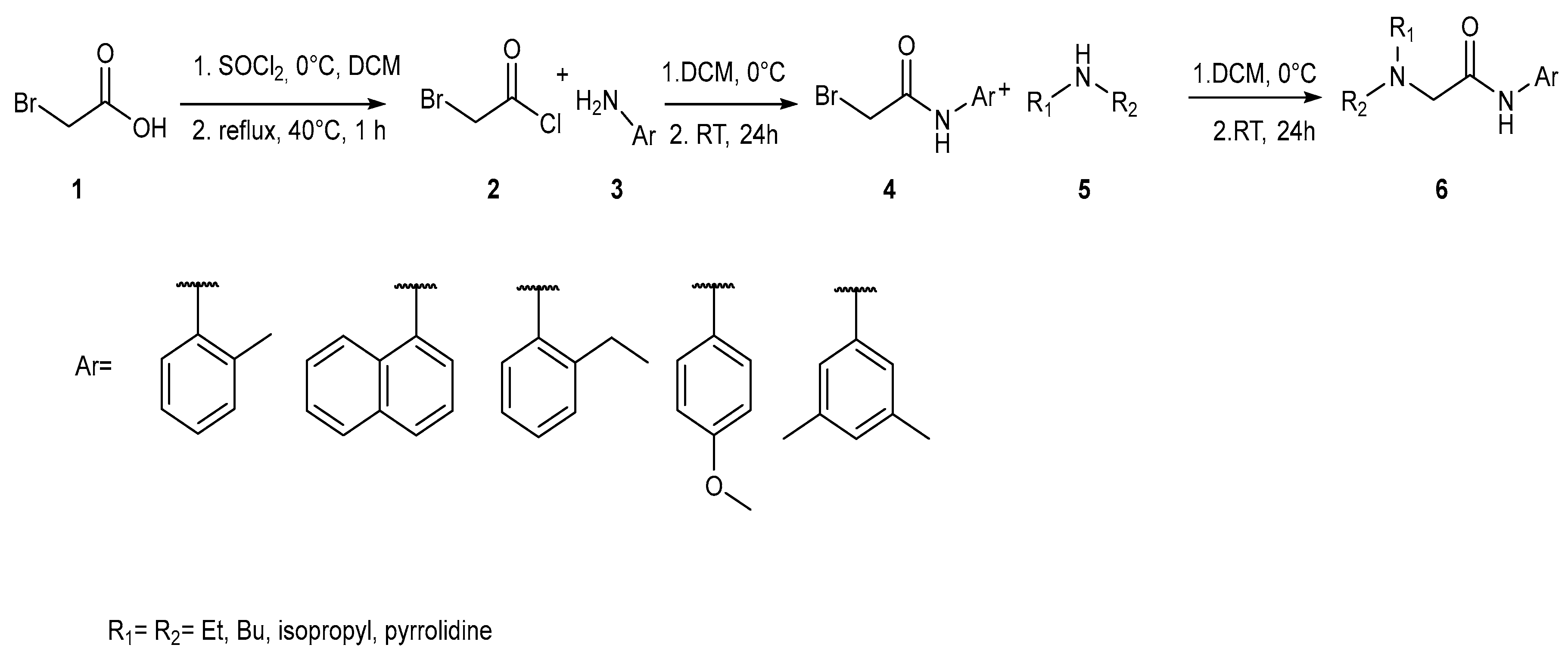
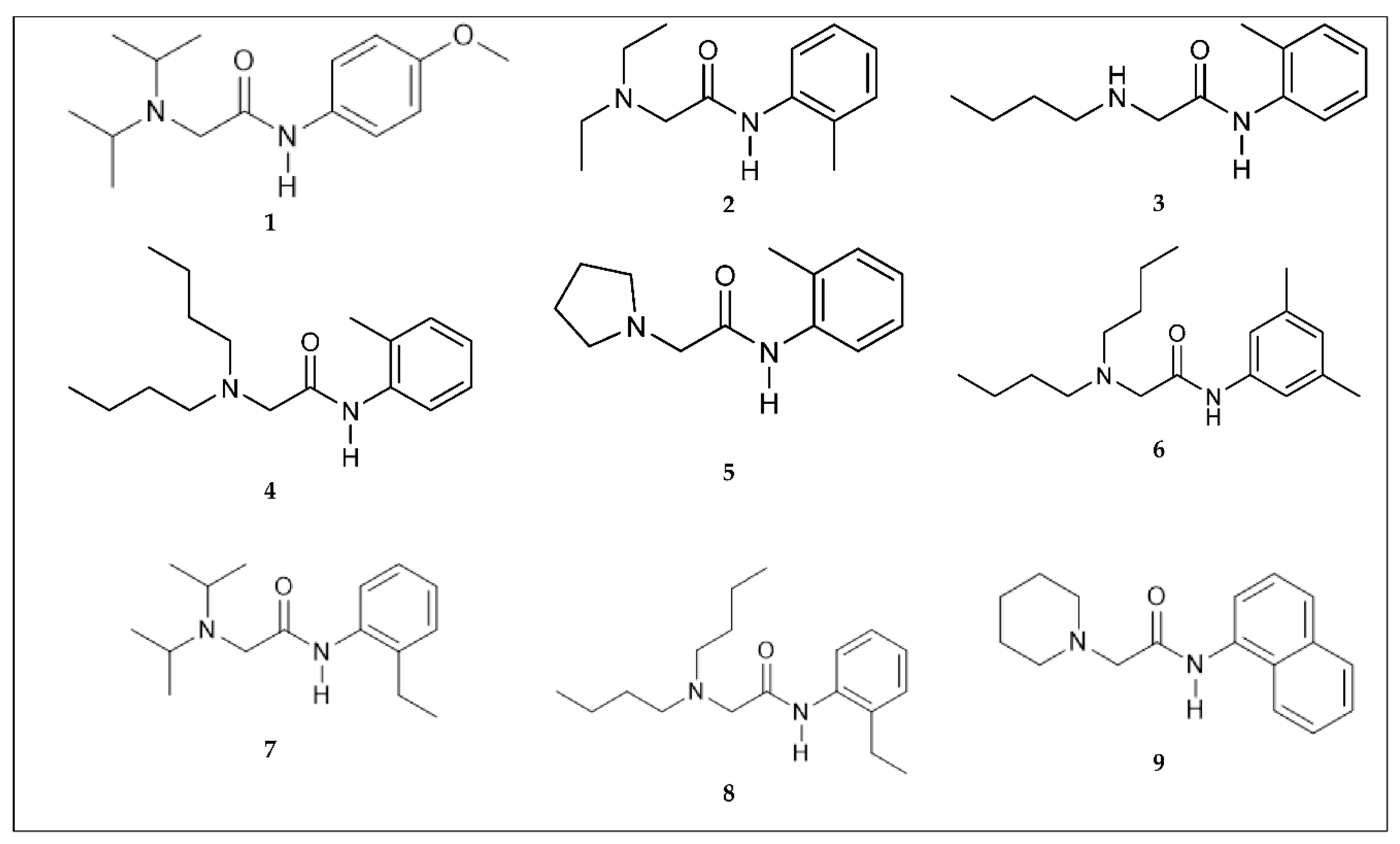
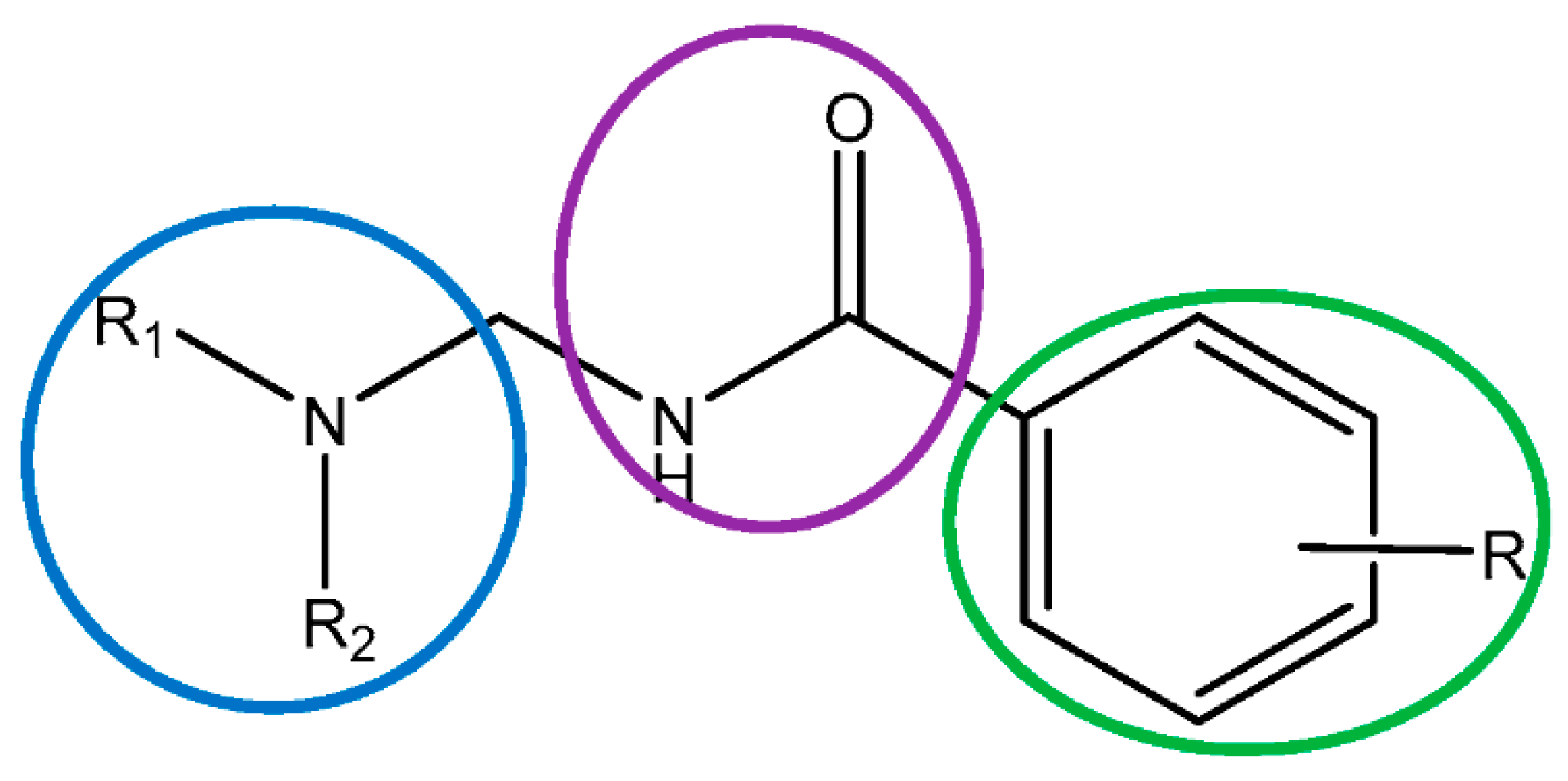
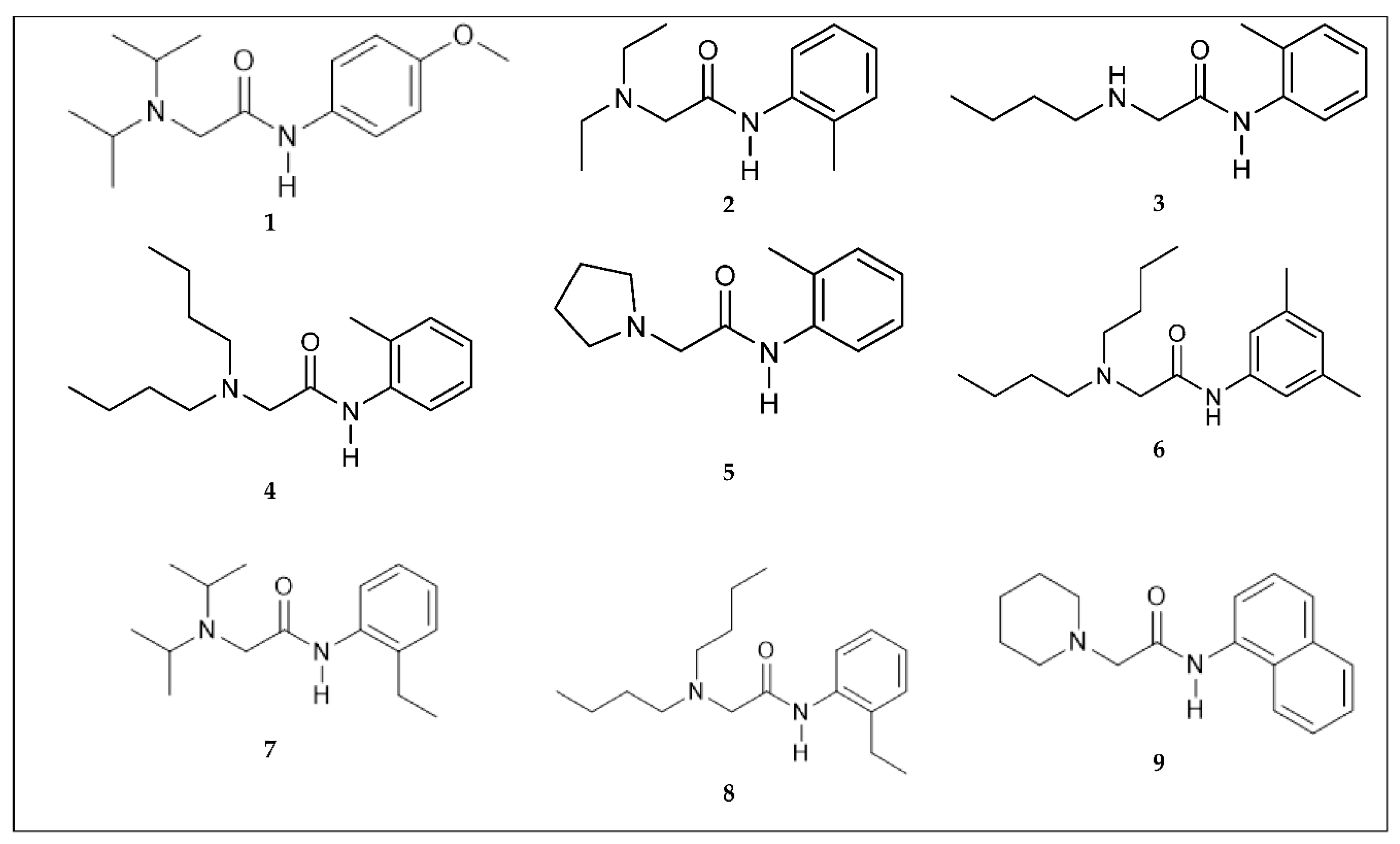
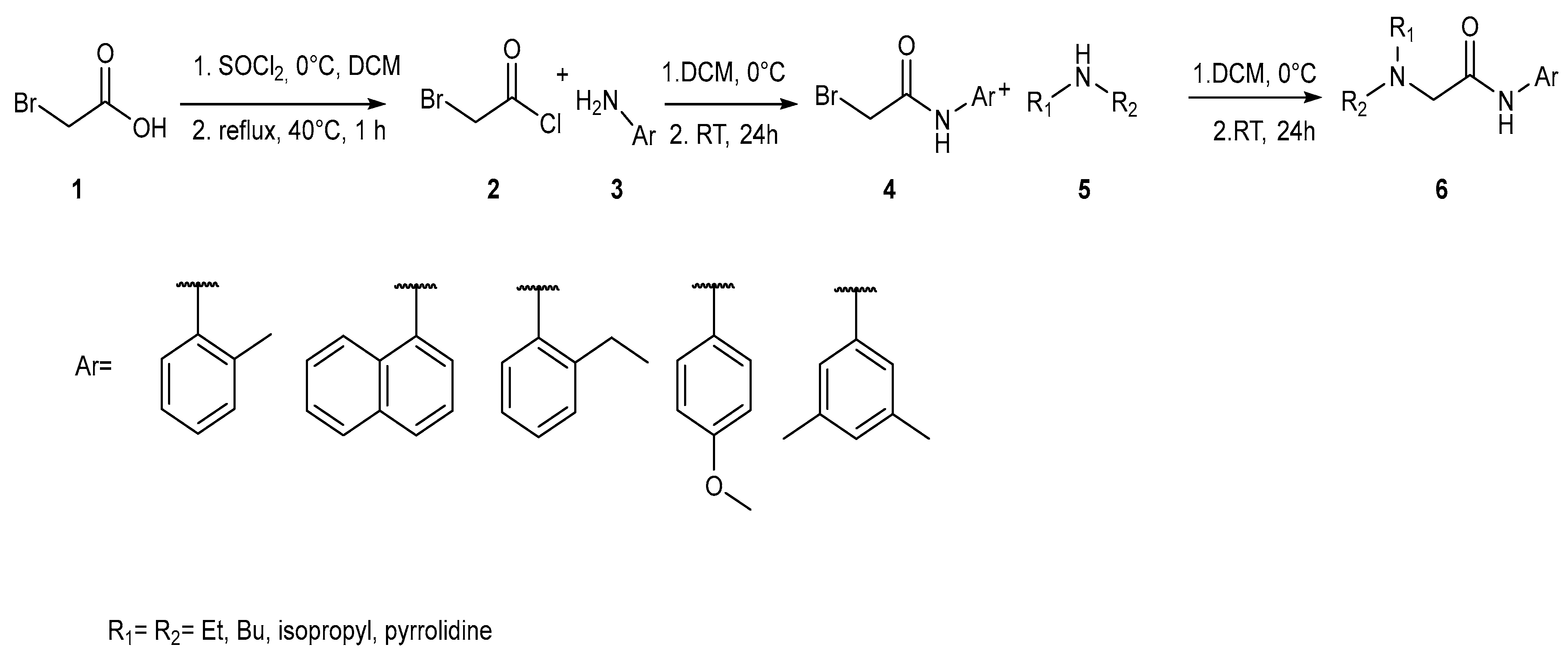
2.1. LAs Peers Design Phase
2.2. Synthesis Phase
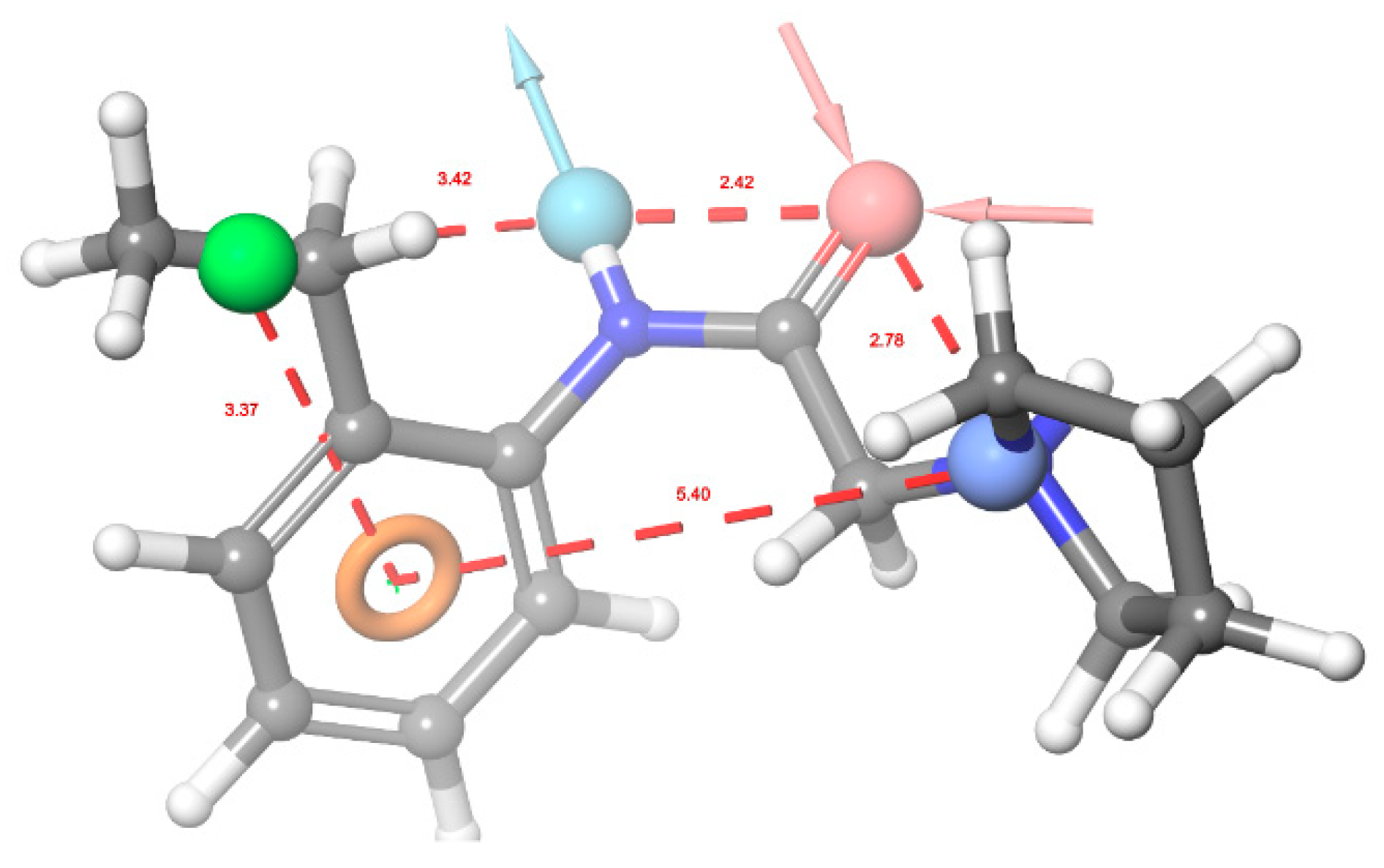
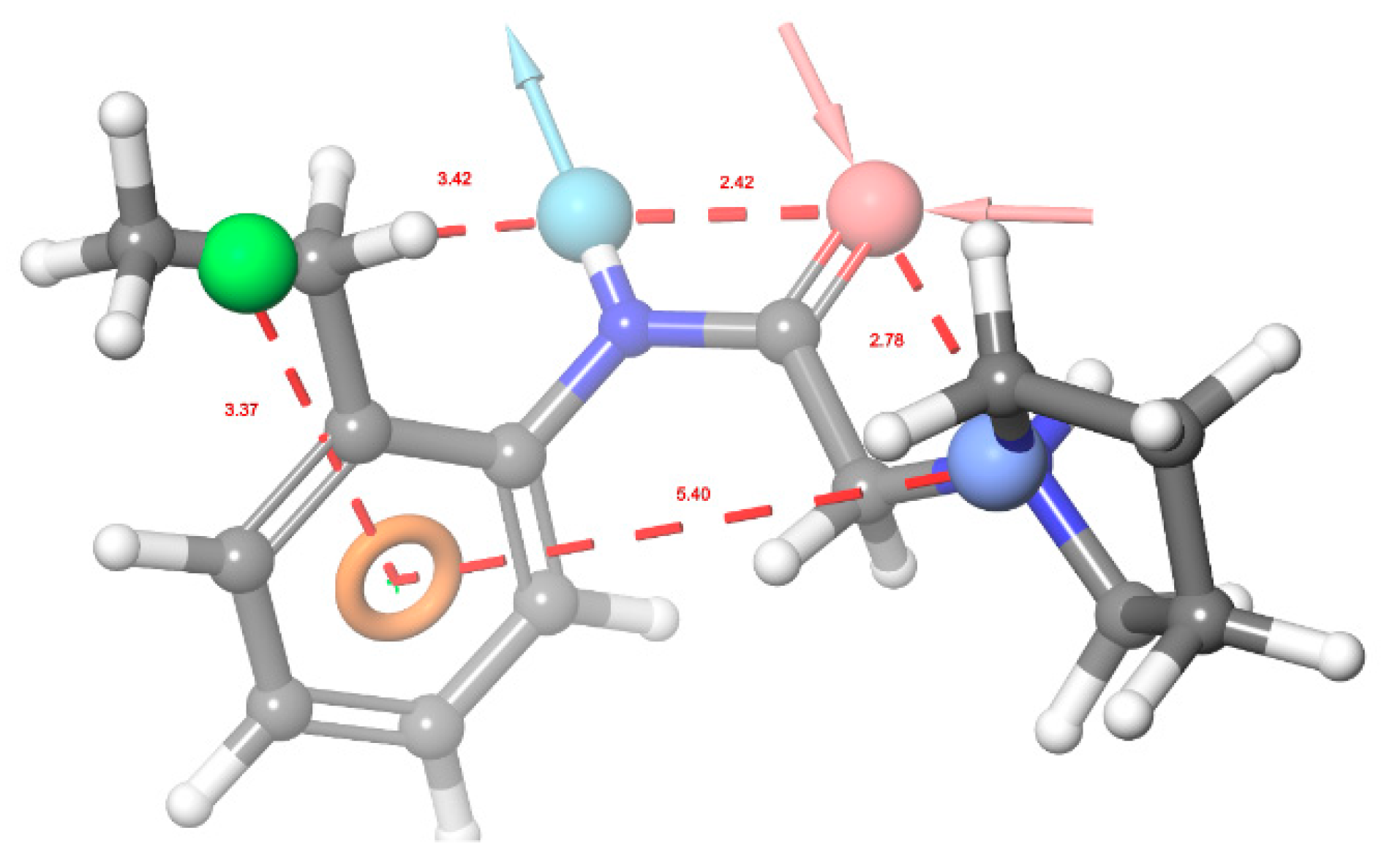
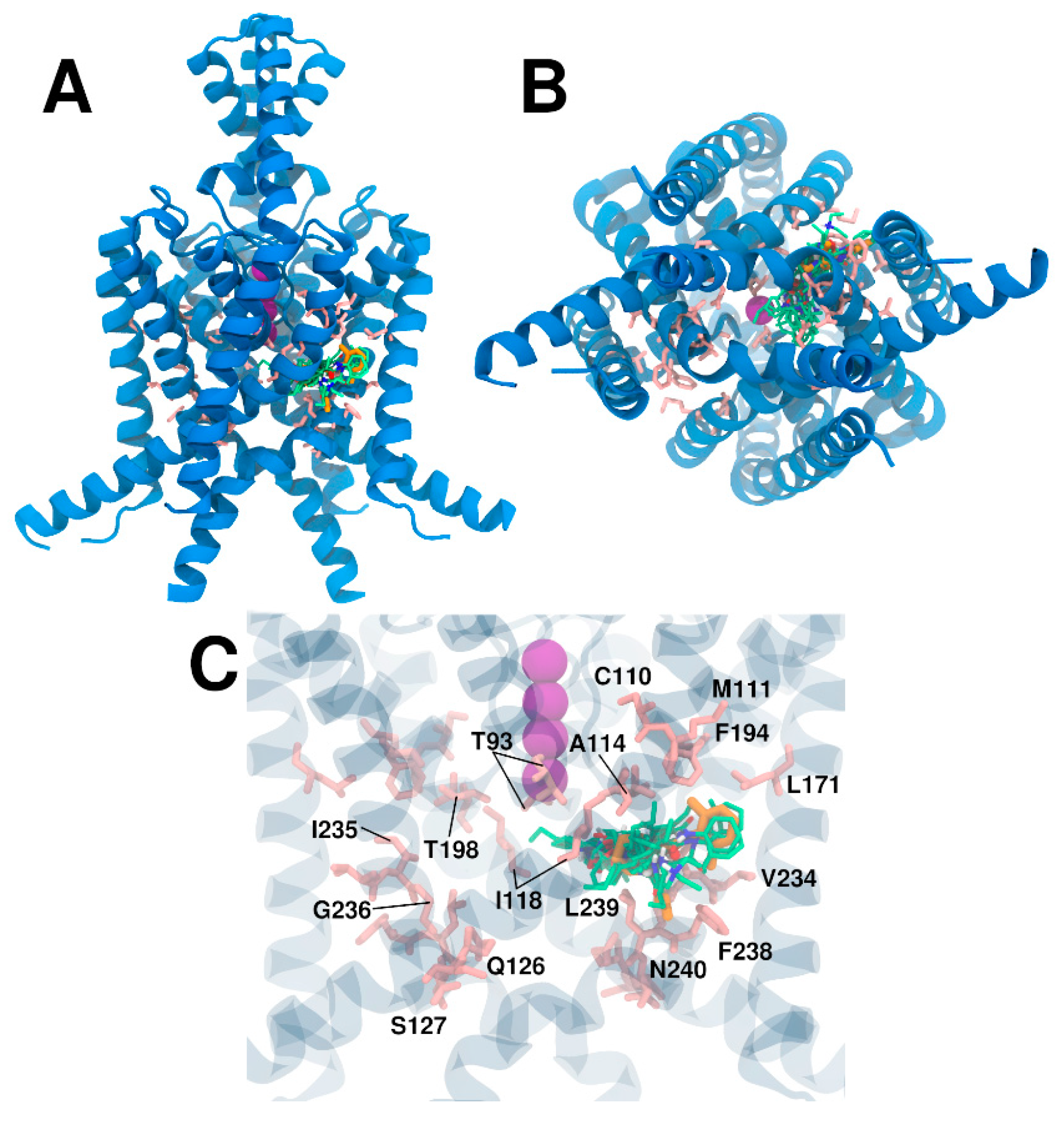
2.3. In Silico Evaluation Phase
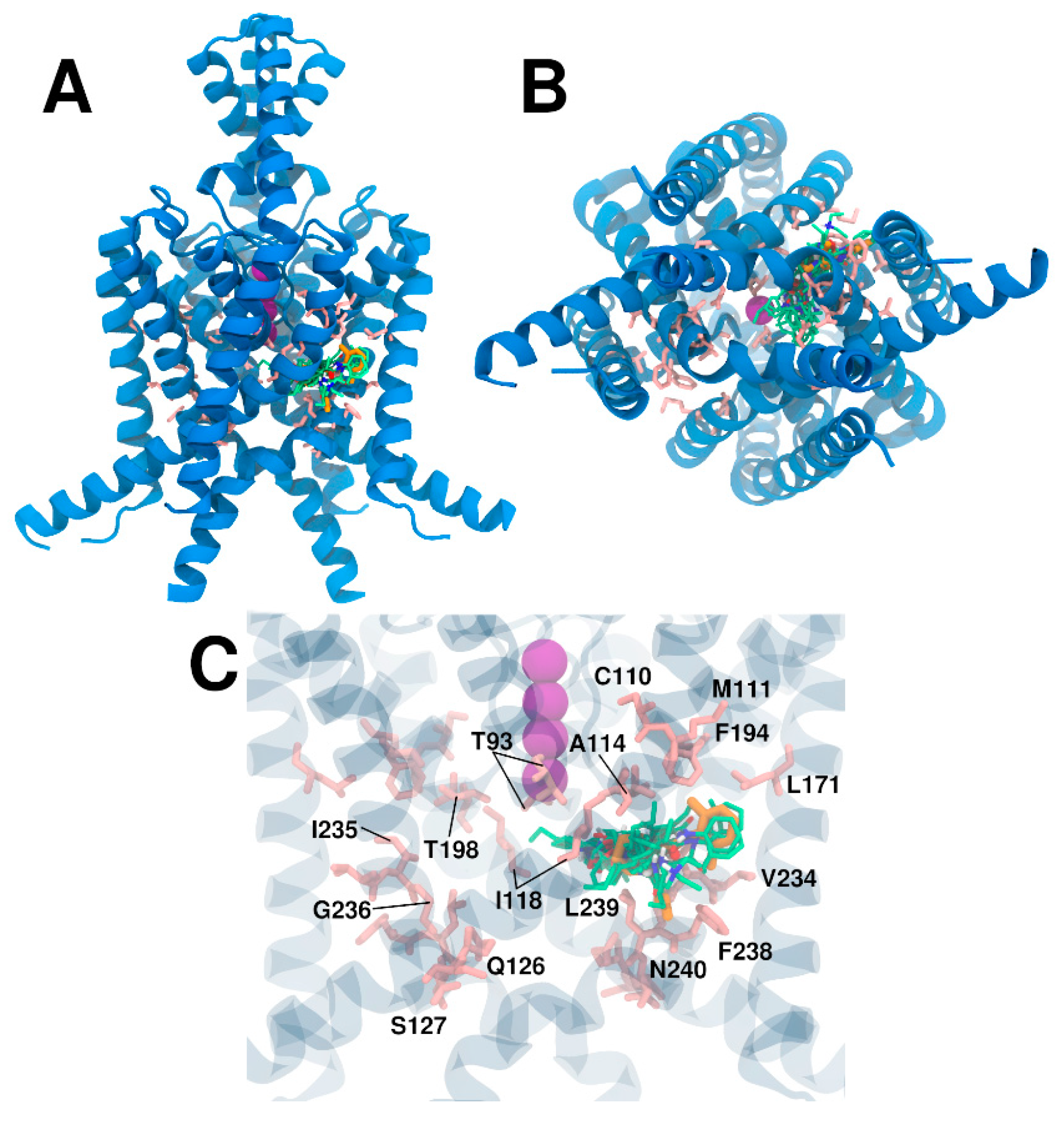
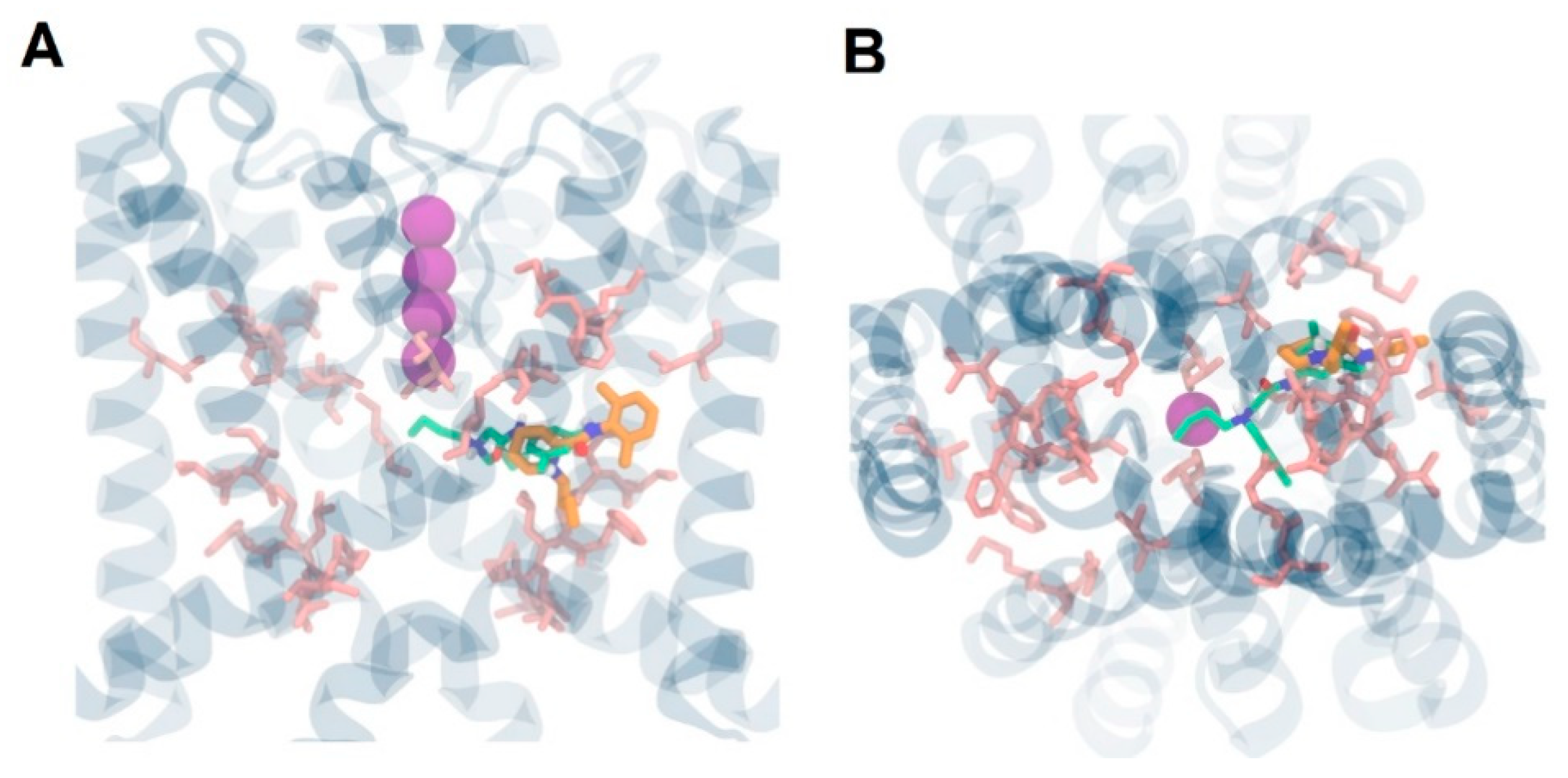
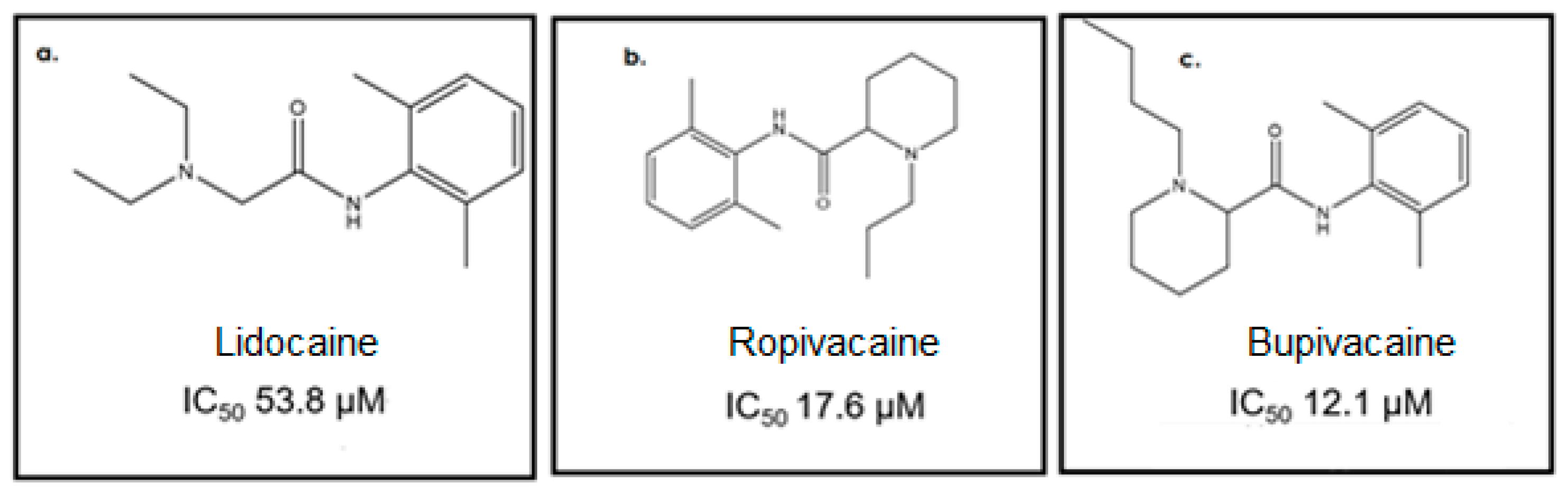
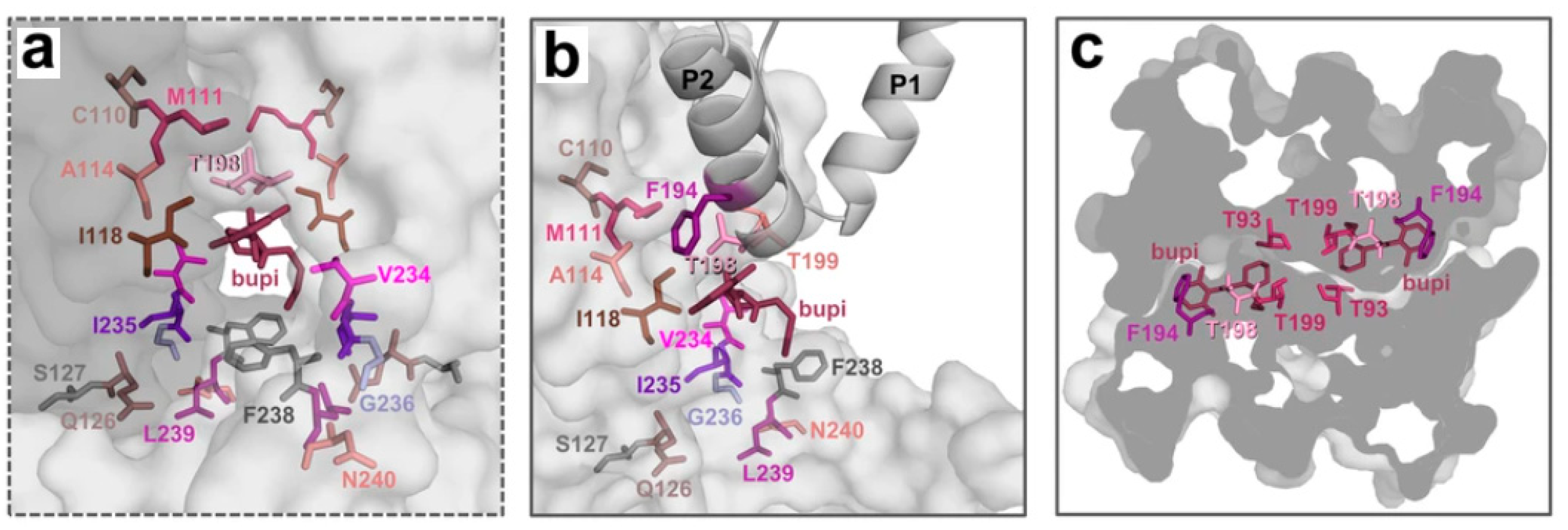
3. Results and Discussion
4. Conclusions
Acknowledgments
References
- Lopes, C.M.B.; Rohács, T.; Czirják, G.; Balla, T.; Enyedi, P.; Logothetis, D.E. PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J. Physiol. 2005, 564, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.N.; Long, S.B. Crystal structure of the human two-pore domain potassium channel K2P1. Science 2012, 335, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirják, G.; Enyedi, P.; et al. TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Kisselbach, J.; Seyler, C.; Schweizer, P.A.; Gerstberger, R.; Becker, R.; Katus, H.A.; Thomas, D. Modulation of K2P 2.1 and K2P 10.1 K(+) channel sensitivity to carvedilol by alternative mRNA translation initiation. Br. J. Pharmacol. 2014, 171, 5182–5194. [Google Scholar] [CrossRef] [PubMed]
- Wiedmann, F.; Kiper, A.K.; Bedoya, M.; Ratte, A.; Rinné, S.; Kraft, M.; Waibel, M.; Anad, P.; Wenzel, W.; González, W.; et al. Identification of the A293 (AVE1231) binding site in the cardiac two-pore-domain potassium channel TASK-1: A common low affinity antiarrhythmic drug binding site. Cell Physiol. Biochem. 2019, 52, 1223–1235. [Google Scholar] [PubMed]
- Calvo, D.; Filgueiras-Rama, D.; Jalife, J. Mechanisms and drug development in atrial fibrillation. Pharmacol. Rev. Am. Soc. Pharmacol. Exp. Ther. 2018, 70, 505–525. [Google Scholar] [CrossRef]
- Schumacher, S.M.; McEwen, D.P.; Zhang, L.; Arendt, K.L.; Van Genderen, K.M.; Martens, J.R. Antiarrhythmic drug-induced internalization of the atrial-specific K+ channel Kv1.5. Circ. Res. 2009, 104, 1390–1398. [Google Scholar] [CrossRef]
- Ravens, U.; Poulet, C.; Wettwer, E.; Knaut, M. Atrial selectivity of antiarrhythmic drugs. J. Physiol. 2013, 591, 4087–4097. [Google Scholar] [CrossRef] [PubMed]
- Ravens, U.; Wettwer, E. Ultra-rapid delayed rectifier channels: Molecular basis and therapeutic implications. In Cardiovascular Research; Oxford Academic: Oxford, UK, 2011; Volume 89, pp. 776–785. [Google Scholar]
- Limberg, S.H.; Netter, M.F.; Rolfes, C.; Rinné, S.; Schlichthörl, G.; Zuzarte, M.; Vassiliou, T.; Moosdorf, R.; Wulf, H.; Daut, J.; et al. TASK-1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell Physiol. Biochem. 2011, 28, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Kiper, A.K.; Rinné, S.; Rolfes, C.; Ramírez, D.; Seebohm, G.; Netter, M.F.; González, W.; Decher, N. Kv1.5 blockers preferentially inhibit TASK-1 channels: TASK-1 as a target against atrial fibrillation and obstructive sleep apnea? Pflugers Arch. Eur. J. Physiol. 2015, 467, 1081–1090. [Google Scholar] [CrossRef]
- Arias, C.; Guizy, M.; David, M.; Marzian, S.; González, T.; Decher, N.; Valenzuela, C. Kvβ1.3 reduces the degree of stereoselective bupivacaine block of Kv1.5 channels. Anesthesiology 2007, 107, 641–651. [Google Scholar] [CrossRef]
- Du, G.; Chen, X.; Todorovic, M.S.; Shu, S.; Kapur, J.; Bayliss, D.A. TASK channel deletion reduces sensitivity to local anesthetic-induced seizures. Anesthesiology 2011, 115, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Longobardo, M.; Delpón, E.; Caballero, R.; Tamargo, J.; Valenzuela, C. Structural determinants of potency and stereoselective block of hKv1.5 channels induced by local anesthetics. Mol. Pharmacol. 1998, 54, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Kiper, A.; Stalke, S.; Marzian, S.; Bedoya, M.; Ramírez, D.; De la Cruz, A.; Peraza, D.A.; Montesinos, J.M.; Ramos, B.A.; Rinné, S.; et al.; Identification of an essential binding site for local anesthetics in the ‘side pockets’ of Kv1 channels Authorea Prepr 2020. Available online: https://www.authorea.com/users/307704/articles/438708-identification-of-an-essential-binding-site-for-local-anesthetics-in-the-side-pockets-of-kv1-channels?commit=53bd58c0cf4a3ec9abcf2b36ee7d1a99bf0ad70c (accessed on 14 September 2020).
- Gonzalez, T.; Longobardo, M.; Caballero, R.; Delpon, E.; Tamargo, J.; Valenzuela, C. Effects of bupivacaine and a novel local anesthetic, IQB-9302, on human cardiac K+ channels. J. Pharmacol. Exp. Therapeut 2001, 296, 573–583. [Google Scholar]
- Valenzuela, C.; Delpón, E.; Tamkun, M.M.; Tamargo, J.; Snyders, D.J. Stereoselective block of a human cardiac potassium channel (Kv1.5) by bupivacaine enantiomers. Biophys. J. 1995, 69, 418–427. [Google Scholar] [CrossRef]
- Kindler, C.H.; Yost, C.S.; Gray, A.T. Local anesthetic inhibition of baseline potassium channels with two pore domains in tandem. Anesthesiology 1999, 90, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Aryal, P.; Abd-Wahab, F.; Bucci, G.; Sansom, M.S.P.; Tucker, S.J. A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Rinné, S.; Kiper, A.K.; Vowinkel, K.S.; Ramírez, D.; Schewe, M.; Bedoya, M.; Aser, D.; Gensler, I.; Netter, M.F.; Stansfeld, P.J.; et al. The molecular basis for an allosteric inhibition of k+-flux gating in k2p channels. Elife 2019, 8, e39476. [Google Scholar] [CrossRef] [PubMed]
- González, W. Proyectos Fondecyt Regular 2019 N°1191133, Talca, Chile, 2019.
- Dror, O.; Shulman-Peleg, A.; Nussinov, R.; Wolfson, H. Predicting Molecular Interactions in silico: I. A Guide to Pharmacophore Identification and its Applications to Drug Design. Curr. Med. Chem. 2005, 11, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D. Pharmacophore-Based Virtual Screening BT—Chemoinformatics and Computational Chemical Biology. In Methods in Molecular Biology; Bajorath, J., Ed.; Humana Press: Clifton, NJ, USA, 2011; pp. 261–298. [Google Scholar] [CrossRef]
- Zhang, H.; Ji, H.; Liu, Z.; Ji, Y.; You, X.; Ding, G.; Cheng, Z. Voltage-dependent blockade by bupivacaine of cardiac sodium channels expressed in Xenopus oocytes. Neurosci. Bull. 2014, 30, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Schwoerer, A.P.; Scheel, H.; Friederich, P. A comparative analysis of bupivacaine and ropivacaine effects on human cardiac SCN5A channels. Anesth. Analg. 2015, 120, 1226–1234. [Google Scholar] [CrossRef]
- Elajnaf, T.; Baptista-Hon, D.T.; Hales, T.G. Potent inactivation-dependent inhibition of adult and neonatal NaV1.5 channels by lidocaine and levobupivacaine. Anesth. Analg. 2018, 127, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, D.V.; Pantsurkin, V.I.; Syropyatov, B.Y.; Kalinina, S.A.; Rudakova, I.P.; Vakhrin, M.I.; Dolzhenko, A.V. Synthesis, local anaesthetic and antiarrhythmic activities of N-alkyl derivatives of proline anilides. Eur. J. Med. Chem. 2013, 63, 144–150. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yu, Z.; Kang, D.; Zhang, J.; Tian, Y.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Design, synthesis and biological evaluation of novel acetamide-substituted doravirine and its prodrugs as potent HIV-1 NNRTIs. In Bioorganic and Medicinal Chemistry; Elsevier Ltd.: Amsterdam, The Netherlands, 2019; Volume 27, pp. 447–456. [Google Scholar]
- Rödström, K.E.; Kiper, A.K.; Zhang, W.; Rinné, S.; Pike, A.C.; Goldstein, M.; Conrad, L.J.; Delbeck, M.; Hahn, M.G.; Meier, H.; et al. A lower X-gate in TASK channels traps inhibitors within the vestibule. Nature 2020, 582, 443–447. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A.; Sheets, M.F.; Hanck, D.A. The sodium channel as a target for local anesthetic drugs. Front. Pharmacol. 2011, 2, 68. [Google Scholar] [CrossRef]
- Liu, L.; Wendt, D.J.; Grant, A.O. Relationship between structure and sodium channel blockade by lidocaine and its amino-alkyl derivatives. J. Cardiovasc. Pharmacol. 1994, 24, 803–812. [Google Scholar] [CrossRef]
- Li, H.L.; Galue, A.; Meadows, L.; Ragsdale, D.S. A molecular basis for the different local anesthetic affinities of resting versus open and inactivated states of the sodium channel. Mol. Pharmacol. 1999, 55, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994, 265, 1724–1728. [Google Scholar] [CrossRef]
- Ramírez, D.; Arévalo, B.; Martínez, G.; Rinné, S.; Sepúlveda, F.V.; Decher, N.; González, W. Side Fenestrations Provide an “anchor” for a Stable Binding of A1899 to the Pore of TASK-1 Potassium Channels. Mol. Pharm. 2017, 14, 2197–2208. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
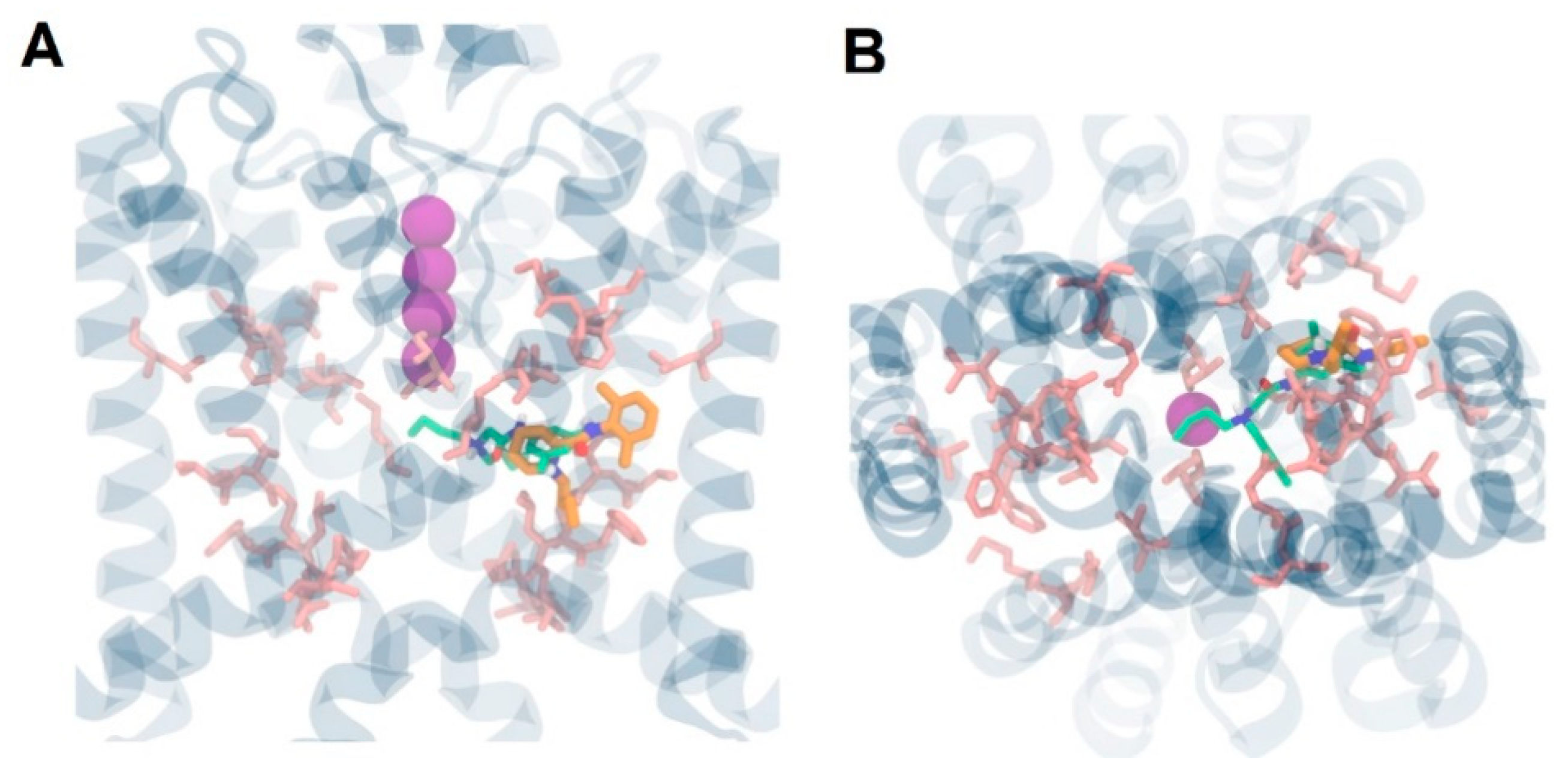{kind=link}
{kind=link}
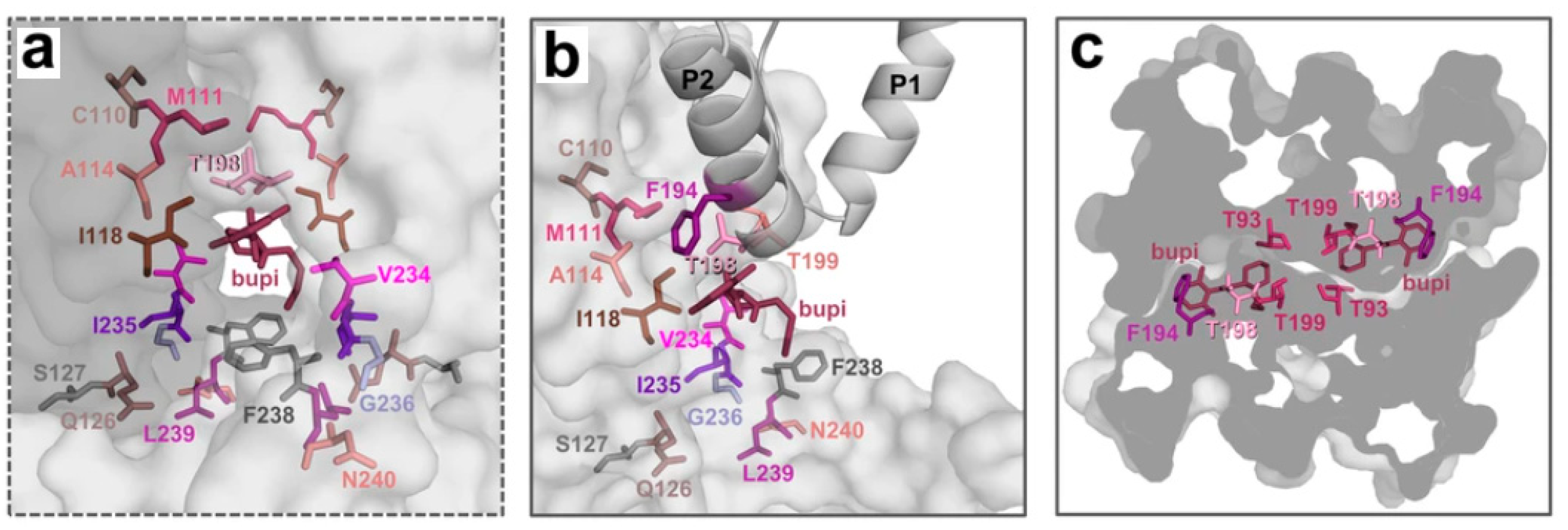
| Compound | IFD Score | Pose | Interactions | Total Interactions | Total EIS |
|---|---|---|---|---|---|
| 6_prot | −1052.16 | 7 | A93, B93, B114, A118, B118, A126, A171, A194, A198, B198, A199, B199, A235, A236, A238, A239 | 16 | 91.85 |
| 6_neut | −1051.13 | 5 | A93, B93, B111, B114, A118, B118, A126, A171, A194, A198, A199, B199, A235, A236, A238, A239 | 16 | 90.45 |
| 9_neut | −1052.34 | 8 | A93, B93, B114, A118, B118, A126, A194, A198, A199, B199, A235, A236, A238, A239 | 14 | 85.42 |
| 8_neut | −1050.38 | 9 | A93, B93, B111, B114, B118, A194, A198, A199, B199, A235, A236, A238, A239 | 13 | 82.14 |
| 5_prot | −1048.26 | 9 | A93, B93, B114, B118, A194, A198, A199, B199, A235, A236, A238, A239 | 12 | 80.17 |
| 1_prot | −1051.92 | 3 | A93, B93, B114, A118, B118, A126, A171, A194, A198, A199, A235, A236, A238, A239 | 14 | 78.26 |
| 3_prot | −1049.29 | 7 | A93, B93, B114, A118, B118, A126, A171, A194, A198, A199, A235, A236, A238, A239 | 14 | 78.26 |
| 4_neut | −1051.27 | 1 | A93, B93, B114, B118, A171, A194, A198, A199, A235, A236, A238, A239 | 12 | 73.01 |
| 3_neut | −1046.00 | 8 | A93, B93, B114, B118, A126, A194, A198, A199, A235, A236, A238, A239 | 12 | 72.98 |
| 5_neut | −1050.12 | 7 | A93, B93, B114, B118, A126, A194, A198, A199, A235, A236, A238, A239 | 12 | 72.98 |
| 7_prot | −1052.30 | 2 | B93, B114, B118, A171, A194, A198, A199, A234, A235, A236, A238, A239 | 12 | 70.60 |
| 2_neut | −1049.67 | 10 | A93, B93, B114, B118, A194, A198, A199, A235, A236, A238, A239 | 11 | 69.94 |
| 9_prot | −1053.36 | 6 | A93, B93, B114, B118, A194, A198, A199, A235, A236, A238, A239 | 11 | 69.94 |
| Bupivacaine | −1051.66 | 1 | B93, B114, B118, A171, A194, A198, B198, A235, A236, A238, A239 | 11 | 62.78 |
| 2_prot | −1050.13 | 9 | B93, B114, B118, A194, A198, A199, A235, A236, A238, A239 | 10 | 59.71 |
| 8_prot | −1051.78 | 4 | B93, B111, B114, B118, A171, A194, A198, A199, A235, A238, A239 | 11 | 59.57 |
| 4_prot | −1050.99 | 1 | B93, B114, B118, A171, A194, A198, A199, A235, A238, A239 | 10 | 57.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camargo-Ayala, L.; Prent-Peñaloza, L.; Bedoya, M.; Gutiérrez, M.; González, W. Rational Design, Synthesis, and In-Silico Evaluation of Homologous Local Anesthetic Compounds as TASK-1 Channel Blockers. Chem. Proc. 2021, 3, 67. https://doi.org/10.3390/ecsoc-24-08416
Camargo-Ayala L, Prent-Peñaloza L, Bedoya M, Gutiérrez M, González W. Rational Design, Synthesis, and In-Silico Evaluation of Homologous Local Anesthetic Compounds as TASK-1 Channel Blockers. Chemistry Proceedings. 2021; 3(1):67. https://doi.org/10.3390/ecsoc-24-08416
Chicago/Turabian StyleCamargo-Ayala, Lorena, Luis Prent-Peñaloza, Mauricio Bedoya, Margarita Gutiérrez, and Wendy González. 2021. "Rational Design, Synthesis, and In-Silico Evaluation of Homologous Local Anesthetic Compounds as TASK-1 Channel Blockers" Chemistry Proceedings 3, no. 1: 67. https://doi.org/10.3390/ecsoc-24-08416
APA StyleCamargo-Ayala, L., Prent-Peñaloza, L., Bedoya, M., Gutiérrez, M., & González, W. (2021). Rational Design, Synthesis, and In-Silico Evaluation of Homologous Local Anesthetic Compounds as TASK-1 Channel Blockers. Chemistry Proceedings, 3(1), 67. https://doi.org/10.3390/ecsoc-24-08416