1. Introduction
Glycosidase inhibitors and other enzyme inhibitors play important roles in the biochemical processing of biopolymers containing carbohydrates. The biological relevance of sulfur containing carbohydrates is gaining substantial attention, especially in medicinal and synthetic chemistry [
1]. On the other hand, selenium chemistry is gaining prominence in organic synthesis. A number of selenium compounds obtained from monosaccharides have shown biological activity [
2]. In our research group, we have synthesized imine sugar derivatives [
3,
4]. Now, our objective is to obtain thio and selenium monosaccharide analogous derivatives, such as
1 and
2 (
Scheme 1).
To our knowledge, no compounds of this type have been previously synthesized. The incorporation of both heteroatoms S or Se and N could improve the possible biological activity.
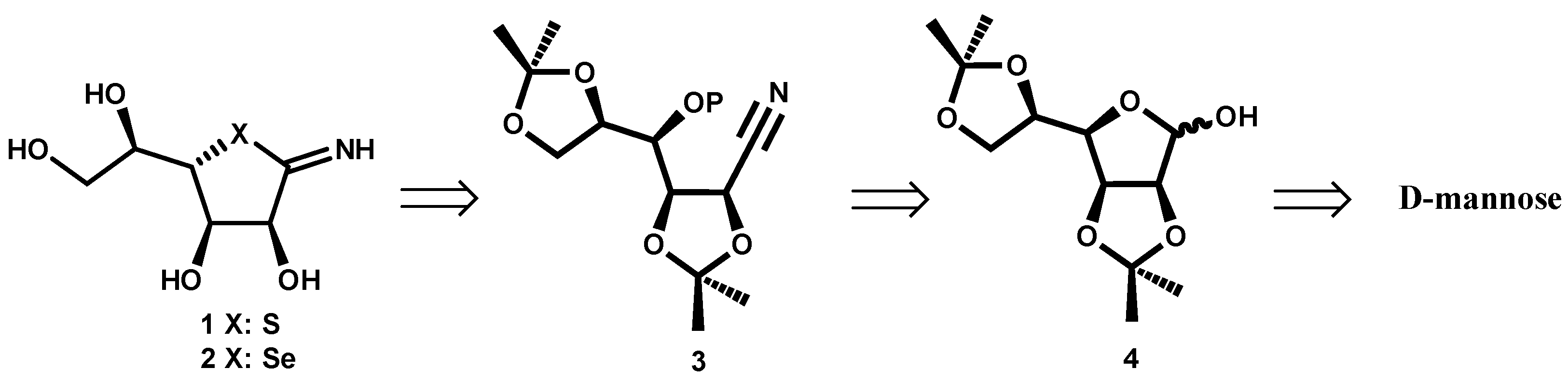
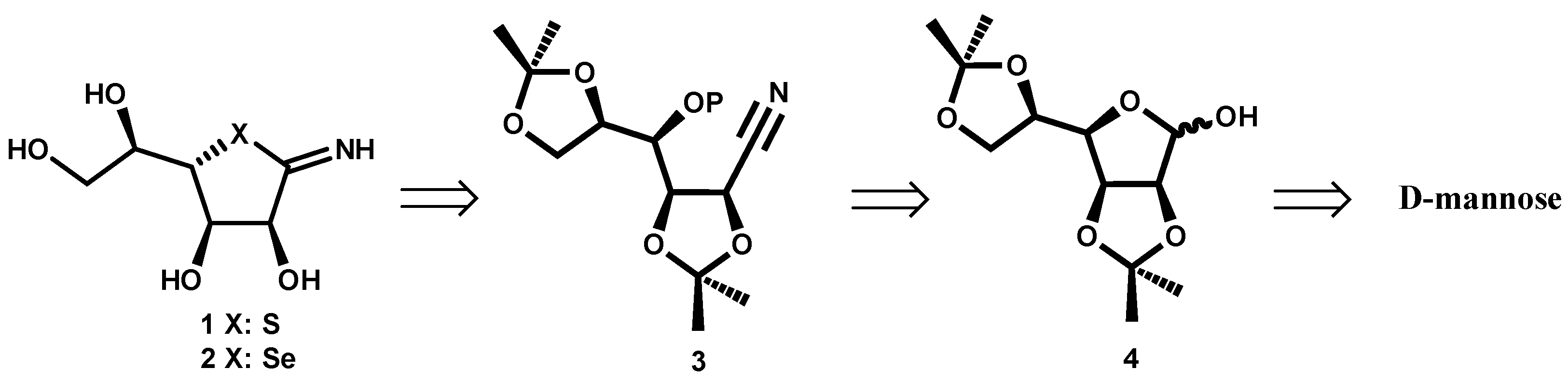
In
Scheme 1 the retrosynthetic route is shown. The target compounds
1 or
2 can be obtained from nitrile
3, with OP being a good leaving group. This intermediate
3 can be formed starting from D-mannose derivative
4. The choice of isopropylidene mannose
4 was based on its stability under the reaction conditions in the following steps and the feasible acid deprotection in the final product.
2. Results and Discussion
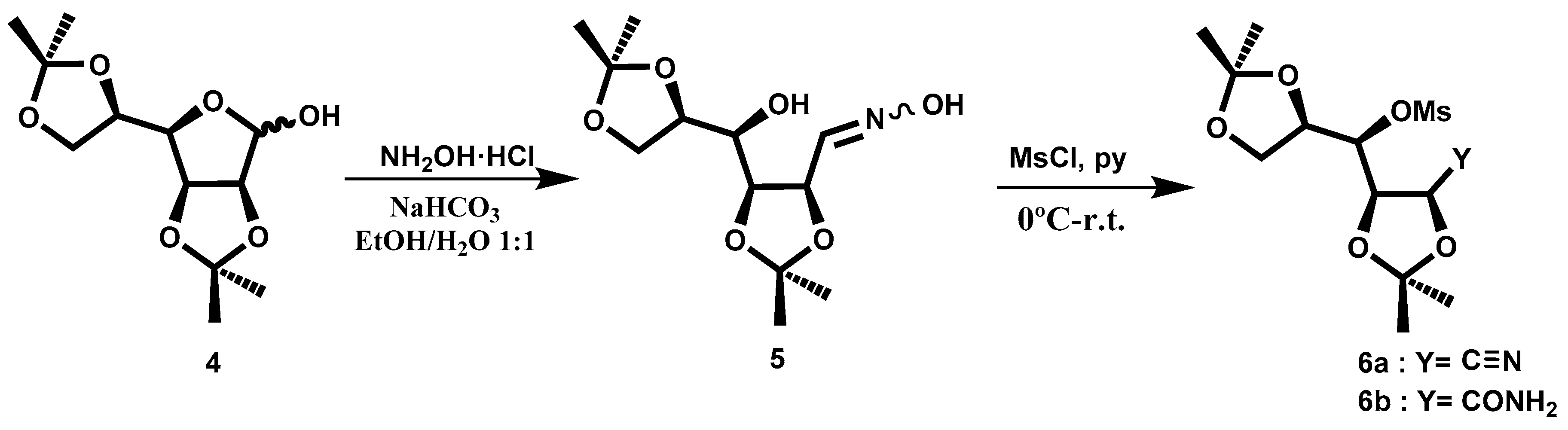
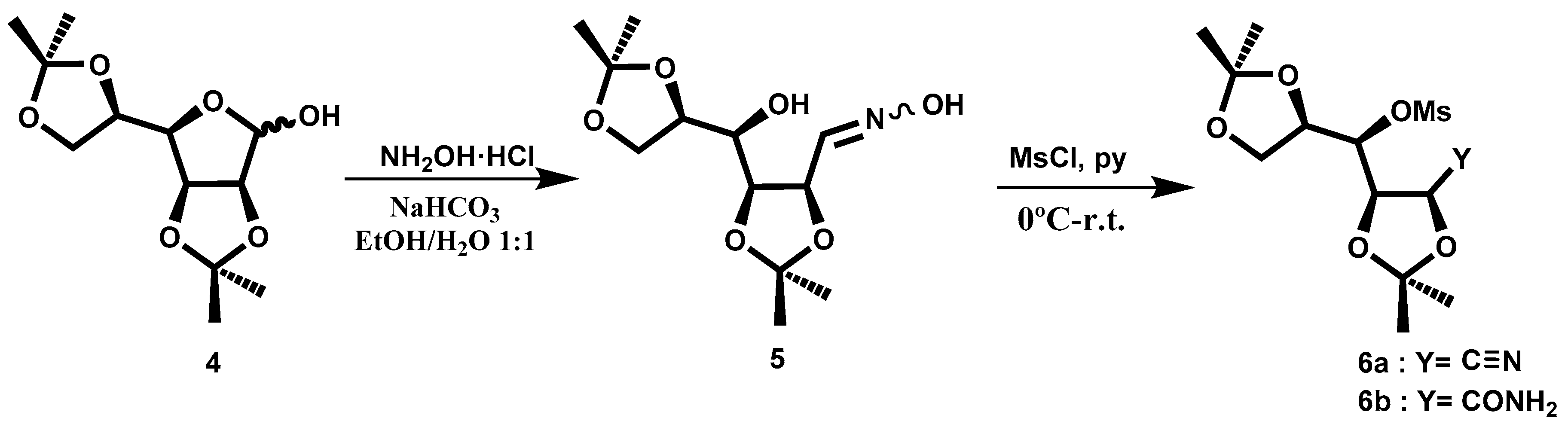
Compound
4 was obtained following the procedure described in the literature [
5]. In order to transform the aldehyde directly into a nitrile group, we tested several procedures. The best choice was the conversion of the oxime
5, which could lead to the nitrile (
Scheme 2). The oxime
5, prepared following the described procedure [
6], was shown to be a mixture of isomers
Z:
E (83:17). The product could be used in the next reaction without purification. To convert the hydroxyl groups into a good leaving group, we tried to introduce the tosyl group, but without good results. Therefore, we decided to test the reaction with MsCl in pyridine. Following the described procedure at room temperature [
6], the principal fraction consisted of a mixture of nitrile
6a and the related amide
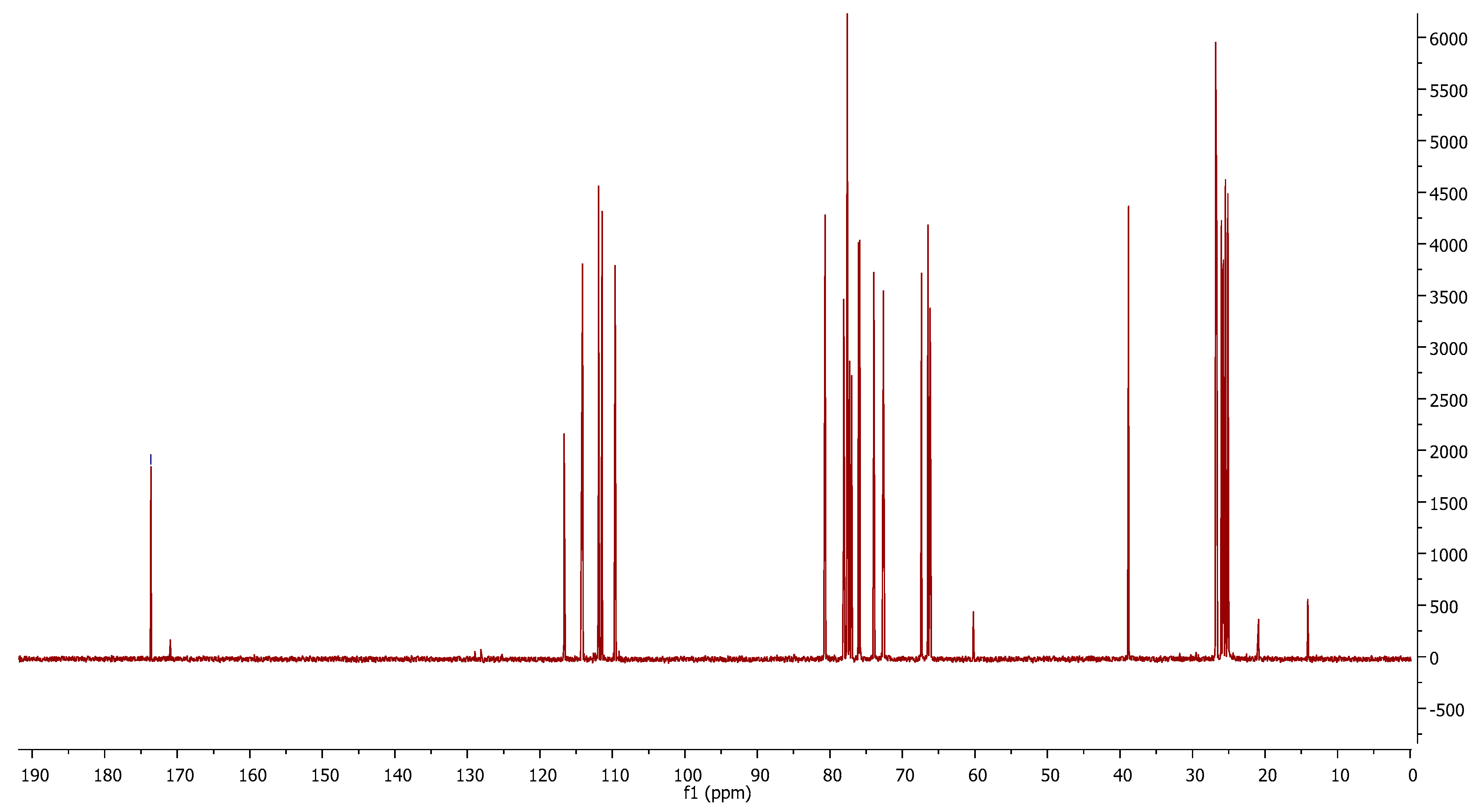
6b, as shown in NMR spectra (
Figure 1). With the aim of increasing the yield of the nitrile, we performed the reaction at a higher temperature, 60 °C, but the obtained yield was lower. The crude reaction was purified by column chromatography.
In the 13C-NMR spectrum, the peaks 173.9 ppm and 116.5 ppm belonging to the amide and nitrile groups, respectively, can be observed. This mixture is used in the following reaction. The amide is transformed into nitrile in the reaction media.
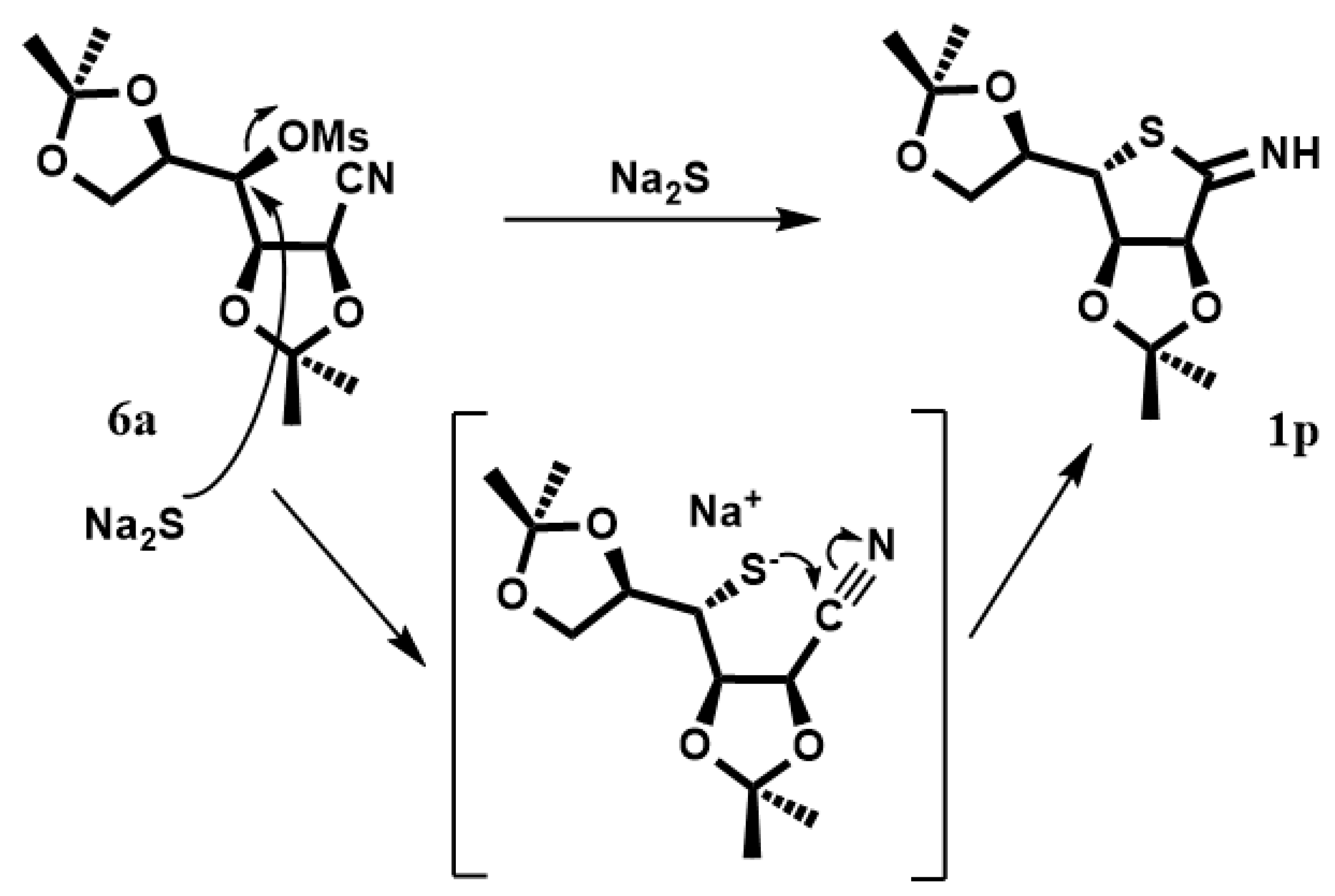
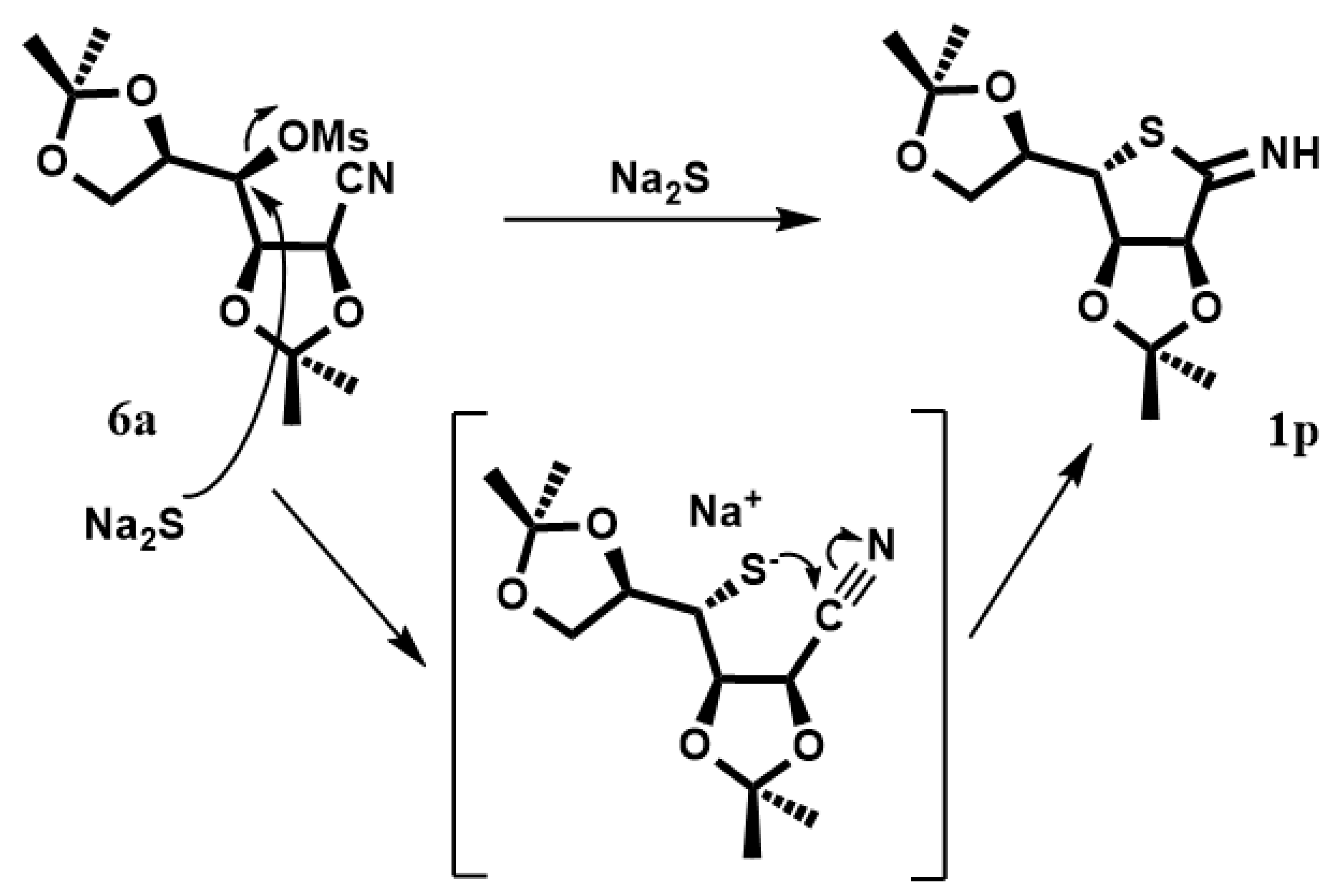
In order to obtain the protected
1p, compound
6 was dissolved in DMF with Na
2S·9H
2O (
Scheme 3). The reaction was heated at 95 °C, giving the desired product. In the proposed mechanism, sulfur anions produce the displacement of the mesylate group with subsequent attack on the nitrile group with cyclization, leading to the compound
1p. Deprotection of
1p in the target compound
1 is in progress.
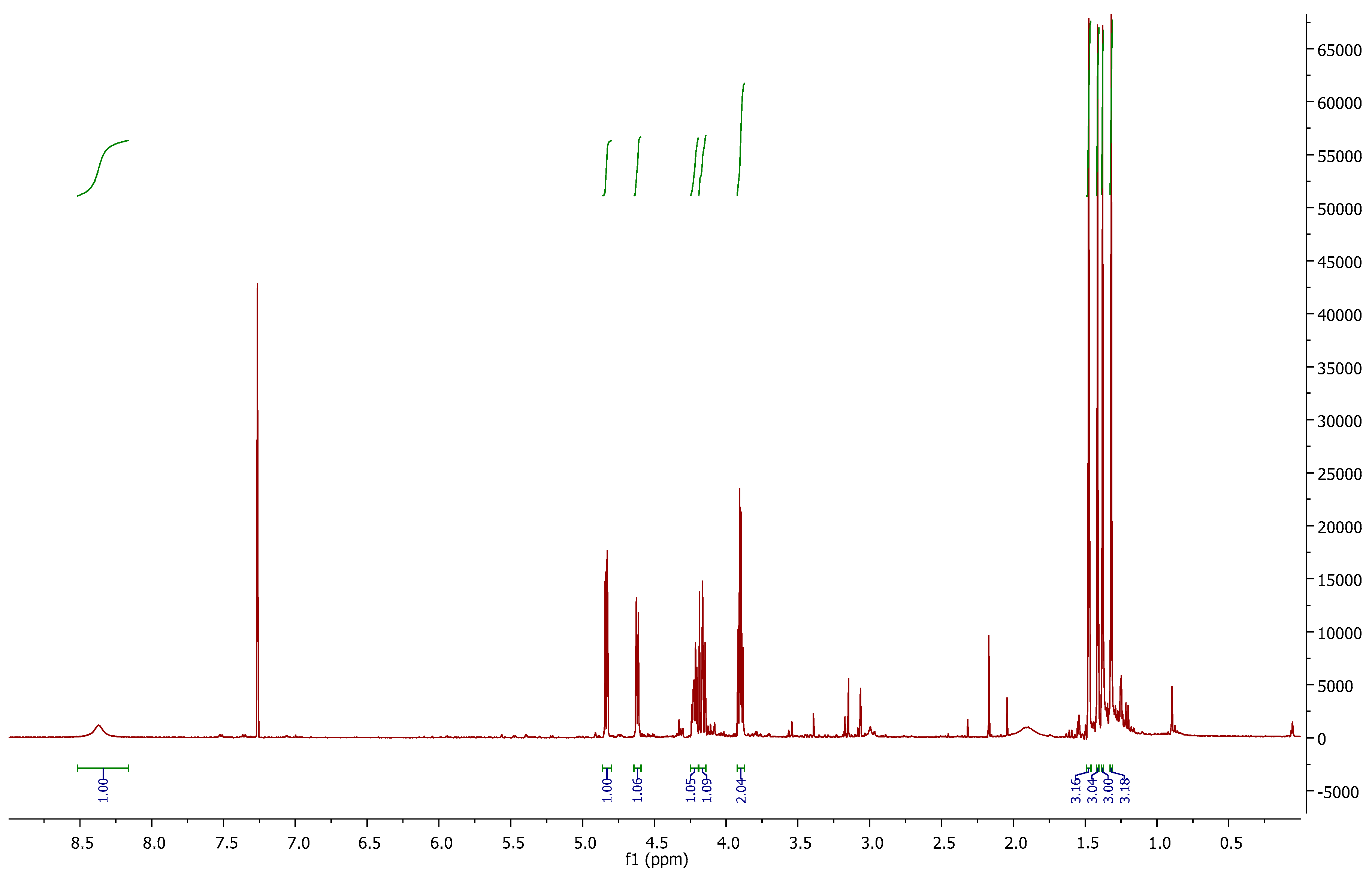
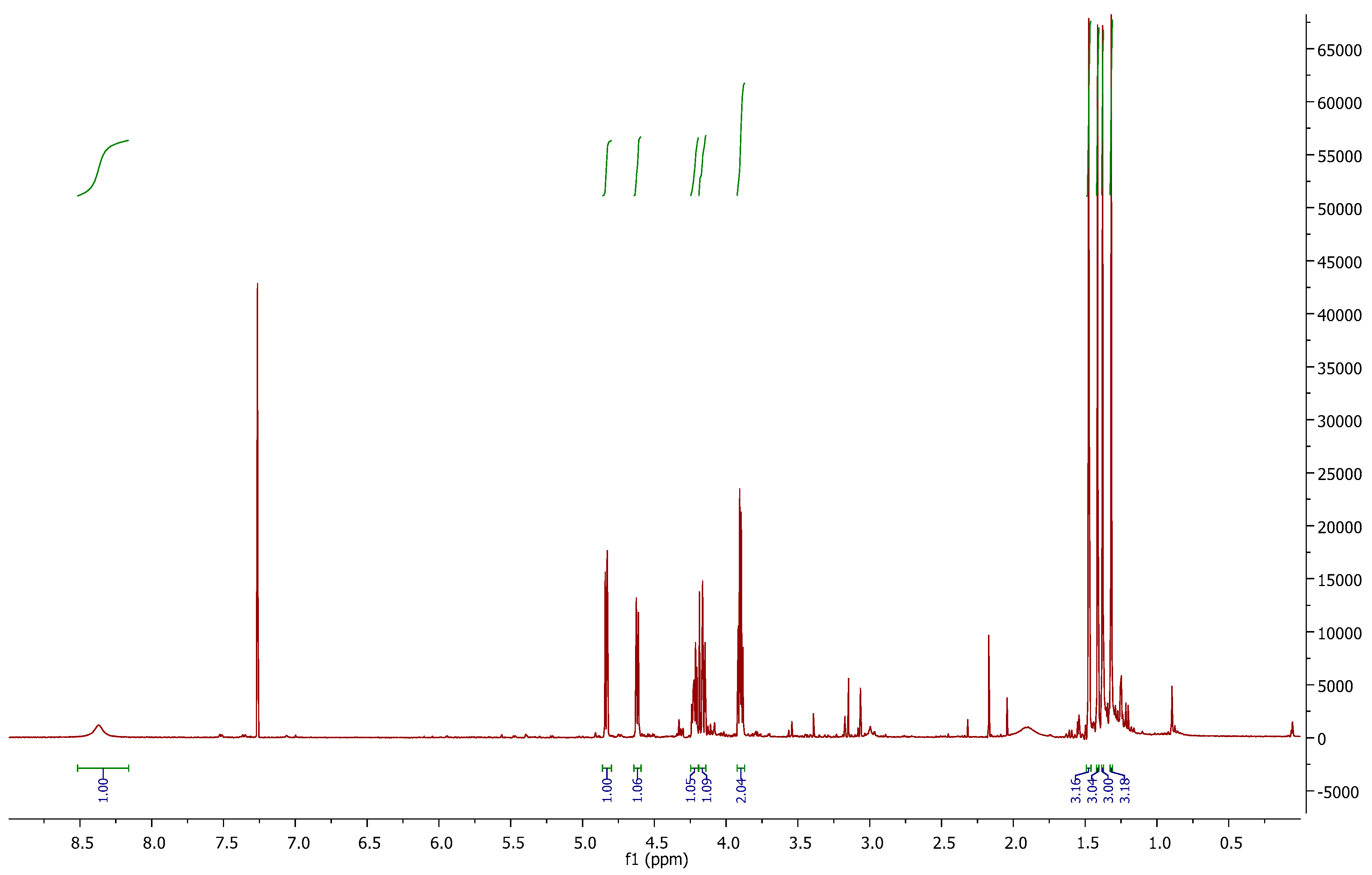
In the
1H-NMR spectrum (
Figure 2), we can observe the disappearance of signal 3.04 ppm, corresponding to the mesyl group of
6. At 4.83 and 4.62 ppm, two “d” are shown with
J = 5.5 Hz, (H-2 and H-3 respectively). The inversion of the configuration at C-4 can be corroborated with
J3,4 = 0 Hz. At 8.37 ppm, a broad singlet is correlated with the C=NH proton.
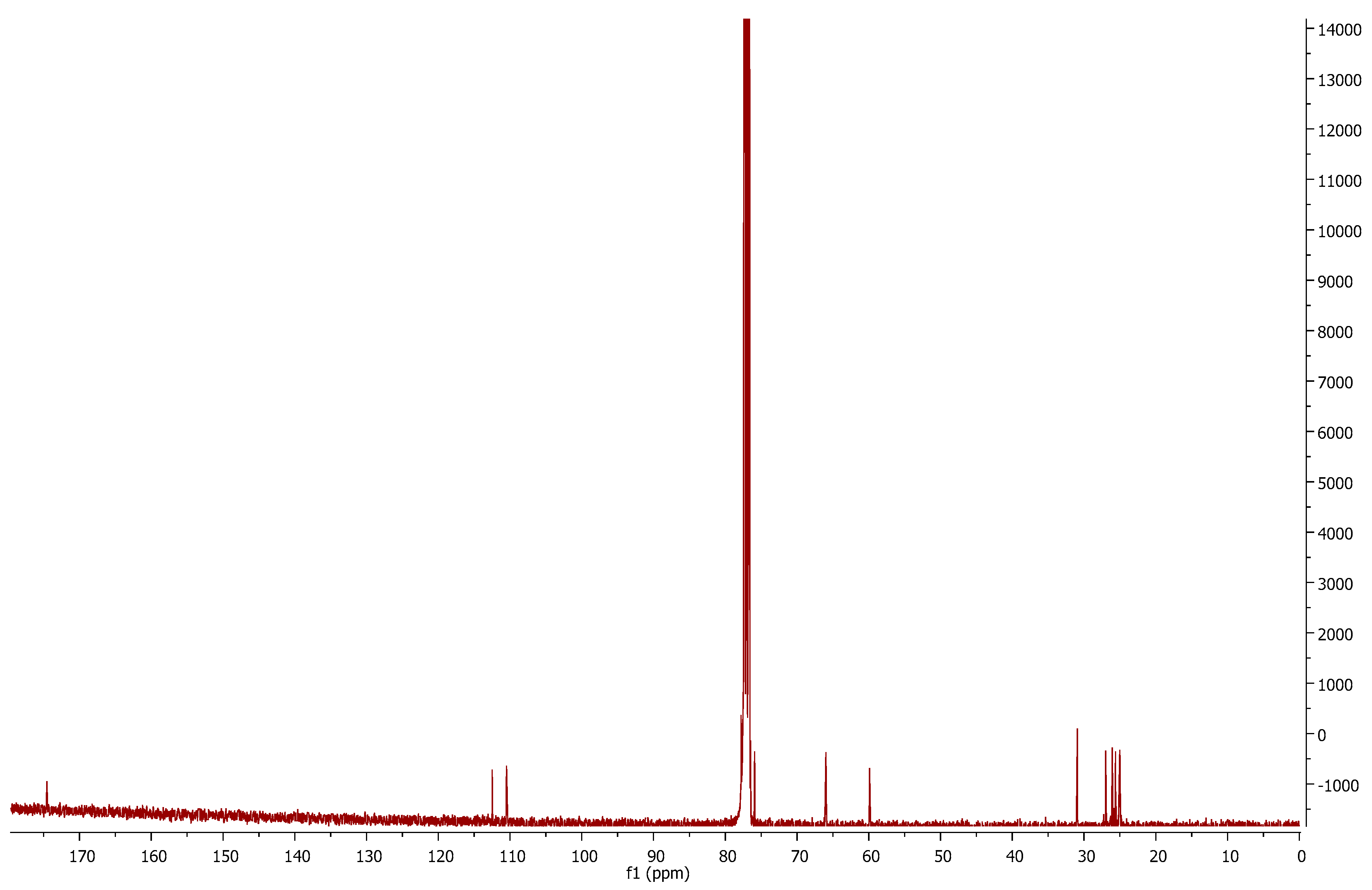
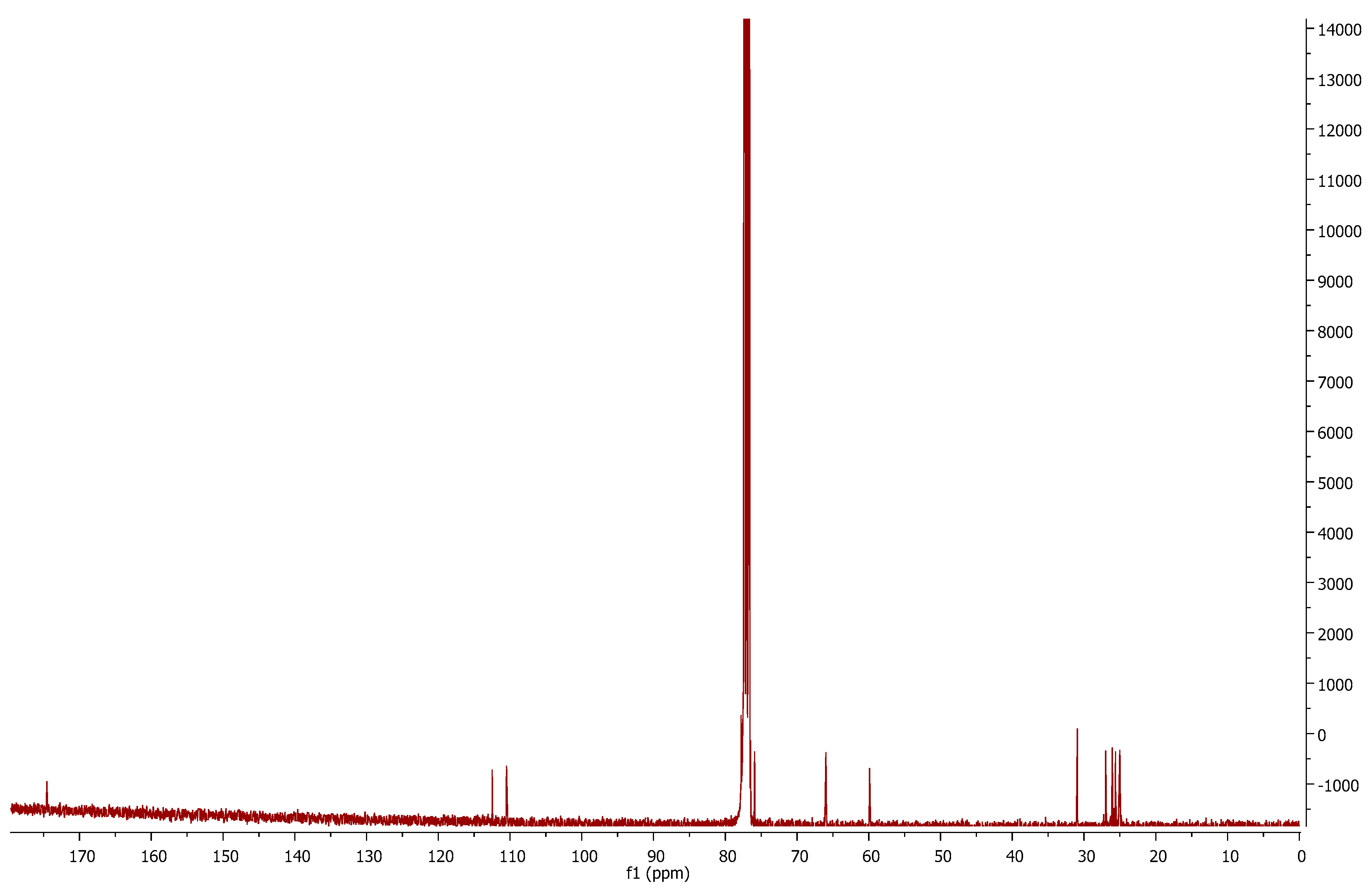
Comparing the
13C-NMR spectra of compound
1p (
Figure 3) and the mesylated
6a, we can observe the disappearance of the peaks corresponding to nitrile
sp carbon (116.5 ppm) and the methyl of the mesyl group (38.8 ppm). This is evidence of the mesylate displacement by the sulfur anion and of further attack to the nitrile carbon. The new structure can be confirmed by a new signal at 174.5 ppm, which is in accordance with imine
sp2 carbon.
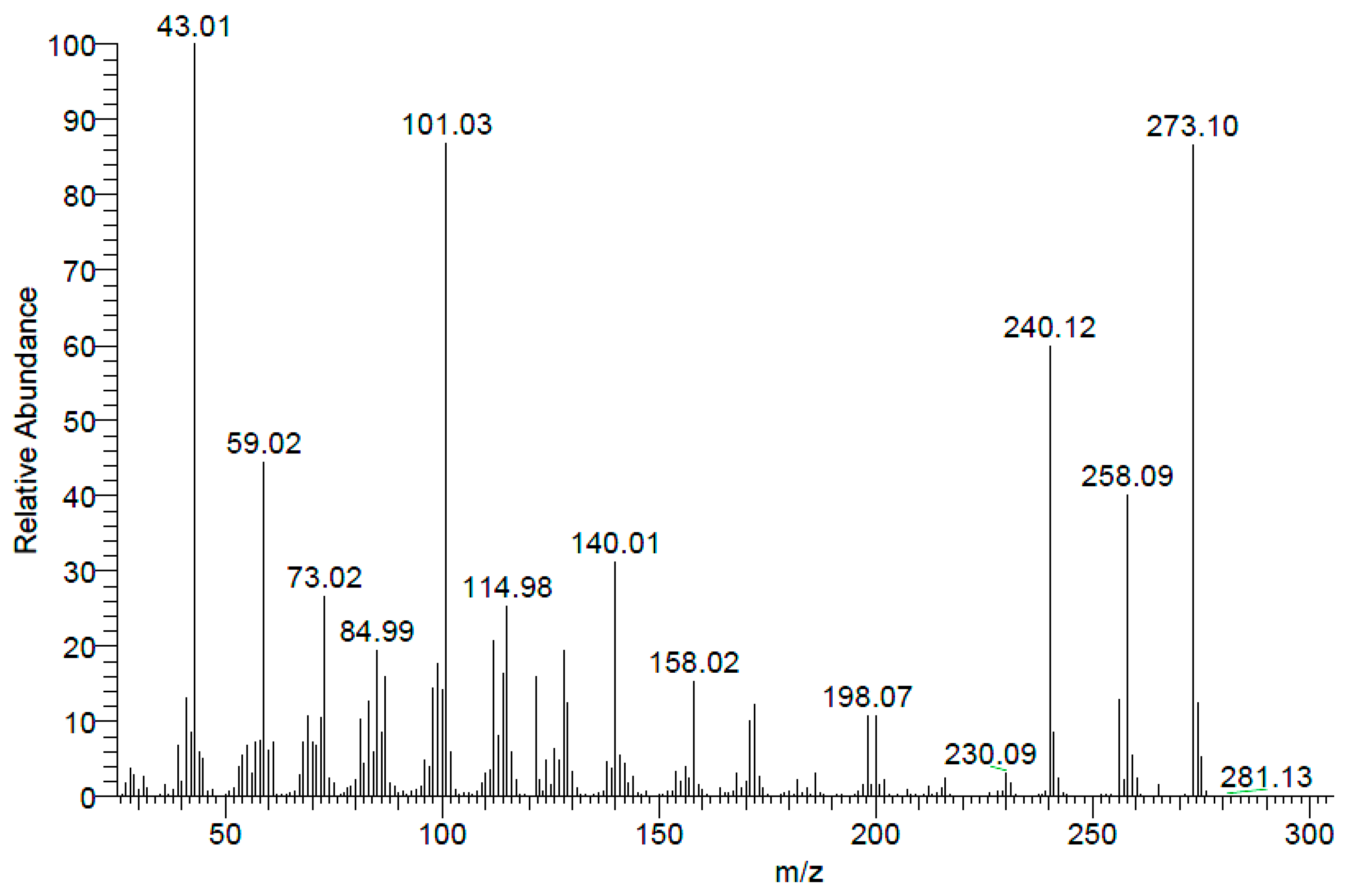
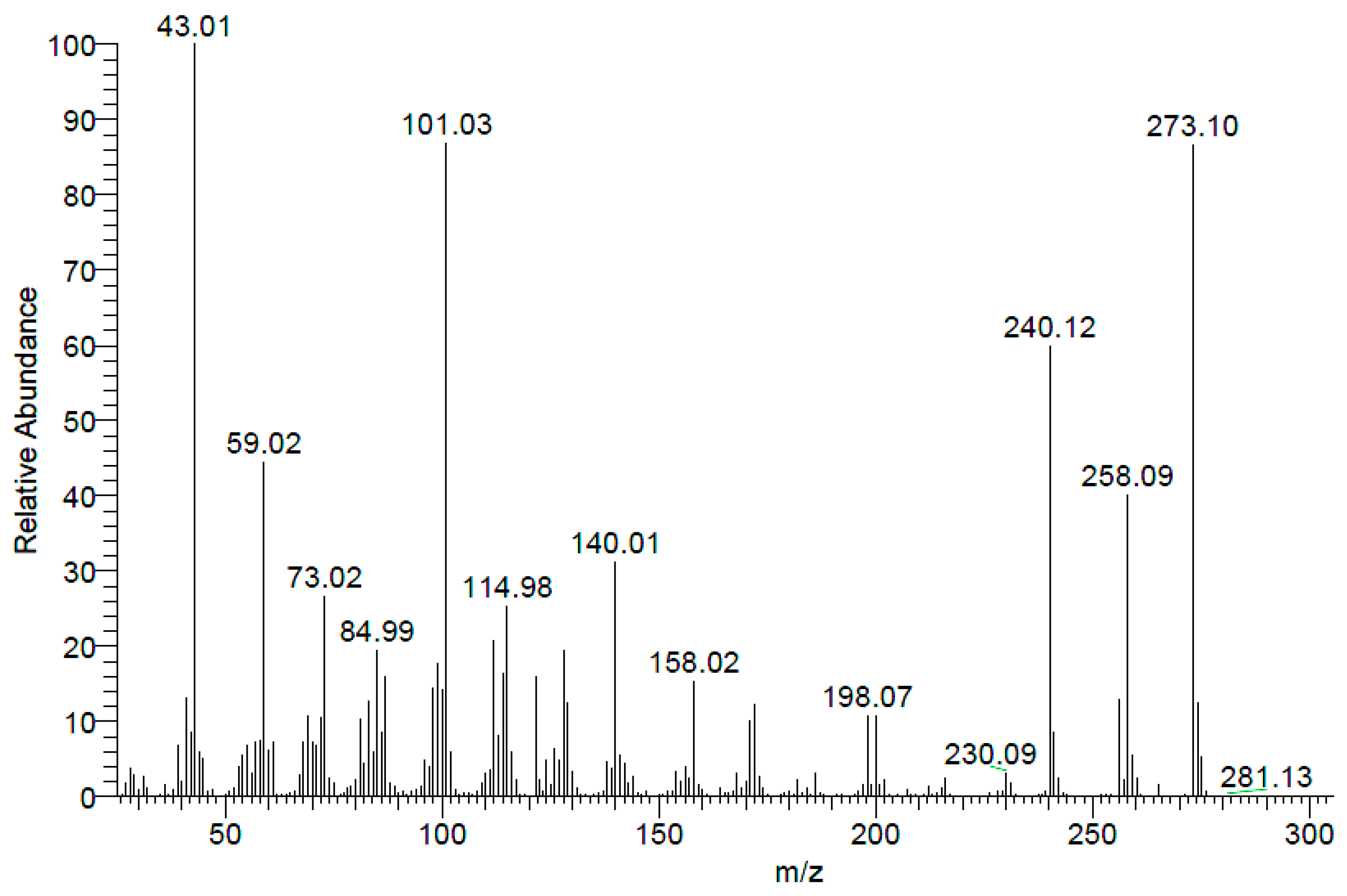
Electron Impact Spectrum (EI) (
Figure 4) showed molecular ion (M
+: 273.10). Additionaly, high resolution mass spectra (ESI) showed (M + H)
+ at 274.11041.
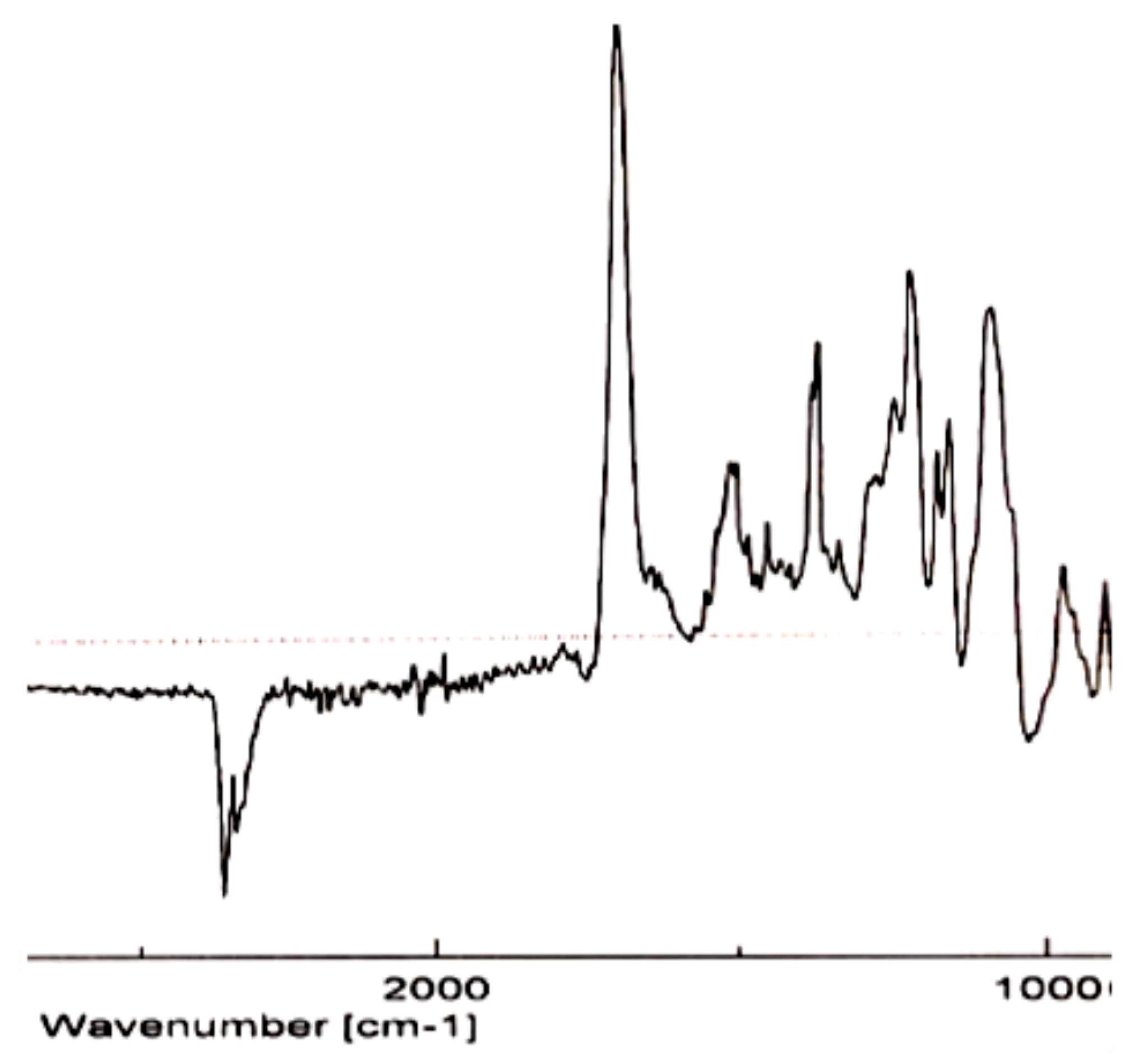
In the infrared spectrum (IR) (
Figure 5), we can observe the band corresponding to C=N tension, which uses to appear at ν
C=N = 1700–1615 cm
−1.
4. Experimental
4.1. Synthesis of 2,3:5,6-di-O-isopropylidene-4-O-metilsulfonil-D-mannononitrile (6)
To a solution of oxime 5 (2.74 g, 9.96 mmol) in pyridine (9.37 mL), in an ice bath while stirring, a cold solution of MsCl (3.22 mL) in pyridine (9.37 mL) was slowly added. The reaction was monitored by TLC, and after 12 h the reaction was quenched with a cold saturated solution of NH4Cl (~30 mL). The reaction product was extracted (3 × 30 mL AcOEt) and the organic phase was washed with saturated aq. CuSO4·5H2O, dried (anh. MgSO4), and evaporated. The crude product was purified (flash chromatography), obtaining nitrile 6a with amide (1.43 g, 43%). Compound 6a had: RF, 0.5 (Hex/AcOEt 2:1). 1H-NMR (CDCl3, 400 MHz, δ ppm): 4.8 (d, J = 4.8 Hz, 1 H, H-2), 4.76 (t, J = 4.75 Hz, 1 H, H-4), 4.28–4.34 (m, 2 H, H-3 y H-6a), 4.13–4.22 (m, 2 H, H-5 y H-6b), 3.04 (s, 3 H, SO2CH3), 1.5, 1.36, 1.26, 1.24 [4s, 4 × 3 H, C(CH3)2]. 13C-NMR (CDCl3, 100 MHz, δ ppm): 116.5 (C-1), 111.9, 111.4 [2 × C(CH3)2], 80.7 (C-2), 77.6 (C-3), 73.9 (C-4), 67.4 (C-6), 66.1 (C-5), 38.8 (SO2CH3), 26.8, 26.0, 25.5, 25.1 [2 × C(CH3)2].
4.2. Synthesis of Thiosugar 1p
To a solution of the obtained mixture 6 (663 mg) in DMF (16 mL), Na2S·9H2O (473 mg) was added and the mixture was heated at 95 °C. The reaction was monitored by TLC, and after 48h, water was added (25 mL). The reaction product was extracted (4 × 25 mL AcOEt) and the organic phase was washed with saturated aq. solution of LiCl and dried (anh. MgSO). Product 1p was purified by column chromatography (150 mg, 30%). Compound 1p had: RF, 0.2 (Hex/AcOEt 3:1). 1H-NMR (CDCl3, 400 MHz, δ ppm): 8.37 (s, 1H, NH), 4.83 (d, J = 5.5 Hz, 1H, H-2), 4.62 (d, J = 5.5 Hz, 1H, H-3), 4.25–4.13 (m, 2H, H-6a y H-6b), 3.90 (dd, J = 8.8, 4.6 Hz, 2H, H-4 y H-5), 1.47, 1.41, 1.38, 1.32 [4s, 4 × 3H, C(CH3)2]. 13C-NMR (CDCl3, 100 MHz, δ ppm): 174.5 (C-1), 112.5, 110.5 [2 × C(CH3)2], 77.3, 77.0, 76.3 (C-2, C-3, C-5), 65.9 (C-6), 59.9 (C-4), 26.9, 26.1, 25.6, 24.9 [2 × C(CH3)2].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}