Abstract
An optically active bicyclo[2.2.0]heptane fragment was introduced in the molecule of new 1′-homonucleosides on a 2- 6-chloro-amino-purine scaffold to obtain 6-substituted carbocyclicnucleozide analogs as antiviral compounds. The synthesis was realized by a Mitsunobu reaction of the base with the corresponding bicyclo[2.2.0]heptane intermediate, and then the nucleoside analogs were obtained by substitution of the 6-chlorime with selected pharmaceutically accepted amines. A molecular docking study of the compounds on influenza, HSV and low active coronavirus was realized. Experimental screening of the compounds on the same viruses is being developed and soon will be finished.
Published: 14 November 2020
1. Introduction
Nucleosides are a recognized class of antiviral and anticancer drugs. The resistance acquired in time and the toxicity are the main factors that motivated the discovery of new more active and selective analogs. The modifications were realized on the nucleobase and/or on the sugar moiety. With guanine and 2-amino-6-substituted purine as nucleobase, recognized carbocyclic nucleoside drugs or active compounds studied in different clinical phases, like carbovir and its prodrug abacavir (with 6-cyclopropylamino substituent), entecavir, lobucavir and cyclohexenyl G, and also recognized acyclic nucleosides like acyclovir, ganciclovir, penciclovir and their valine esters valaciclovir, valganciclovir and famciclovir became recognized as milestone compounds in the treatment of antiviral and anticancer diseases [1,2].
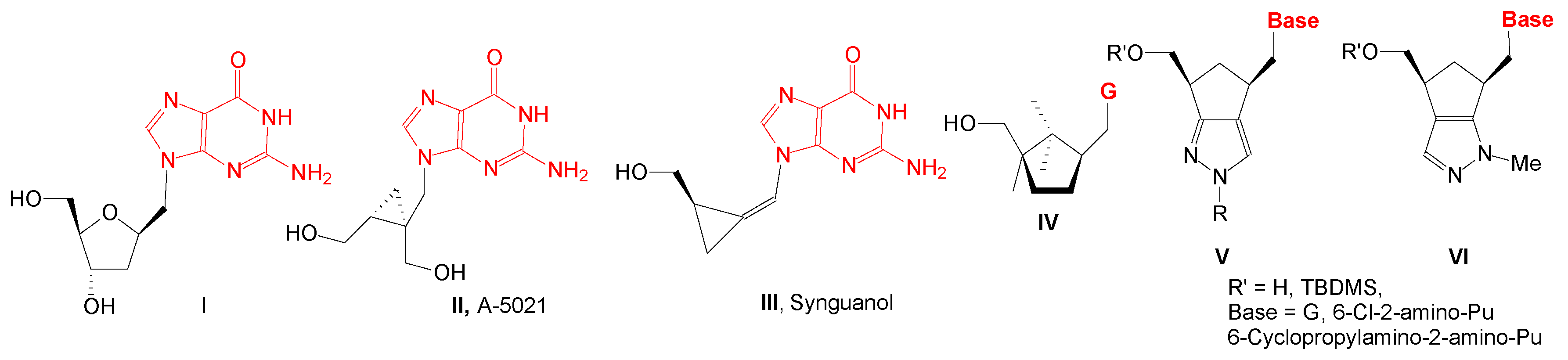
1′-Homonucleosides, due to the methylene group between nucleobase and sugar moiety, are structurally a class of compounds closer to acyclic nucleosides than nucleosides. In this class, there are also compounds with guanine, 6-chloro-2-aminopurine or 6-substituted-2-aminopurine as nucleobase with potential antiviral or anticancer activity (Figure 1): compound II has antiherpetic activity, compound III has activity against HCMV and Epstein–Barr virus, compound IV, with a 2,2,3-trimethylcyclopentanol, is active against HIV-1 and HIV-2, and compound V, with a cyclopenta[c]pyrazole moiety, is very active against VZV/TK- strain.
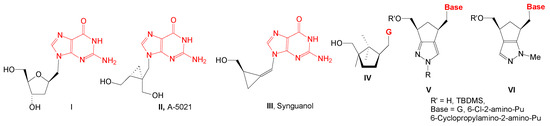
Figure 1.
1′Homonucleosides with guanine and 6-substituted-2-aminopurine as nucleobases.
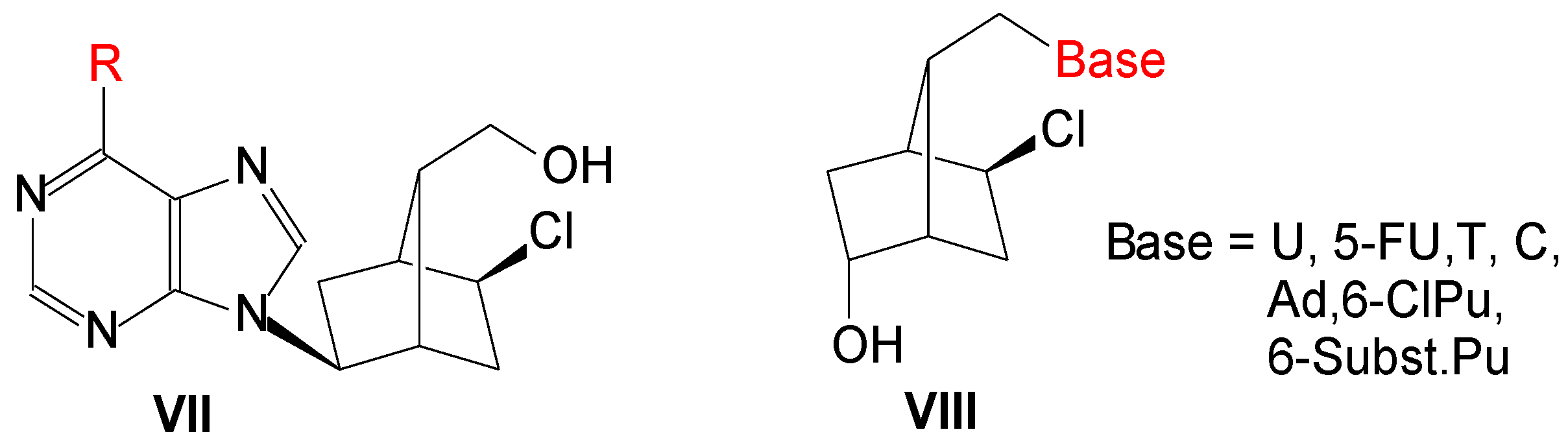
Previously we used an optically active bicyclo[2.2.1]heptane moiety to obtain new L-type carbocyclic nucleosides, VII (Figure 2), and some of them presented antiviral activity against influenza virus or coxsackievirus B4 [3]. Then new HSV-1 1′-homocarbanucleoside analogs, VIII, were synthesized with nucleobase: U, 5-FU, T, C, Ad, 6-Cl-purine and 6-substituted purine. Two compounds had lower IC50 (15 ± 2 and 21 ± 4 μM) and one equal to that of acyclovir (IC50: 28 ± 4 μM). In the present paper, we present the synthesis, molecular docking study and antiviral activity of a number of compounds VIII, in which base is 2-amino-6-chloropurine, guanine, 2,6-diaminopurine and 2-amino-6-substitutedpurine.
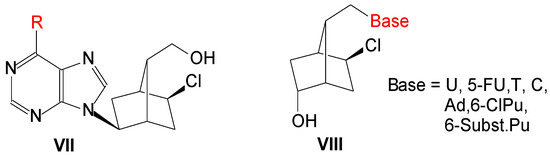
Figure 2.
L-Carbanucleosides and 1′-homocarbanucleosides with a bicyclo[2.2.1]heptane skeleton.
2. Results
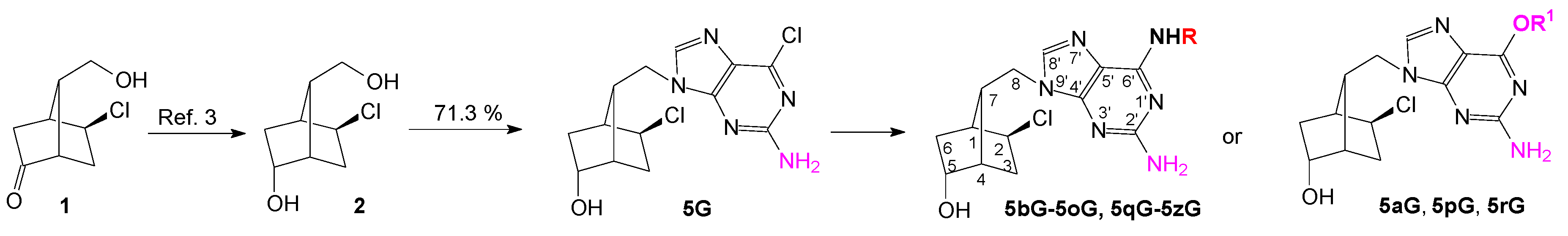
Synthesis of the new 1′-homocarbanucleosides with an optically active bicyclo[2.2.1]heptane fragment as sugar moiety and guanine, 6-O-alkyl-guanine, 2,6-diaminopurine and 2-amino-6-N-substitutedpurine as nucleobase started from diol 2, was obtained crystallized by NaBH4 reduction of the keto-compound 1 [3]. The unprotected diol 2 was used in the Mitsunobu reaction with 2-amino-6-chloropurine, taking into account that the primary alcohol will react more quickly than the secondary one. Indeed, the primary alcohol reacted selectively to give the key nucleoside intermediate 5G isolated by simple crystallization in 71.3% yield (Scheme 1). For comparison, 6-chloropurine reacted with diol 2 in the same Mitsunobu reaction in 67.6% yield [4,5].
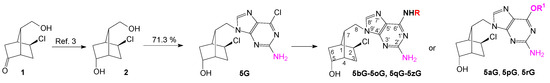
Scheme 1.
Synthesis of new 1′-homocarbanucleoside analogs with guanine and 2-amino-6-substituted purine as nucleobase.
The following 1′-homocarbocyclic nucleosides were synthesized by substitution of the chlorine atom with ammonia, with primary or secondary pharmacological amines, with methoxide or ethoxide or by substitution with hydroxyl (acid hydrolysis) to the guanine, all in good yield [6]. Twenty-four new compounds were obtained, fully characterized and used for antiviral screening against influenza, herpes simplex virus and low active coronavirus.
A molecular docking study was realized using CLC Drug Discovery Workbench Software, on 24 compounds to obtain accurate predictions about the structure and interactions of the studied compounds in complex with a protein/enzyme receptor to evaluate the biological activity.
In this study, some protein/enzyme receptors that were imported from a protein data bank were used (http://www.rcsb.org/:PDB, accessed on September 2020):
Herpes simplex type-1 thymidine kinase (PDB ID 2KI5).
Wild-type influenza N2 neuraminidase (PDB ID 4H52).
SARS coronavirus main protease (PDB ID:3TNT).
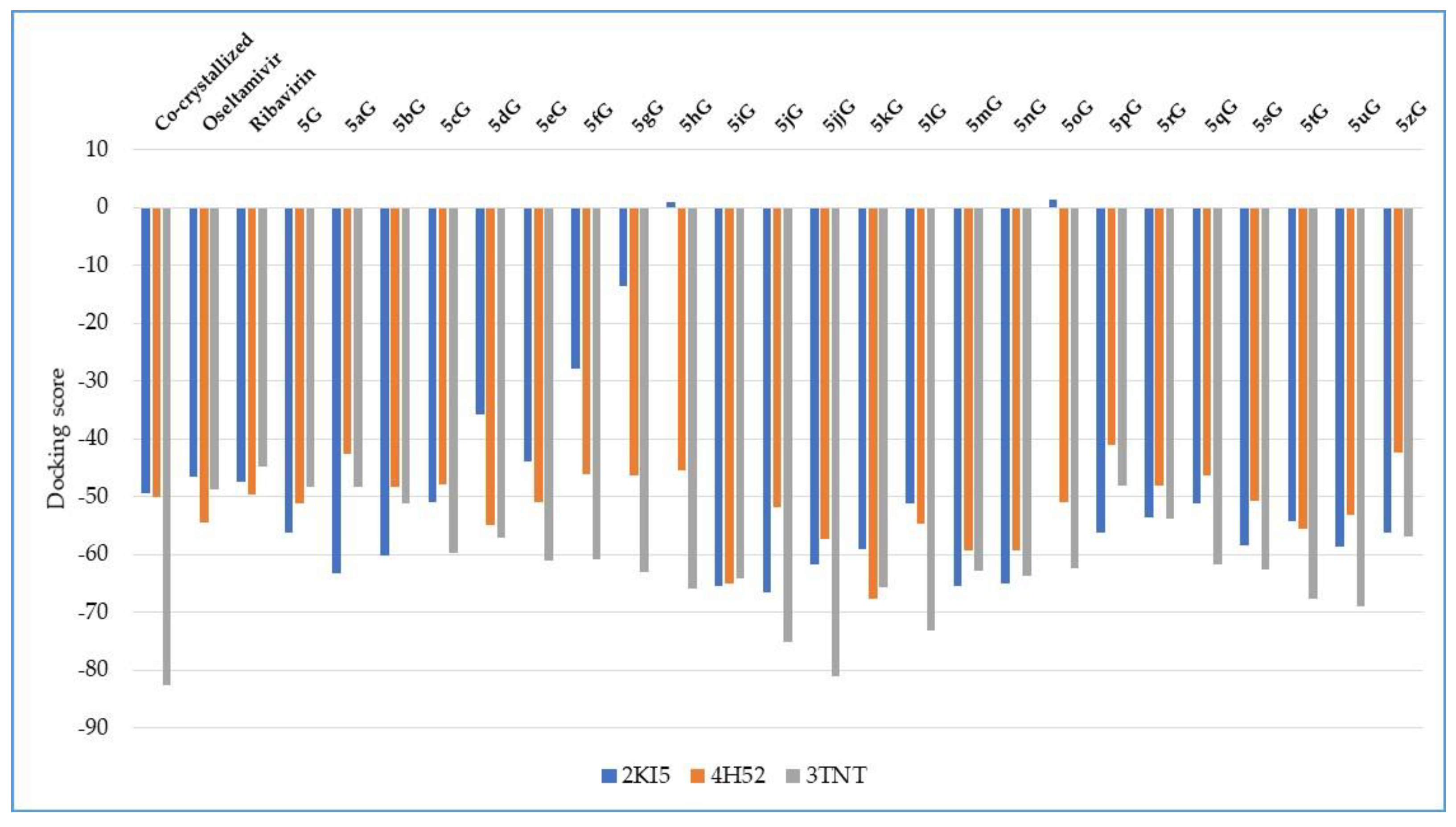
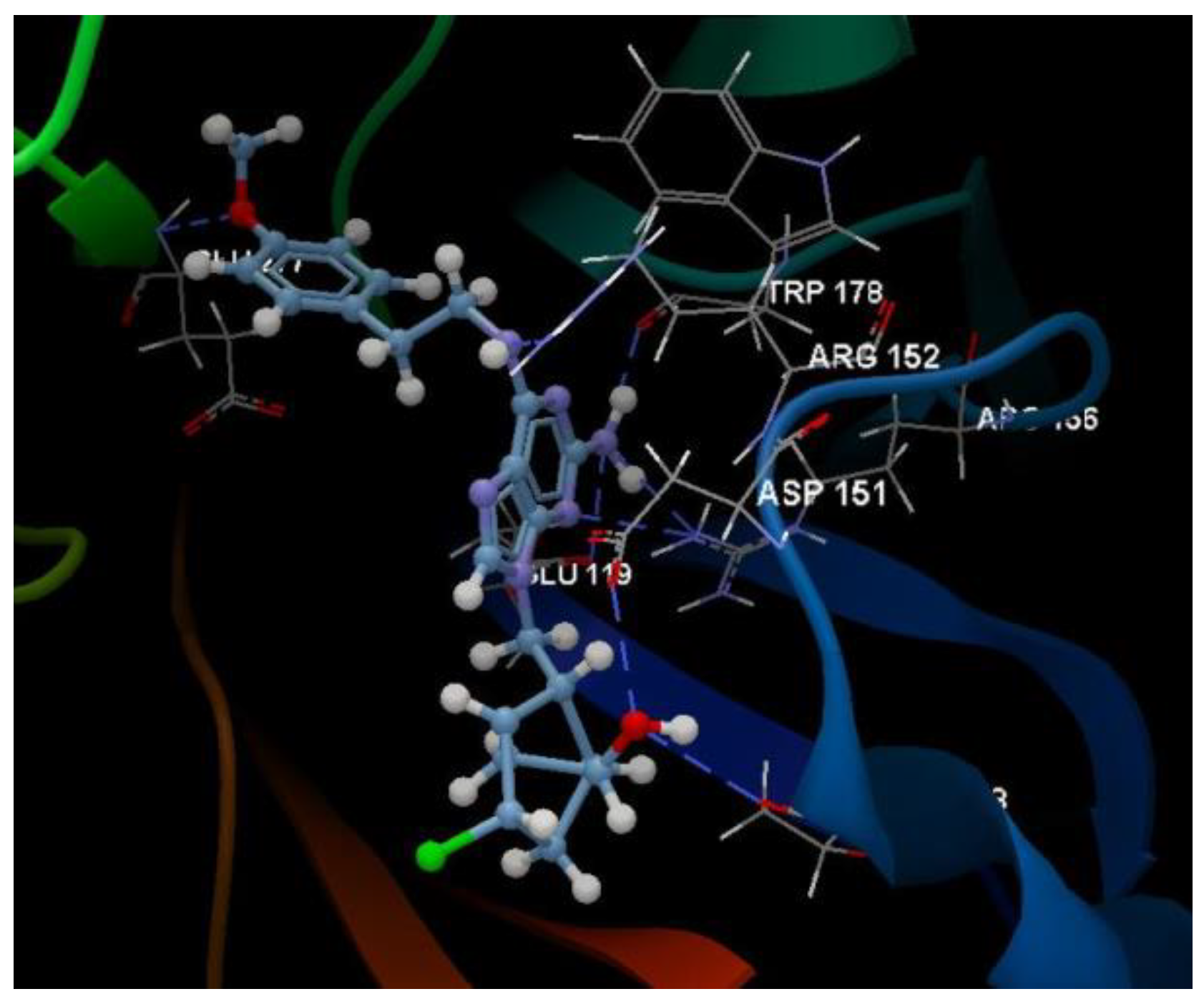
Docking evaluation against herpes simplex type-1 thymidine kinase: docking studies were performed to achieve accurate predictions on the optimized conformations for both ligand and protein target to form a stable complex. All ligands were docked on the crystal structure of the herpes simplex type-1 thymidine kinase (PDB ID: 2KI5). The docking pose of the co-crystallized AC2 interacting with amino acid residues of the active site is shown in Figure 3. The oseltamivir and ribavirin were taken as reference ligands to compare the docking results of all the studied compounds. The docking studies revealed that the 5jG compound has the best docking score −66.42 (RMSD: 0.55) (Table 1, Figure 4).
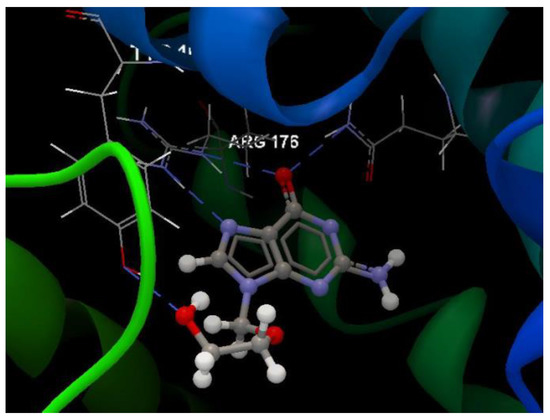
Figure 3.
Hydrogen bond (blue dotted lines) between AC2 and amino acid residues from the binding site of 2KI5.

Table 1.
Docking score of ligands.
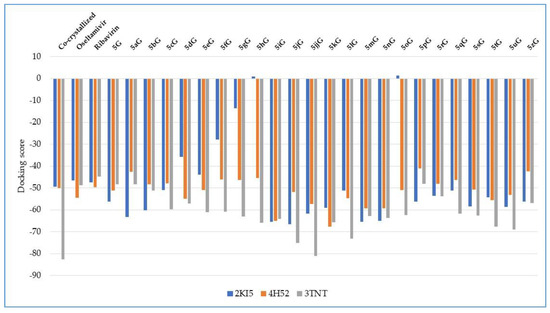
Figure 4.
Docking score of the compounds, comparative with the docking score of the co-crystallized and with the docking score of the reference drugs oseltamivir and ribavirin.
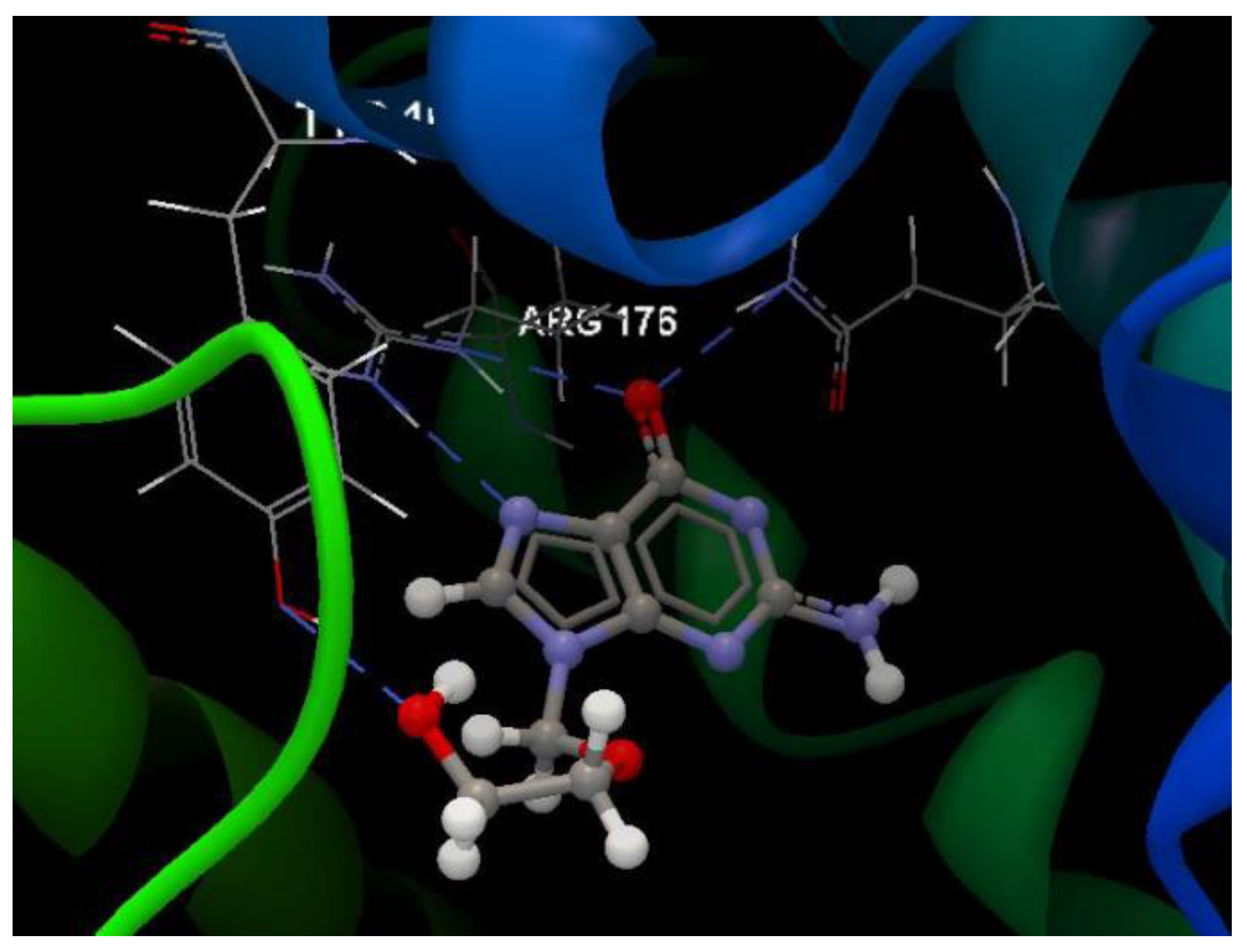
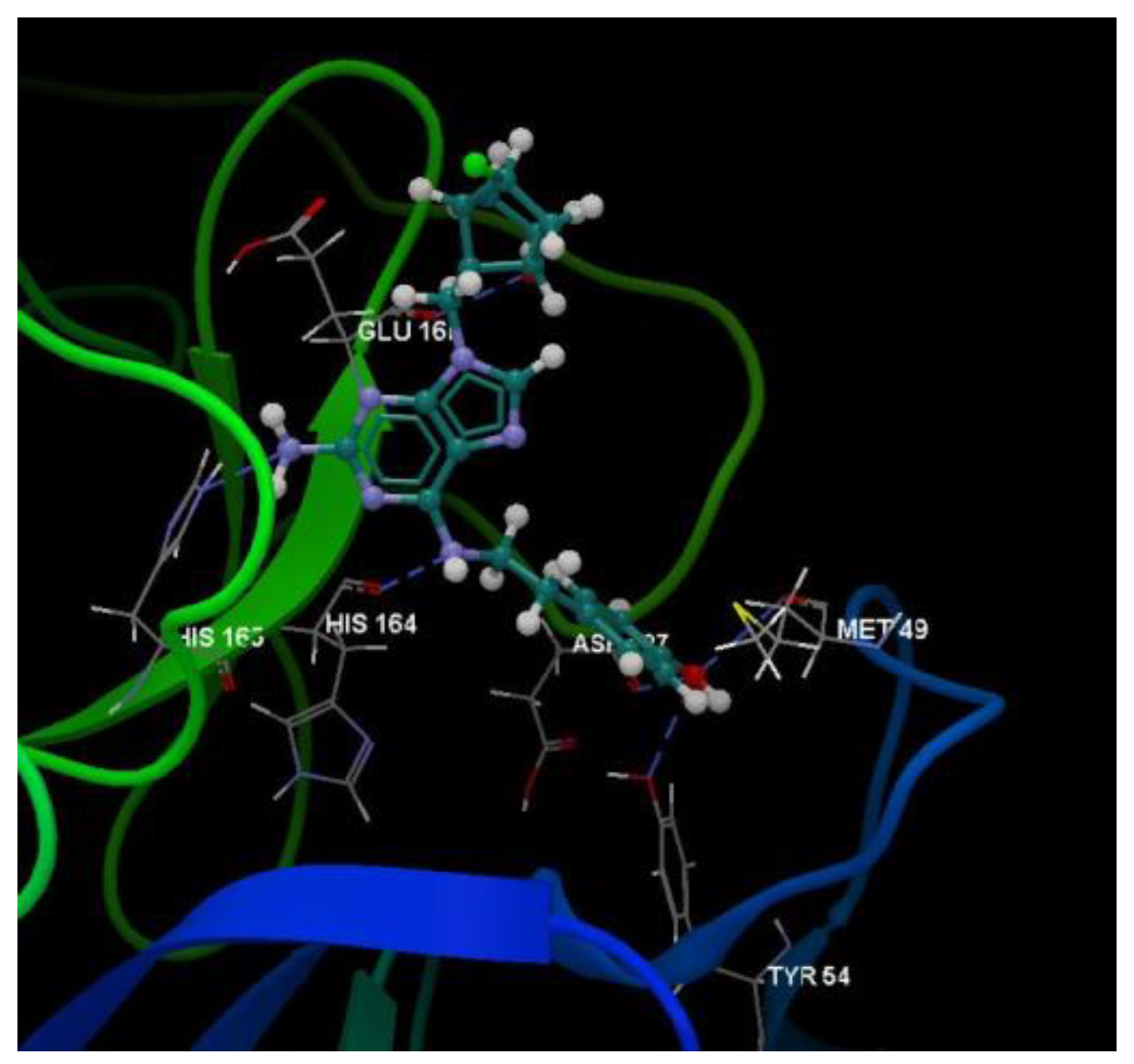
The docking pose of the 5jG compound is shown in Figure 5.
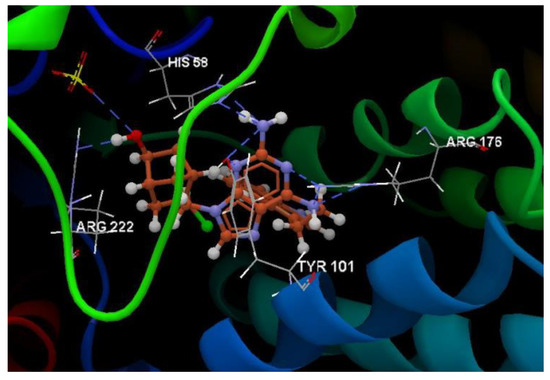
Figure 5.
Hydrogen bond (blue dotted lines) between 5jG compound and amino acid residues from the binding site of 2KI5.
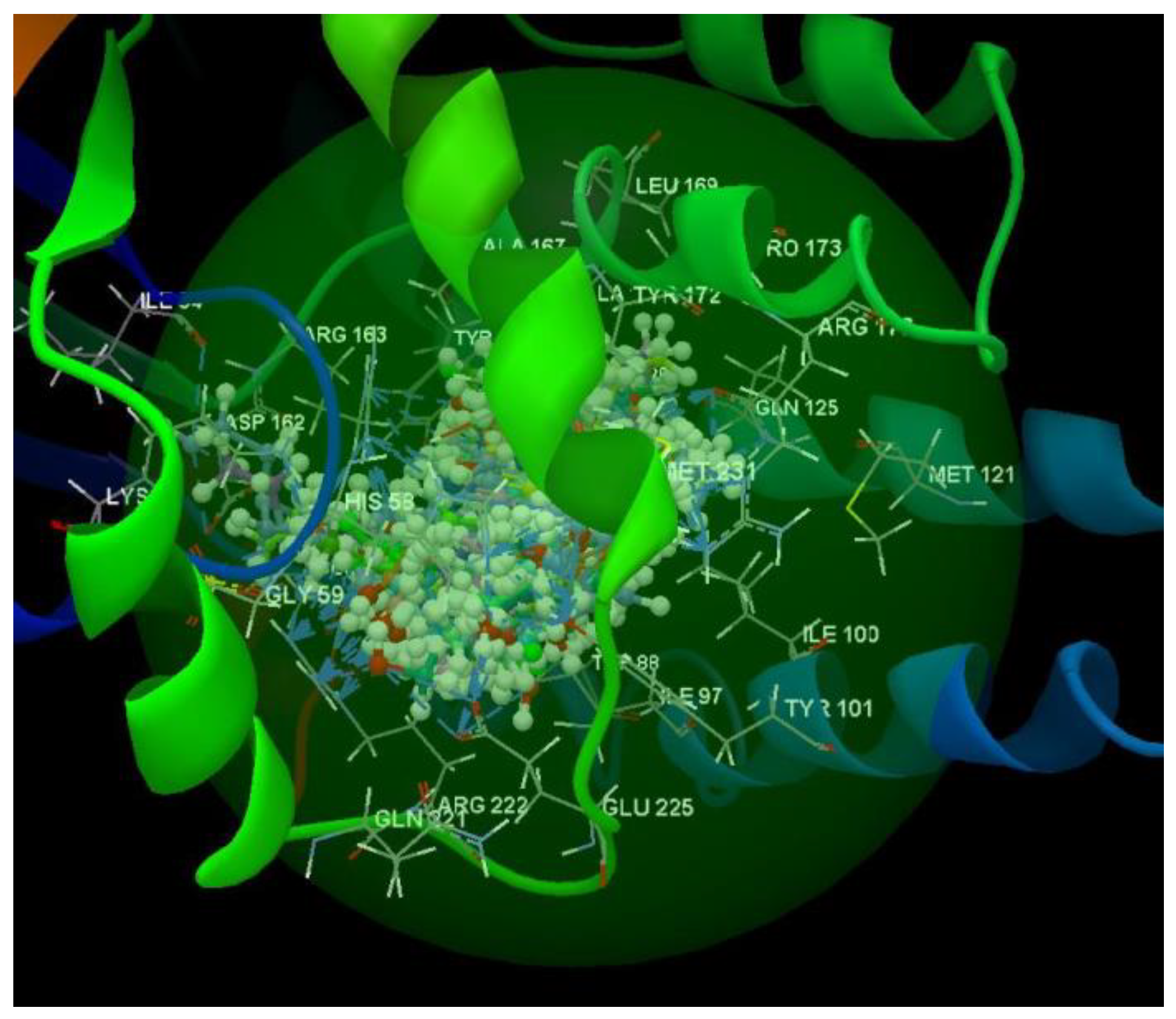
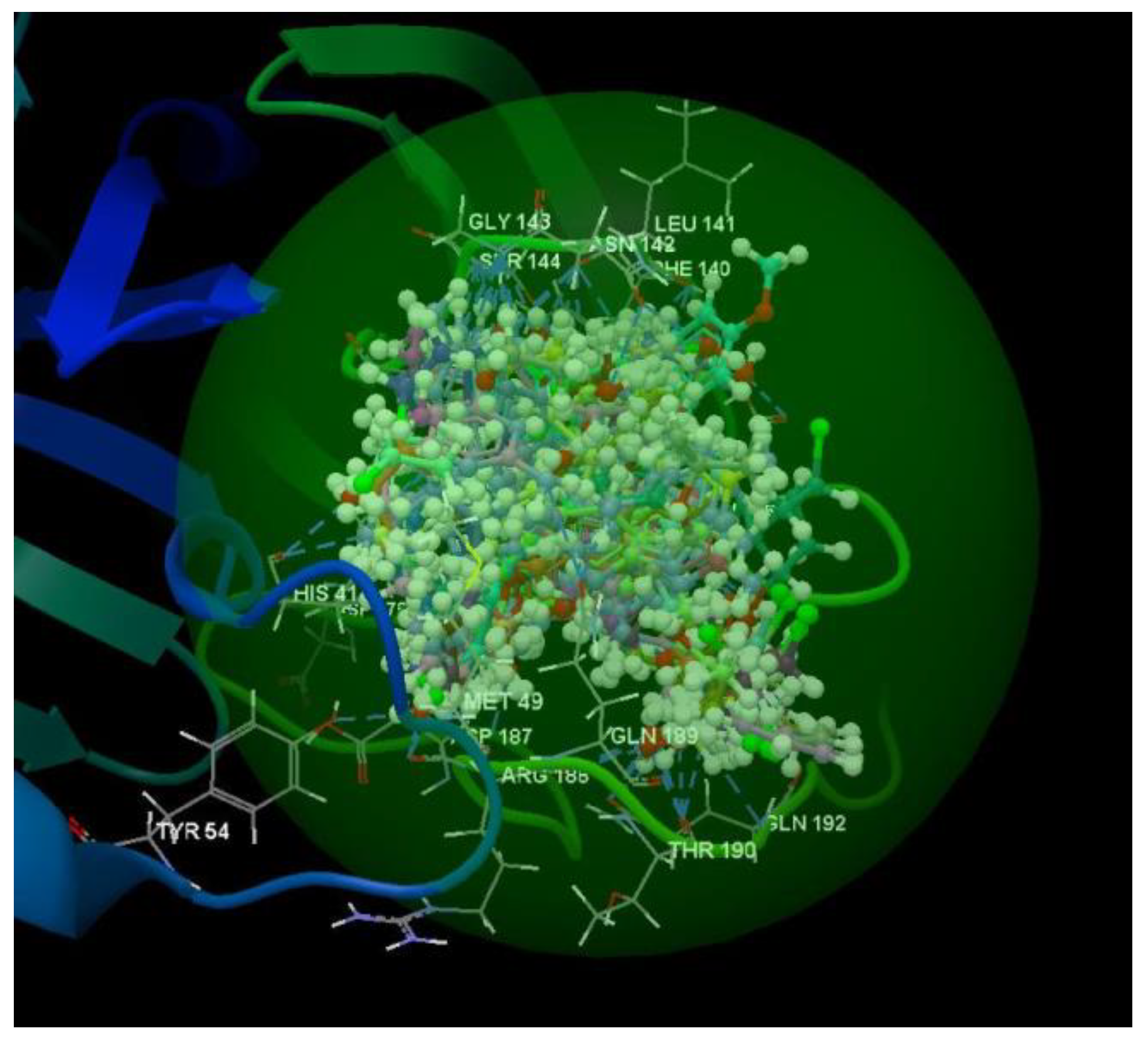
After analyzing the data obtained from the docking study, it was observed that all the compounds were placed in the same binding site of 2KI5 as the co-crystallized (Figure 6).
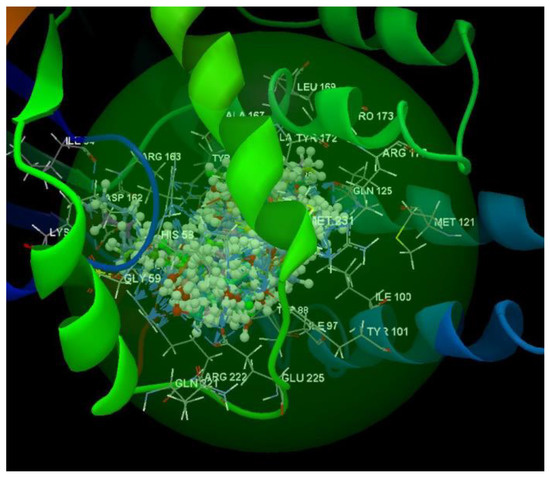
Figure 6.
Docking pose of the co-crystallized AC2, of the oseltamivir and ribavirin and of the studied compounds in the binding site of 2KI5.
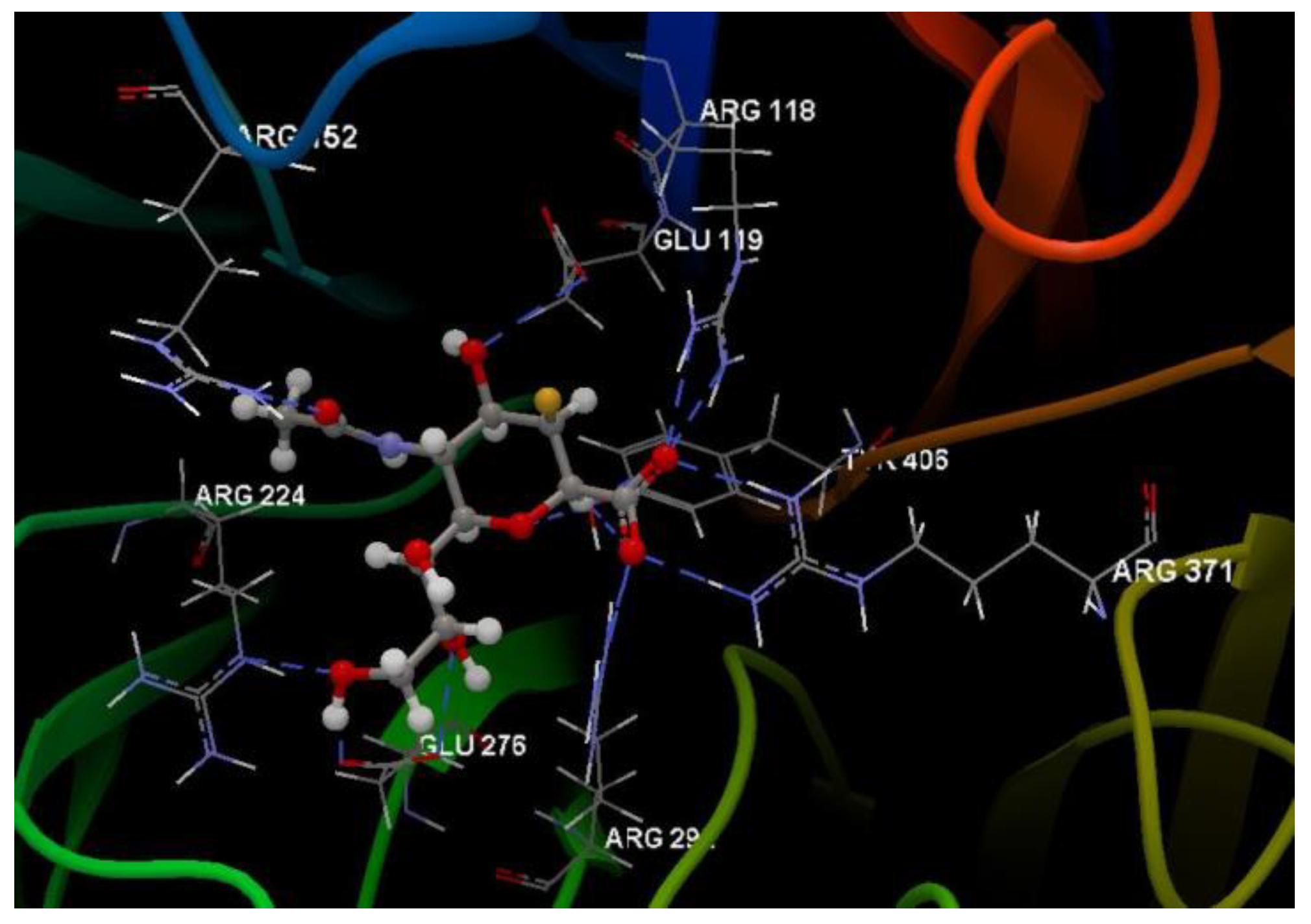
Docking evaluation against wild-type influenza N2 neuraminidase: docking studies were performed to achieve accurate predictions on the optimized conformations for both ligand and protein target to form a stable complex. All ligands were docked on the crystal structure of the wild-type influenza N2 neuraminidase (PDB ID: 4H52). The docking pose of the co-crystallized FSI A 508 interacting with amino acid residues of the active site is shown in Figure 7. Oseltamivir and ribavirin were taken as reference ligands to compare the docking results of all the studied compounds. The docking studies revealed that the 5kG compound has the best docking score −67.57 (RMSD: 0.58) (Table 1, Figure 4). The docking pose of the 5kG compound is shown in Figure 8.
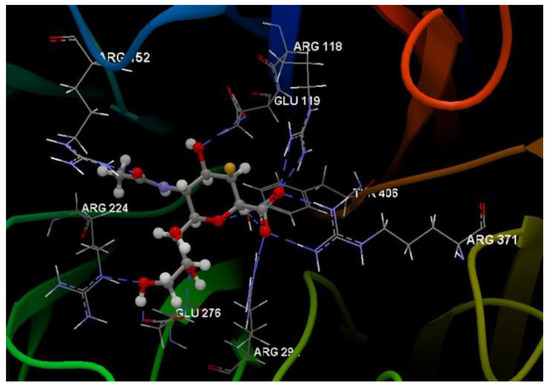
Figure 7.
Hydrogen bond (blue dotted lines) between FSI A 508 and amino acid residues from the binding site of 4H52.
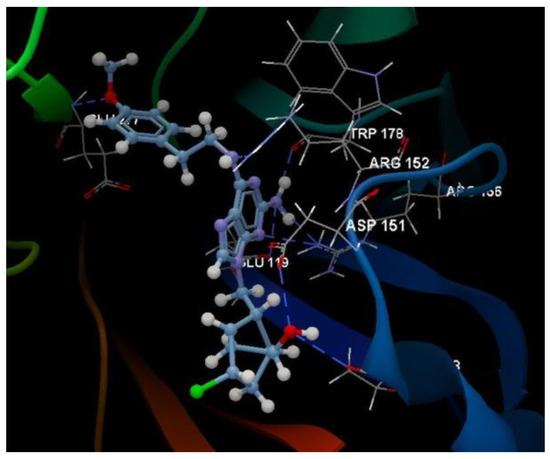
Figure 8.
Hydrogen bond (blue dotted lines) between the 5kG compound and amino acid residues from the binding site of 4H52.
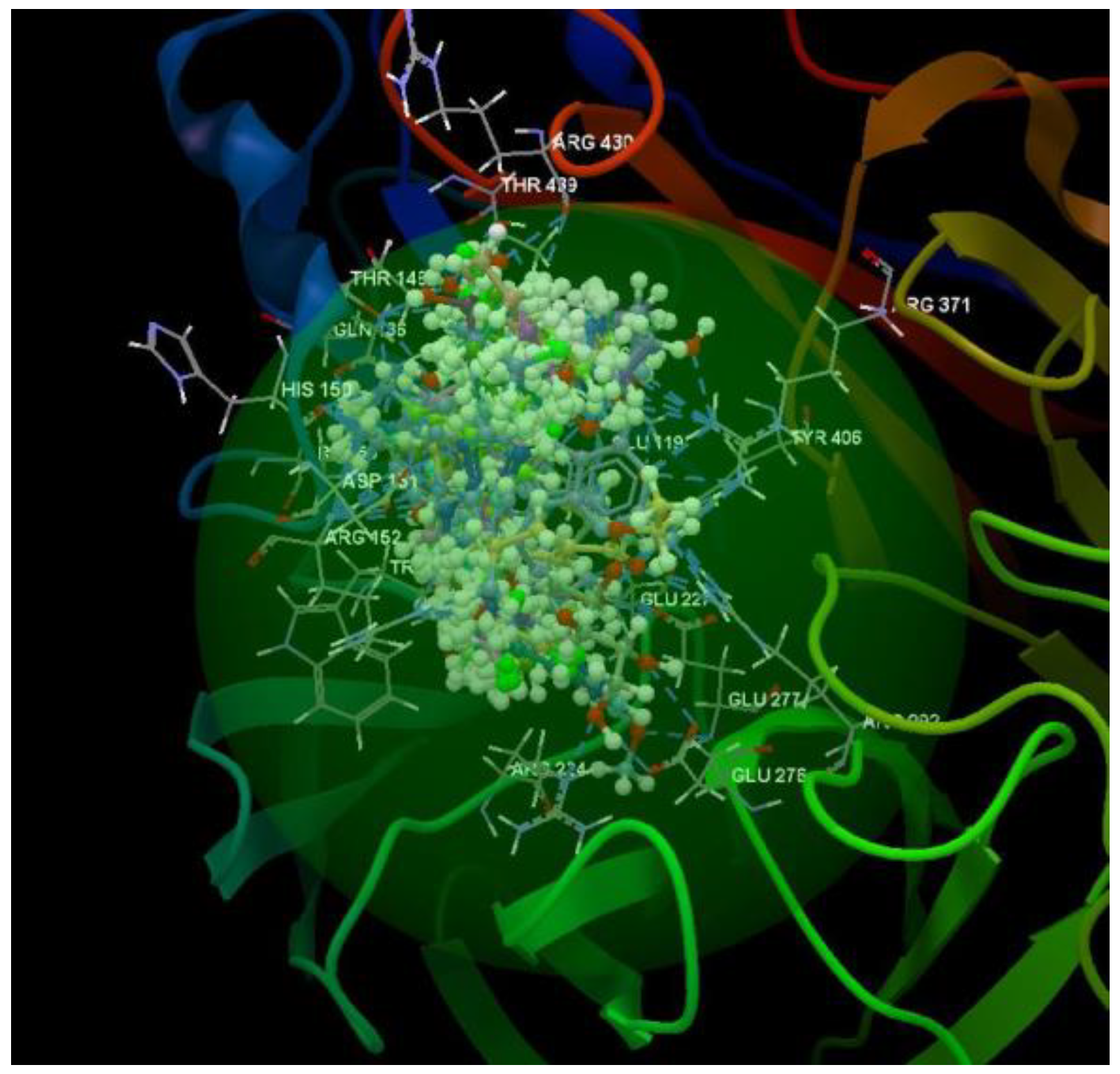
After analyzing the data obtained from the docking study, it was observed that all the compounds were placed in the same binding site of 4H52 as the co-crystallized (Figure 9).
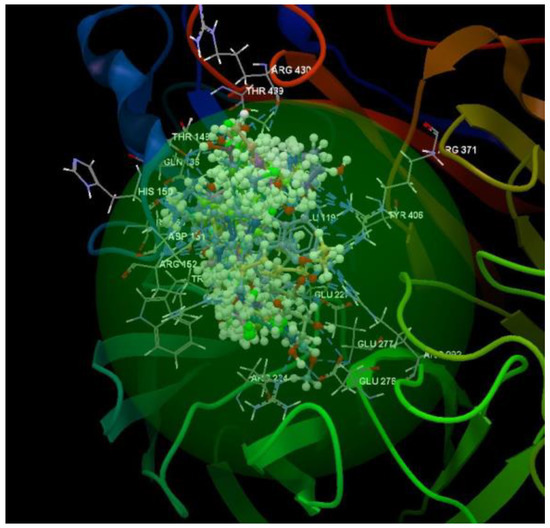
Figure 9.
Docking pose of the co-crystallized FSI A 508, of the oseltamivir and ribavirin and of the studied compounds in the binding site of 4H52.
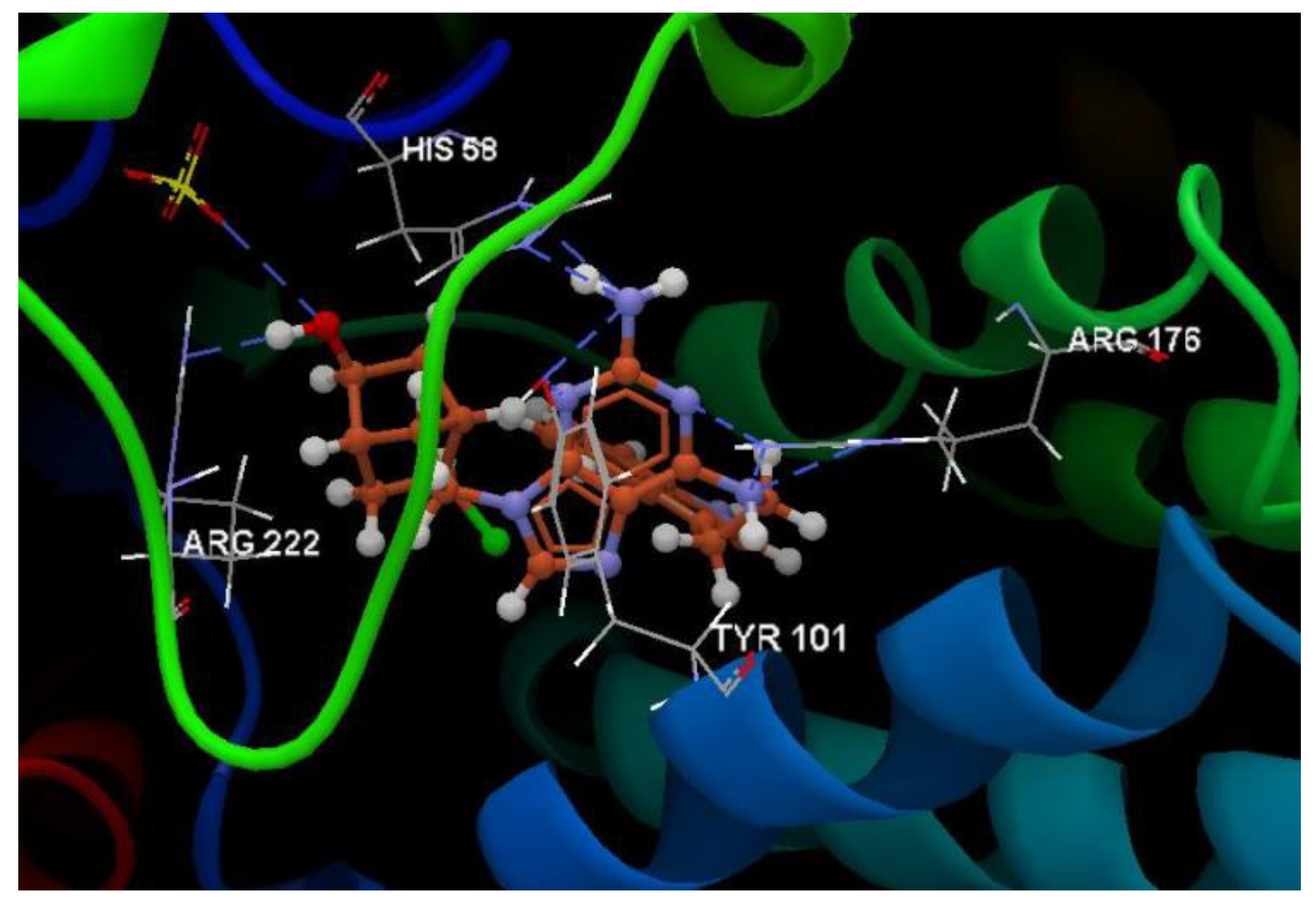
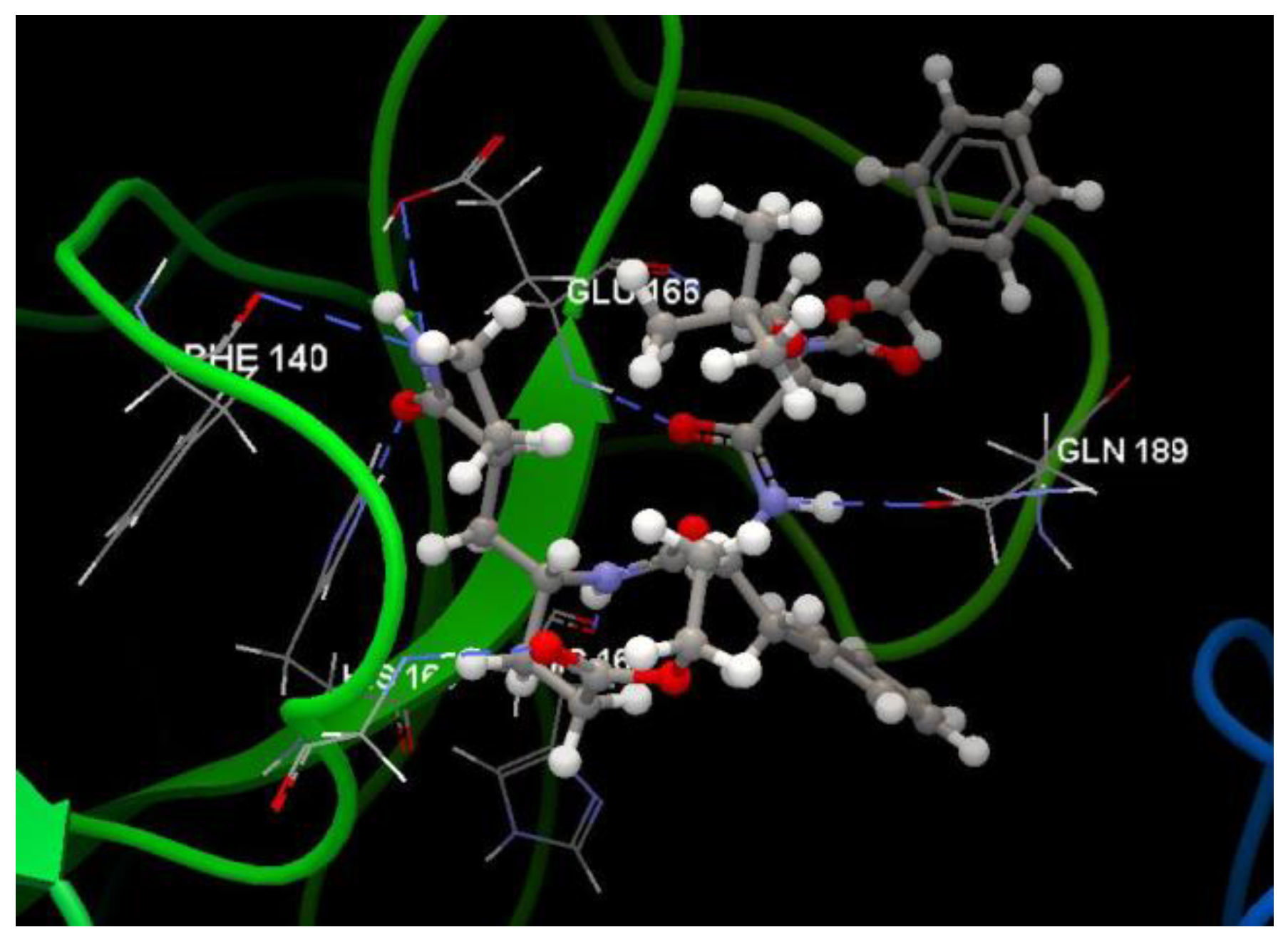
Docking evaluation against SARS coronavirus main protease: docking studies were performed to achieve accurate predictions on the optimized conformations for both ligand and protein target to form a stable complex. All ligands were docked on the crystal structure of the SARS coronavirus main protease (PDB ID: 3TNT). The docking pose of the co-crystallized G85A 501 interacting with amino acid residues of the active site is shown in Figure 10. Oseltamivir and ribavirin were taken as reference ligands to compare the docking results of all the studied compounds. The docking studies revealed that the 5jjG compound has the best docking score −81.11 (RMSD: 1.90) (Table 1, Figure 4). The docking pose of the 5jjG compound is shown in Figure 11.
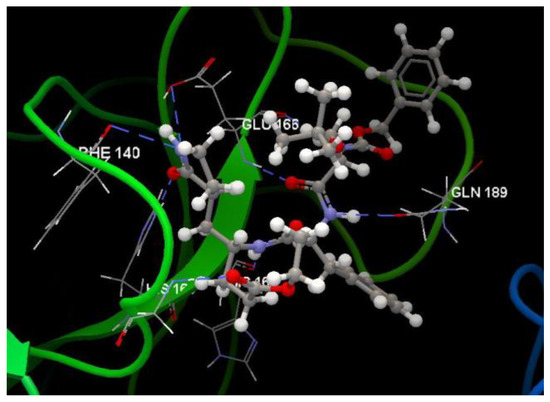
Figure 10.
Hydrogen bond (blue dotted lines) between G85A 501 and amino acid residues from the binding site of 3TNT.
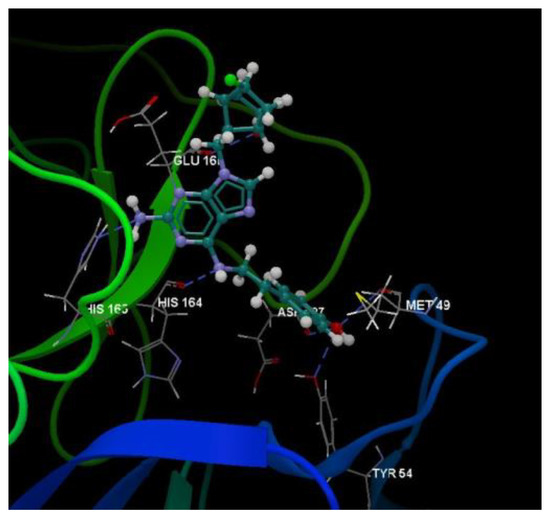
Figure 11.
Hydrogen bond (blue dotted lines) between the 5jjG compound and amino acid residues from the binding site of 3TNT.
After analyzing the data obtained from the docking study, it was observed that all the compounds were placed in the same binding site of 3TNT as the co-crystallized (Figure 12).
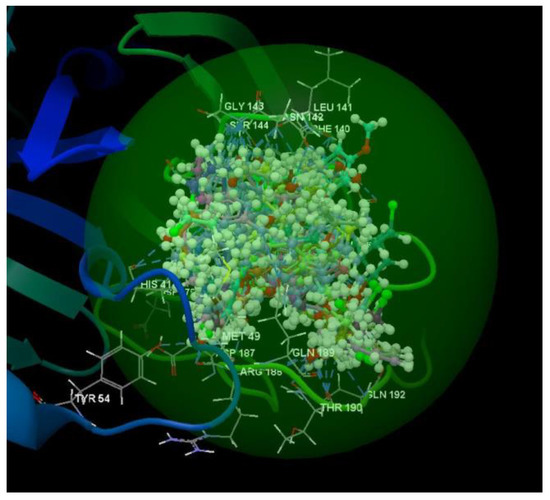
Figure 12.
Docking pose of the co-crystallized G85A 501, of the oseltamivir and ribavirin and of the studied compounds in the binding site of 3TNT.
Important molecular properties—molecular weight, flexible bonds, the number of hydrogen bond donors, the number of hydrogen bond acceptors and log P—were calculated (Table 2). These parameters can predict if a molecule possesses properties that might turn it into an orally active drug, according to Lipinski’s rule of five. The number of violations of the Lipinski rules allows one to evaluate drug likeness for a molecule. According to the data presented in Table 2, only the 5lG compound failed with respect to the Lipinski rules (Lipinski violation is 1, hydrogen bond donors >5).
Table 2.
Calculated properties of ligands.
The compounds were screened against influenza virus and two compounds, 5fG and 5gG, had SI of 25 and 23 (IC50 = 8 µM and 12.8 µM) and two SI of 10 (5kG, IC50 = 24 µM and 5sG, IC50 = 29 µM). The screening of the compounds against HSV and low active coronavirus is being developed and soon will be finished.
In conclusion, a number of 24 new 1′-homonucleoside with a 2-amino-6-substituted purine as nucleobase and an optically active bicyclo[2.2.0]heptane scaffold were synthesized, and a molecular docking study on three viruses and an experimental screening of the compounds against influenza virus were realized.
3. Patents
Tanase, C.; Pintilie, L. New 1′-homocarbanucleoside analogs with a constrained bicyclo[2.2.0]heptane fragment and 2-amino-6-substituted purine as nucleobase. Patent request A/00290/27.05.2020.
Author Contributions
Conceptualization, C.I.T.; methodology, C.I.T.; molecular docking L.P.; IR analysis, M.M.; NMR spectroscopy, C.D. and A.H.; antiviral screening, V.V.Z., A.V. and E.S.; writing—original draft preparation, C.I.T. and L.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Education and Research, UEFISCDI, grant number NUCLEU: PN 19-41 01 01.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Balzarini, J.; Naesens, L.; De Clercq, E. New antivirals—Mechanism of action and resistance development. Curr. Opin. Microbiol. 1998, 1, 535–546. [Google Scholar] [CrossRef]
- De Clerck, E.; Neyts, J. Antiviral Agents Acting as DNA or RNA Chain Terminator. In Handbook of Experimental Pharmacology 189; Kräusslich, H.-G., Bartenschlager, R., Eds.; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008; pp. 54–60. [Google Scholar]
- Tănase, C.; Drăghici, C.; Cojocaru, A.; Galochkina, A.V.; Orshanskaya, J.R.; Zarubaev, V.V.; Shova, S.; Enache, C.; Maganu, M. New carbocyclic N6-substituted adenine and pyrimidine nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; synthesis, antiviral, anticancer activity and X-ray crystallography. Bioorg. Med. Chem. 2015, 23, 6346–6354. [Google Scholar] [CrossRef] [PubMed]
- Tănase, C.; Drăghici, C.; Hanganu, A.; Pintilie, L.; Maganu, M.; Volobueva, A.; Sinegubova, E.; Zarubaev, V.; Neyts, J.; Jochman, D.; et al. New HSV-1 Anti-Viral 1′-Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Fragment as A Glycoside Moiety. Molecules 2019, 24, 2446. [Google Scholar] [CrossRef] [PubMed]
- Tănase, C.; Pintilie, L.; Mihai, E. New 1′-homocarbonucleoside Analogs with a Constrained Bicyclo[2.2.1]heptane Fragment. Patent Request A/00316/30.05, 2019. [Google Scholar]
- Tanase, C.; Pintilie, L. New 1′-homocarbanucleoside Analogs with a Constrained bicyclo[2.2.0]heptane Fragment and 2-amino-6-substituted Purine as Nucleobase. Patent request A/00290/27.05, 2020. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).