Abstract
The relationship between properties of aromatic polyamides and the structure and pKa of substituted diamine monomers allows for modeling and designing the synthesis of polymers with special properties for different uses (dialysis membranes, ion exchange systems, to thermo-resistant materials and impact protection devices). Nuclear Magnetic Resonance is a versatile analytical tool for such purposes. Based on the obtained equations (pKa = 13.1512 − 1.67039 × δ (ppm)), and chemical shift (δ, ppm) = −7.0593 − (37.57628 × φ) the reactivity of the monomers can be calculated and selectively modify some properties of polyamides. Aromatic diamines were synthetized according to established protocols and statistical-molecular studies were developed using Hyperchem pack-2005 and Mopac-6.
1. Introduction
The relationship between properties of aromatic polyamides and the structure of monomers allows for modeling and designing the synthesis of polymers with special properties [1,2,3] for different uses (dialysis membranes and ion exchange systems to thermo-resistant materials and impact protection devices). A singular dependence is observed for aromatic diamines, specifically with their basicity (pKa), which constitutes one of the fundamental criteria for evaluating their reactivities or reaction capacity under catalytic conditions.
Subbotin and Tacoronte [4] demonstrated the direct dependence between the magnitude of the chemical shifts of the protons of amino groups (NH2−) and the pKa values. Based on the equation found by them and on the experimental data of the chemical shift values (δ, ppm), the basicity of the primary amino groups, which characterize the reactivity of the monomers at the initial moment of the interphasic polycondensation, can be calculated.
This idea constituted the methodological basis for the study of the influence of various factors on the reactivity (or basicity) of the reacting centers in aromatic diamines with acidic ionogenic substituents (−COOH and −SO3H). Such substituents must influence the electronic density of the nitrogen atom of the amino group: first, due to its acceptor nature, and second, as a consequence of possible associative processes of intra- and intermolecular hydrogen bonding [5].
To evaluate this influence, the 1H-NMR spectra of amino-benzoic and aminosulfonic acids and their salts in dimethylacetamide (DMAA) were studied, specifically the values of the chemical shifts (ppm, δ) of the NH2− group because these substituted derivatives constitute simple models of monomers for polyamidation processes in order to obtain thermo-resistant polyamides with excellent properties for membranes. In parallel, for validating the scope of the proposal, we considered evaluating the significance of the net charge on the nitrogen atom, the reaction center in polyamidation processes, when aromatic diamine-type monomers are used.
2. Materials and Methods
The study of the molecules was carried out from molecular designs prepared by the HYPERCHEM-2005 program, and a first optimization of molecular geometry with molecular mechanics was carried out. The quantum mechanical calculations were performed at the semi-empirical level using the AM1 formalism, always setting the PRECISE option that lowers the gradient norm to 0.01 kcal/A (always also using the EF option). To perform the calculation, the Mopac package version 6.0 (MOPAC-6) was used. In this way the net charge of the nitrogen atom in each of the amino groups was calculated. With this result, chemical shift values (δ, ppm) of the hydrogen atom bonded to the nitrogen atom, the amino group (−NH2) can be obtained, from the charge values of the same reported by the MOPAC-6 program, and PM3 method.
To find a model that satisfactorily explains a dependency (structural correlation) between the chemical shift and the charge on the nitrogen atom, we use the ORIGIN program in its version 6.1 (2001–2005) and the STATGRAPHICS program in its version 3.1 (2004–2006).
The synthesis and molecular characterization processes are described in [6]. The recording of the 1H-NMR spectra was developed in a 200 MHz TESLA equipment, at 25 °C in dimethylacetamide with 2% CaCl2 or 2% LiCl, using as reference standard TMS (tetramethylsilane, 0.0 ppm, from 0.0 to 12.0 ppm).
The synthetic processes of polysubstituted aromatic diamines, grosso modo, included the use of intermediates from the textile dye industry such as diazo-derivatives and azo-derivatives with various amino (NH2−) or aminoacetoxy (AcONH−) groups or were generated via synthetic methodologies through a sequence of steps that included nitration reactions under eco-sustainable conditions, the insertion of acid ionogenic groups or their precursors, reduction reactions via catalytic hydrogenation and precipitation-isolation for molecular characterization via infrared spectroscopy (FTIR, 4300–500 cm−1, in KBr tablets), thermogravimetry and nuclear magnetic resonance (250 MHz, 0.5 mg analytical sample in CaCl2-dimethylformamide, 12–0.0 ppm, and dimethylsulfoxide-d6).
All the aromatic diamines synthesized are solids with variable coloration depending on the degree of substitution and type of functional groups, and were characterized by their physicochemical properties. Handling procedures, on a laboratory scale, were rigorously considered, given the toxicity and potential carcinogenicity of such organic systems [6,7].
3. Results and Discussion
Grosso modo, two basic conceptual positions were used for evaluating the reactivity (basicity, pKa) of the substituted aromatic diamines, used as monomers for polyamidation.
- Procedure 1—Correlation of chemical shift value (H-NH) vs. net charge on nitrogen of the amine group.
- Procedure 2—Chemical shift value-basicity correlation (reactivity or pKa).
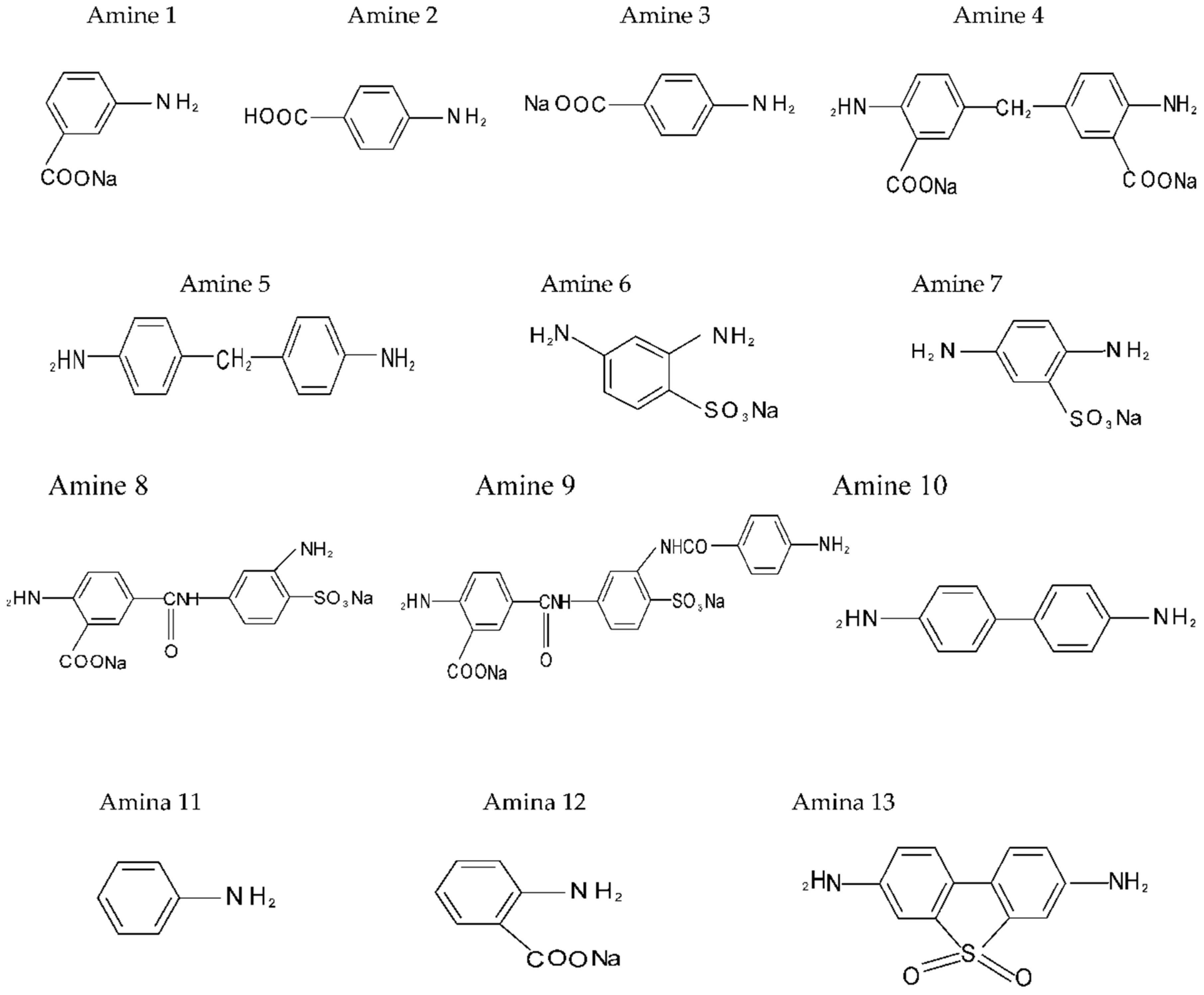
The aromatic diamines under study are depicted in Figure 1 depending on the procedure used to evaluate the structure–activity correlation: influence of the chemical shift (ppm, 1H-NMR) of the protons (H) bound to the atom of nitrogen in the amino group (NH2−) on the basicity or reactivity of the aromatic diamine type monomers; and assessment of the net charge correlation on the nitrogen atom vs. chemical shift (δ, ppm) and basicity. All the diamines under study were synthesized according to [6], under conditions of heterogeneous phase transfer catalysis in aqueous-organic systems of maximum miscibility.
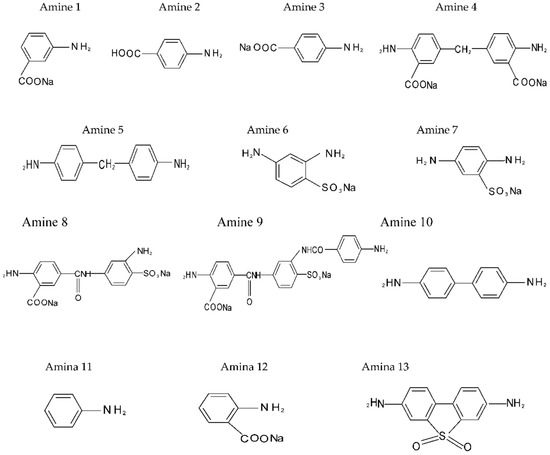
Figure 1.
Aromatic diamines (1–13) evaluated according to procedure 1.
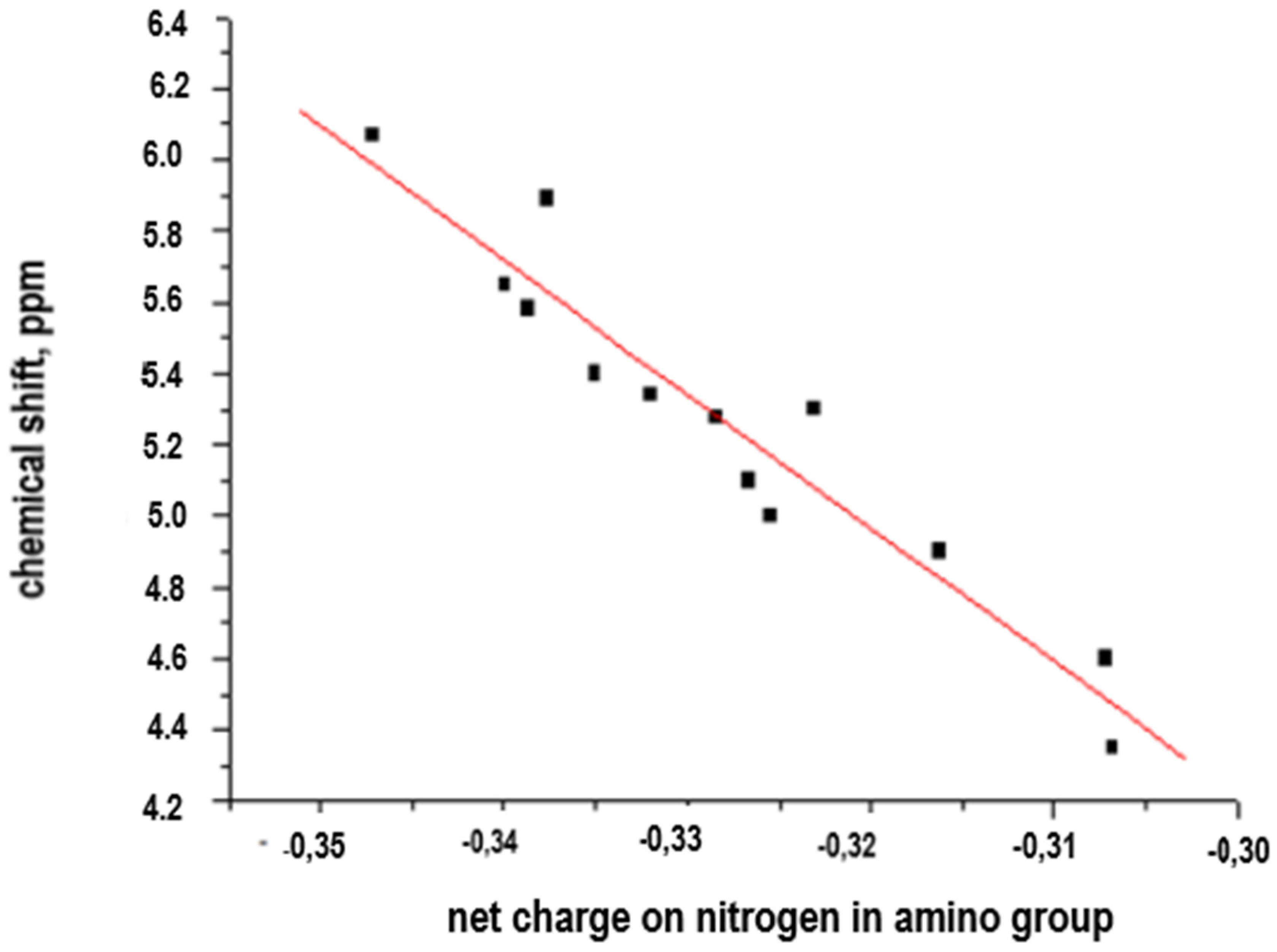
The results of the chemical shift correlation of aromatic amines vs. net charge on the nitrogen atom in NH2− are shown in Table 1 and Figure 2.

Table 1.
Chemical shift correlation of aromatic amines vs. net charge on the nitrogen atom in NH2−.
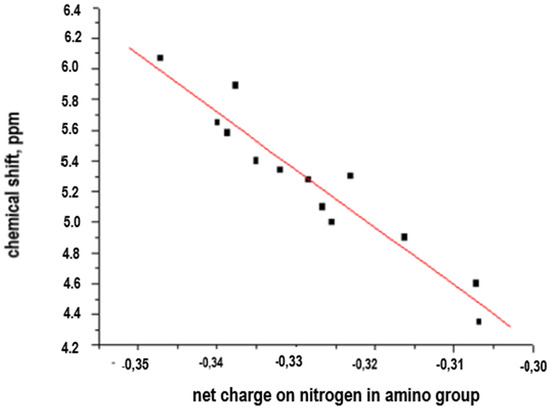
Figure 2.
Linear relationship of chemical shift data vs. net charge.
A preliminary analysis of the data presented in the Table 1 and evidence of a potential linear correlation allows us to consider the applicability of a simple system of equations (y = ax + b) and to assess the direct correlation between chemical shift and net charge. The obtained linearity is described in Equation (1).
The Equation (1) that describes the linear correlation of the chemical shift (δ, ppm) vs. net charge on the nitrogen atom in the amino groups is:
Chemical shift (δ, ppm) = −7.0593 − 37.57628 × Net Charge (φ) on the nitrogen atom in NH2−.
Correlation coefficient = −0.957361.
R-Squared = 91.654 %.
p-value = 0.0000.
Chemical shift (δ, ppm) = −7.0593 − (37.57628 × φ)
Equation (1): Linear correlation of the chemical shift (δ, ppm) vs. net charge on the nitrogen atom in the amino groups.
Considering that the value reported in the ANOVA table is less than 0.01, there is a statistically significant structural relationship between chemical shift and net charge for a 99% confidence level. The R-Squared value of the model explains the 91.654% of the variability of the chemical shift, which allows us to consider this conceptualization valid in order to correlate chemical shift data vs. net charge on reactive center in polyamidation processes from aromatic diamines as monomers for interphasic polycondensation.
The graphic representation of the correlation study for the poly-substituted aromatic diamines represented in Figure 1 is shown in Figure 2.
The observed linearity confirms the veracity of the hypothesis about the existence of a direct structure–properties correlation, and its potential applicability in polycondensation processes to determine the reactivity associated with the amino groups (−NH2−) of the aromatic-monomer diamines and potential variability of properties of aromatic polyamides with ionogenic groups (−COOH, −SO3H) and bridging groups (−CH2−, −NH−, −S−, −SO2−, −CONH−) depending on the reactivity of these amino groups (chemical shift, δ, in ppm, net charge on the nitrogen atom for each aromatic diamine). This linearity allows us to consider a satisfactory predictive character for this correlation.
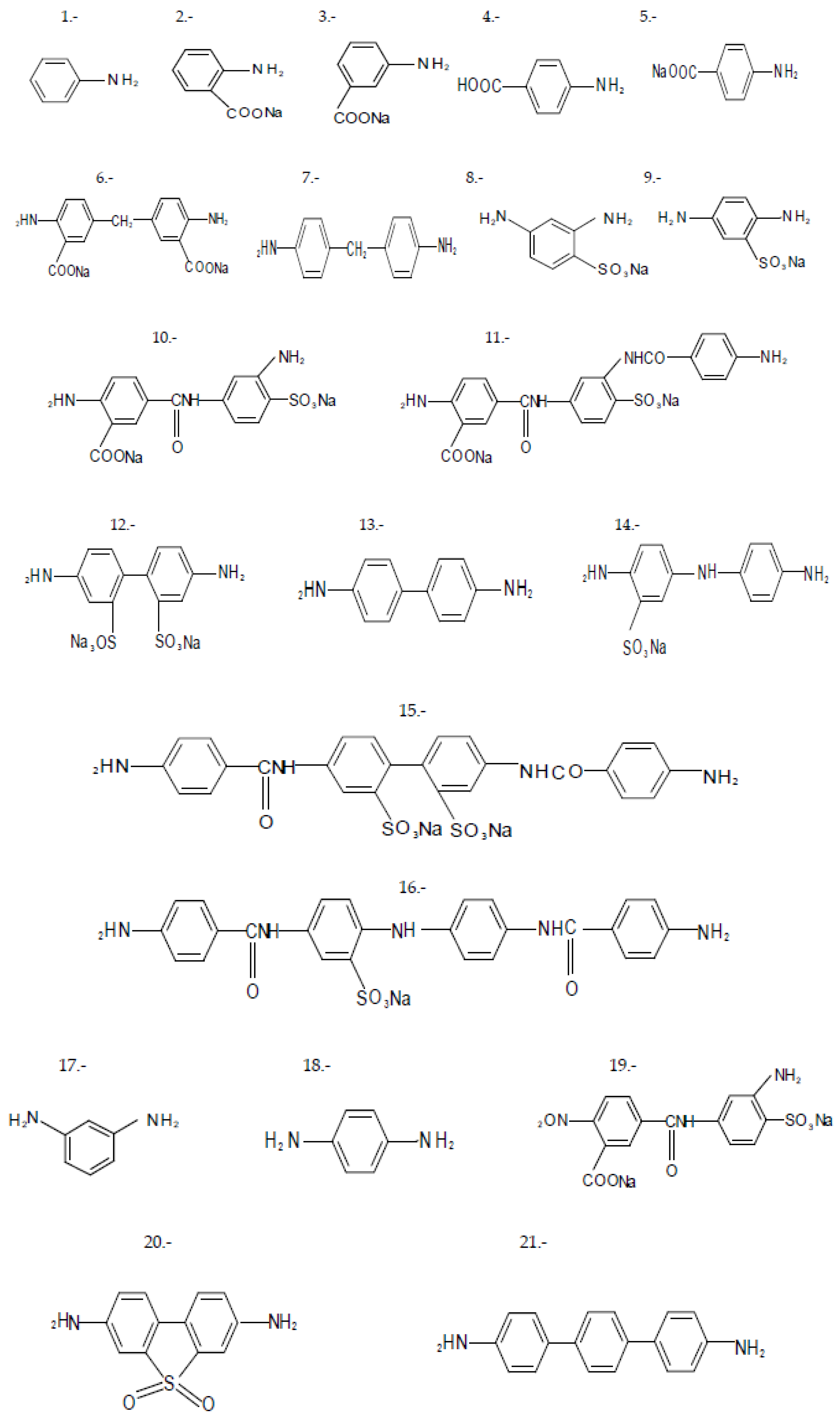
In a second approach, to evaluate the correlation between the structure and the properties of substituted aromatic diamines, the potential dependence between the chemical shift (δ, ppm) and the acidity constant (pKa) of a group of aromatic amines, which defines the reactivity of these, in interphasic polycondensation processes, was evaluated. In this case, used poly-substituted aromatic diamines with ionogenic groups (carboxyl and sulfonic, or in the form of alkaline salts), are represented in Figure 3. The results are shown in Table 2 and Figure 4.
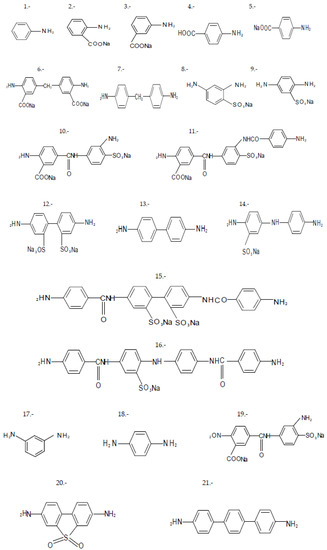
Figure 3.
Poly-susbtituted aromatic diamines used according to procedure 2.
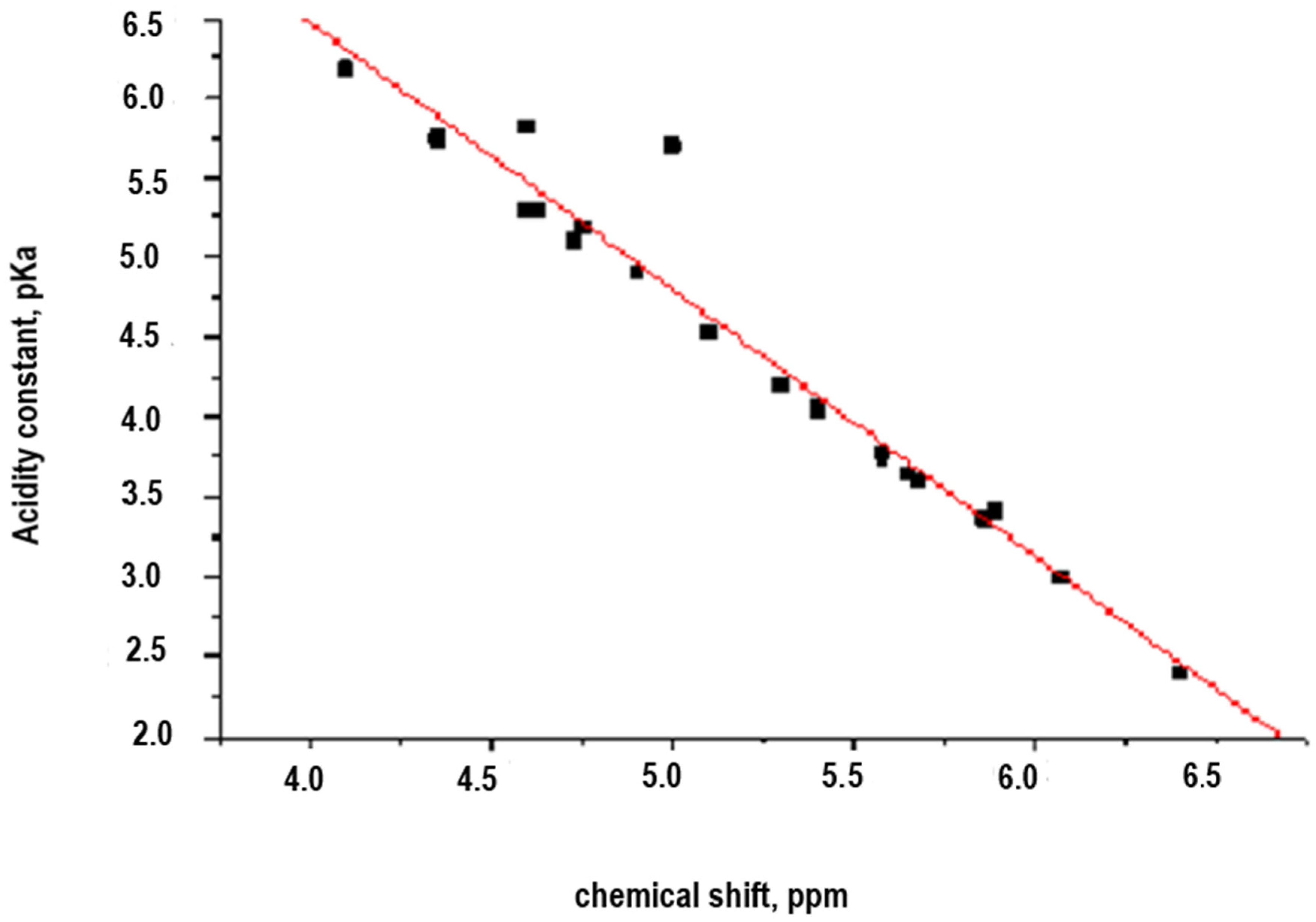
Table 2.
pKa values vs. chemical shift (δ, ppm) of poly-substituted aromatic diamines.
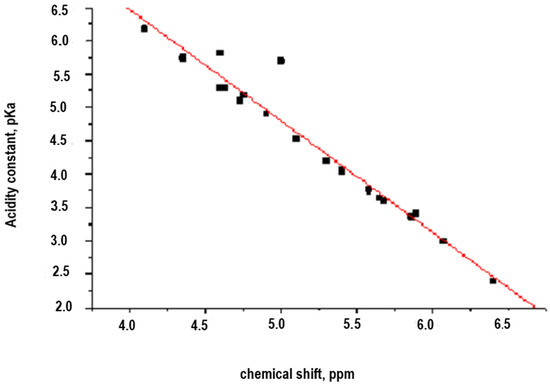
Figure 4.
Representation of the correlation pKa vs. chemical shift (δ, ppm, see Table 2).
To find a linear correlation between the pKa and the chemical shift (δ, ppm) of the hydrogen atom bonded to the nitrogen atom of the amino group in a series of aromatic amines (Table 2), the Statgraphics program was used in its version 2.1, because it is easy to manipulate and has a satisfactory degree of freedom and robustness, which allows rapid optimization. The results of the analysis are shown below and the correlation is expressed in Equation (2)
Regression analysis (linear model) y = a + bx
- Dependent variable: pKa (basicity or reactivity).
- Independent variable: chemical shift, (δ, ppm) of the proton signals (−NH2).
| Parameter | Estimated | Standar Deviation (S) | p Value |
| Intercept | 13.1512 | 0.492256 | 0.0000 |
| Slope | −1.67039 | 0.094198 | 0.0000 |
Correlation coefficient: −0.974017.
R-Squared: 94.8709%.
The Equation (2) for the model is
pKa = 13.1512 − 1.67039 × δ (chemical shift, in ppm)
Equation (2): Linear correlation of the pKa vs. the chemical shift (δ, ppm).
The R-squared value indicates that the model explains the 94.8709% of the variability of the acid constant (pKa). The correlation coefficient of −0.974017 indicates that there is a strong relationship between the variables for a confidence level of 99%.
To evaluate the predictive capacity and potential applicability of this model (procedure 2), the pKa values of some aromatic diamines were determined. The results are shown in Table 3.
Table 3.
Prediction of acidity constants (pKa) for a group of aromatic amines (whose pKa is not reported in the literature) using the equation obtained and the values of the chemical shift for the hydrogen atom bonded to the nitrogen of the amino group –NH2.
The real value of the pKa of amine 13, determined by the equation, is 4.63, but determined experimentally is 4.53, which is considered within the predicted confidence interval. In the case of amine 12, the real value is 5.30, which only differs by 0.117 from the theoretical value (5.417) and which is also considered within the predicted confidence interval. In the case of the predictions for amine 20 and amine 21, a comparison cannot be made, since the experimental value is not reported in the literature; which supports the possibility of using this procedure and model to predict the potential basicity (pKa), and consequently, reactivity, of these substituted aromatic diamines with ionogenic groups, in poliamidation processes for obtaining ionogenic polyamides with specific properties.
4. Conclusions
Using a simple procedure based on the recording of the nuclear magnetic resonance spectra, NMR-1H, and on the basis of the chemical shifts δ, in ppm, and the net charge on the nitrogen atom in the amino groups, -NH2- the pKa (basicity or reactivity) of a series of polysubstituted aromatic amines and aromatic diamines with ionogenic groups (−COOH and −SO3H, and their alkaline salts) which are used in interphasic polycondensation processes can be determined. The equation to determine the basicity, or pKa, (Equation (2)) has a satisfactory predictive capacity and allows one to quickly evaluate the reactivity of monomeric diamines. Currently, research is being carried out on structural optimization models for powerful QSAR studies.
Author Contributions
Individual contributions are as follows: J.E.T.M. conceptualization and methodology, investigation and spectral data analysis; M.T.C.P. writing and preparation, review and editing. J.C.S. preparation of the manuscript and data base; C.B.V. statistical analysis and correlation studies. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no government, academic or external funding.
Acknowledgments
To Technical University of Esmeraldas, for facilitating computational support during a pandemic.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Calderón, V. Síntesis, Caracterización, Propiedades y Aplicaciones de Nuevas Poliamidas Aromáticas con Subestructuras Benzoéter Corona y Benzopodandos en la Cadena Lateral. Ph.D. Thesis, Universidad de Burgos, Burgos, Spain, 2007. [Google Scholar]
- Herrero, M. Desarrollo de Nanocompuestos de Poliamida/Sepiolita para Aplicaciones Industriales. Ph.D. Thesis, Universidad de Valladolid, Valladolid, Spain, 2018. [Google Scholar]
- Coreño-Alonso, J.; Méndez-Bautista, M.T. Relación estructura-propiedades de polímeros. Educ. Quím. 2010, 21, 291–299. [Google Scholar] [CrossRef]
- Subbotin, V.; Tacoronte, J. Benceno-Sulfonic Amino Acids as Monomers for Thermoresistant Polyamides. URSS Patent No. 1527853, 8 August 1989. [Google Scholar]
- Tacoronte, J.; Guitis, S.S.; Subbotin, B.; Fedotov, Y.; Tereojina, L. Basicity Basicidad of aromatic aminoderivatives of sulfonic acids in acylation reactions. Monomers for thermoresistant polymers. Compend. Sci. Rep. Russ. Inst. Polym. Technol. 1988, 1988, 132–135. (In Russian) [Google Scholar]
- Tacoronte, J. Synthesis and Evaluation of Reactivity (pKa) of Sulphonic Aromatic Diamines, Monomers for Polyamidation in Heterphasic Conditions. Ph.D. Thesis, León Tolstoi University, Kazan, Russia, 1989. (In Russian). [Google Scholar]
- Benigni, R.; Giuliani, A.; Franke, R.; Gruska, A. Quantitative Structure−Activity Relationships of Mutagenic and Carcinogenic Aromatic Amines. Chem. Rev. 2000, 100, 3697–3714. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).