
Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19 †



Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of Mpro Sequences
2.2. Sequence Alignment and Multiple Sequence Comparisons
|
| Identities 294/306 (96%) |
| SARS Mpro 2AMD SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDTVYCPRHVICTAEDMLNPNYEDLLIR 65 |
| COVID-19 Mpro YP_009725301 ...................................................................................................V........................S.................................... 60 |
| SARS Mpro 2AMD KSNHSFLVQAGNVQLRVIGHSMQNCLLRLKVDTSNPKTPKYKFVRIQPGQTFSVLACYNG 125 |
| COVID-19 Mpro YP_009725301........N............................................................................V.K.........A........................................................... 120 |
| SARS Mpro 2AMD SPSGVYQCAMRPNHTIKGSFLNGSCGSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGK 185 |
| COVID-19 Mpro YP_009725301..............................................................F.................................................................................................N 180 |
| SARS Mpro 2AMD FYGPFVDRQTAQAAGTDTTITLNVLAWLYAAVINGDRWFLNRFTTTLNDFNLVAMKYNYE 245 |
| COVID-19 Mpro YP_009725301...........................................................................V....................................................................................... 240 |
| SARS Mpro 2AMD PLTQDHVDILGPLSAQTGIAVLDMCAALKELLQNGMNGRTILGSTILEDEFTPFDVVRQC 305 |
| COVID-19 Mpro YP_009725301..............................................................S.................................................AL............................................. 300 |
| SARS Mpro 2AMD SGVTFQ 311 |
| COVID-19 Mpro YP_009725301 ...... 306 |
|
| Identities 157/310 (51%) |
| MERS Mpro 5C3N SGLVKMSHPSGDVEACMVQVTCGSMTLNGLWLDNTVWCPRHVMCPADQLSDPNYDALLIS 60 |
| COVID-19 Mpro YP_009725301.........FR........AF...........K........G...............TT..............DV.......Y............I.......TSEDMLN.........ED..........R 60 |
| MERS Mpro 5C3N MTNHSFSVQKHIGAPANLRVVGHAMQGTLLKLTVDVANPSTPAYTFTTVKPGAAFSVLAC 120 |
| COVID-19 Mpro YP_009725301 KS.......N......L.......---AGNVQ........I.......S.......NCV........K.......T........K.......K......K......VRIQ.......QT..... 117 |
| MERS Mpro 5C3N YNGRPTGTFTVVMRPNYTIKGSFLCGSCGSVGYTKEGSVINFCYMHQMELANGTHTGSAF 180 |
| COVID-19 Mpro YP_009725301...........S.......S........VYQCA...........F............N............FNIDYDCVS..........H........PT......V.......A.......TDL 177 |
| MERS Mpro 5C3N DGTMYGAFMDKQVHQVQLTDKYCSVNVVAWLYAAILNGCAWFVKPNRTSVVSFNEWALAN 240 |
| COVID-19 Mpro YP_009725301 E.......NF........P.......V....R....TA....AAG.....TTIT......L............VI.....DR.....LNRFT....TLND.....LV....MKY 237 |
| MERS Mpro 5C3N QFTEFVGTQSVDM---LAVKTGVAIEQLLYAIQQLY-TGFQGKQILGSTMLEDEFTPEDV 296 |
| COVID-19 Mpro YP_009725301 NY-.....PLTQDH......ILGP.....SAQ......I.....VLDMCASLKE.....LQN.....MN.....RT...........AL..............F.. 296 |
| MERS Mpro 5C3N NMQIMGVVMQ 306 |
| COVID-19 mpro YP_009725301 VR.CS..TF. 306 |
2.3. Docking
2.4. Predictive ADME Studies
2.5. Toxicity
3. Results and Discussions
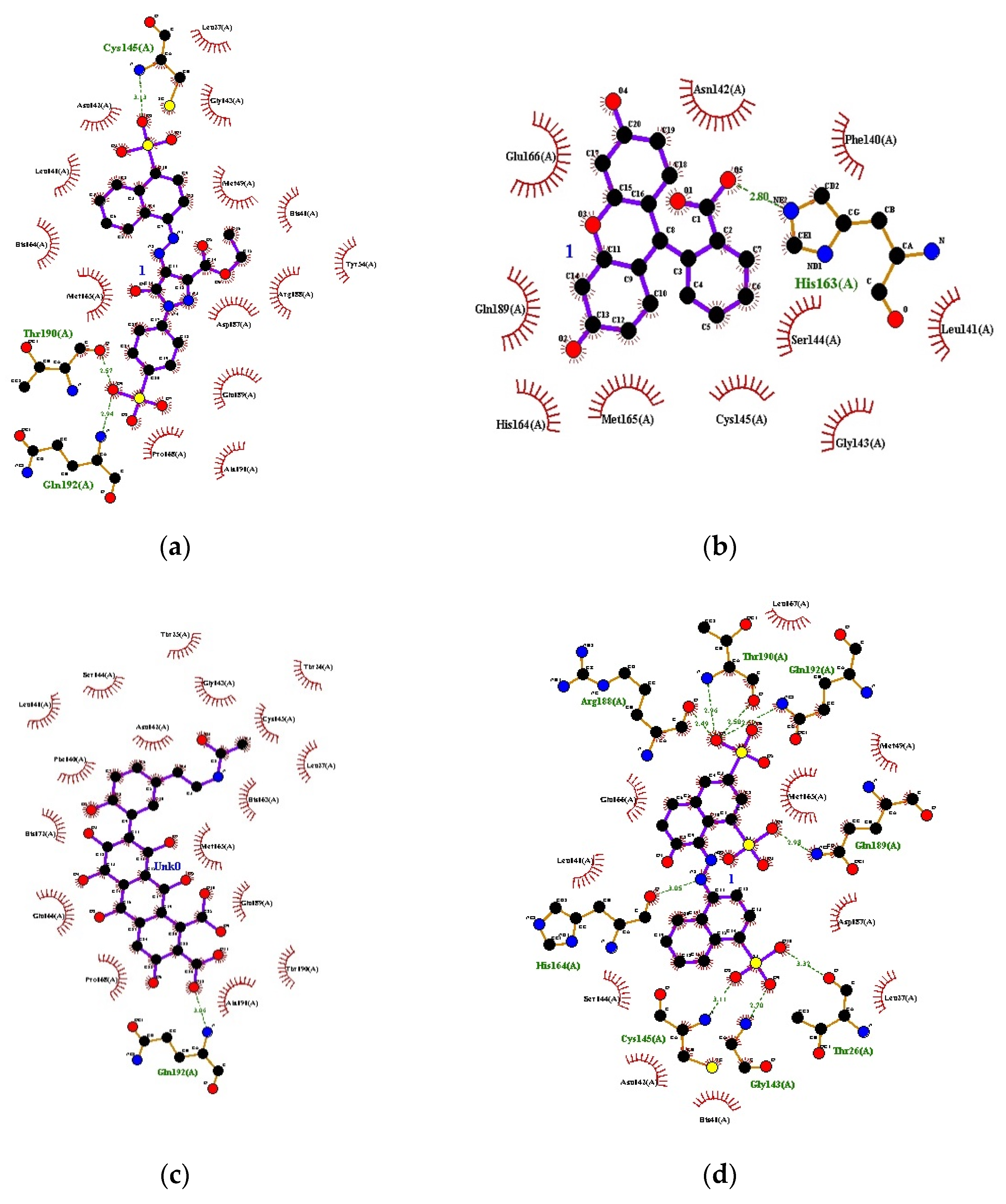
3.1. Docking
3.2. Predictive ADME Studies
- ⮚
- Molecular weight (mol MW) (150–650)
- ⮚
- Octanol/water partition coefficient (Log Po/w) (−2–6.5)
- ⮚
- Hydrogen bond donor (≤5)
- ⮚
- Hydrogen bond acceptor (≤10)
- ⮚
- Human oral absorption percentage (≥80% is high, ≤25% is poor)
3.3. Toxicity
- ⮚
- Class I—1800 (30 µg/kg bw/d)
- ⮚
- Class II—540 (9 µg/kg bw/d)
- ⮚
- Class III—90 (1.5 µg/kg bw/d)
4. Conclusions
Author Contributions
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Li, W.; Wong, S.-K.; Li, F.; Kuhn, J.H.; Huang, I.-C.; Choe, H.; Farzan, M. Animal Origins of the Severe Acute Respiratory Syndrome Coronavirus: Insight from ACE2-S-Protein Interactions. J. Virol. 2006, 80, 4211–4219. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.; Van Boheemen, S.; Bestebroer, T.; Osterhaus, A.; Fouchier, R. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Desenclos, J.C.; Van der Werf, S.; Bonmarin, I.; Levy-Bruhl, D.; Yazdanpanah, Y.; Hoen, B. Introduction of SARS in France, March–April, 2003. Emerg. Infect. Dis. 2004, 10, 195. [Google Scholar] [CrossRef] [PubMed]
- Guarner, J. Three Emerging Coronaviruses in Two Decades. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral pro- teases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Altaher, A.A.; Alnazawi, M. Molecular Dynamics and Inhibition of MERS CoV Papain-like Protease by Small Molecule Imidazole and Aminopurine Derivatives. Lett. Drug Des. Discov. 2019, 16, 584–591. [Google Scholar] [CrossRef]
- Li, Y.-H.; Hu, C.-Y.; Wu, N.-P.; Yao, H.; Li, L. Molecular Characteristics, Functions, and Related Pathogenicity of MERS-CoV Proteins. Engineering 2019, 5, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.swissadme.ch (accessed on 15 October 2020).
- Patlewicz, G.; Jeliazkova, N.; Safford, R.J.; Worth, A.P.; Aleksiev, B. An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR QSAR Environ. Res. 2008, 19, 495–524. [Google Scholar] [CrossRef] [PubMed]
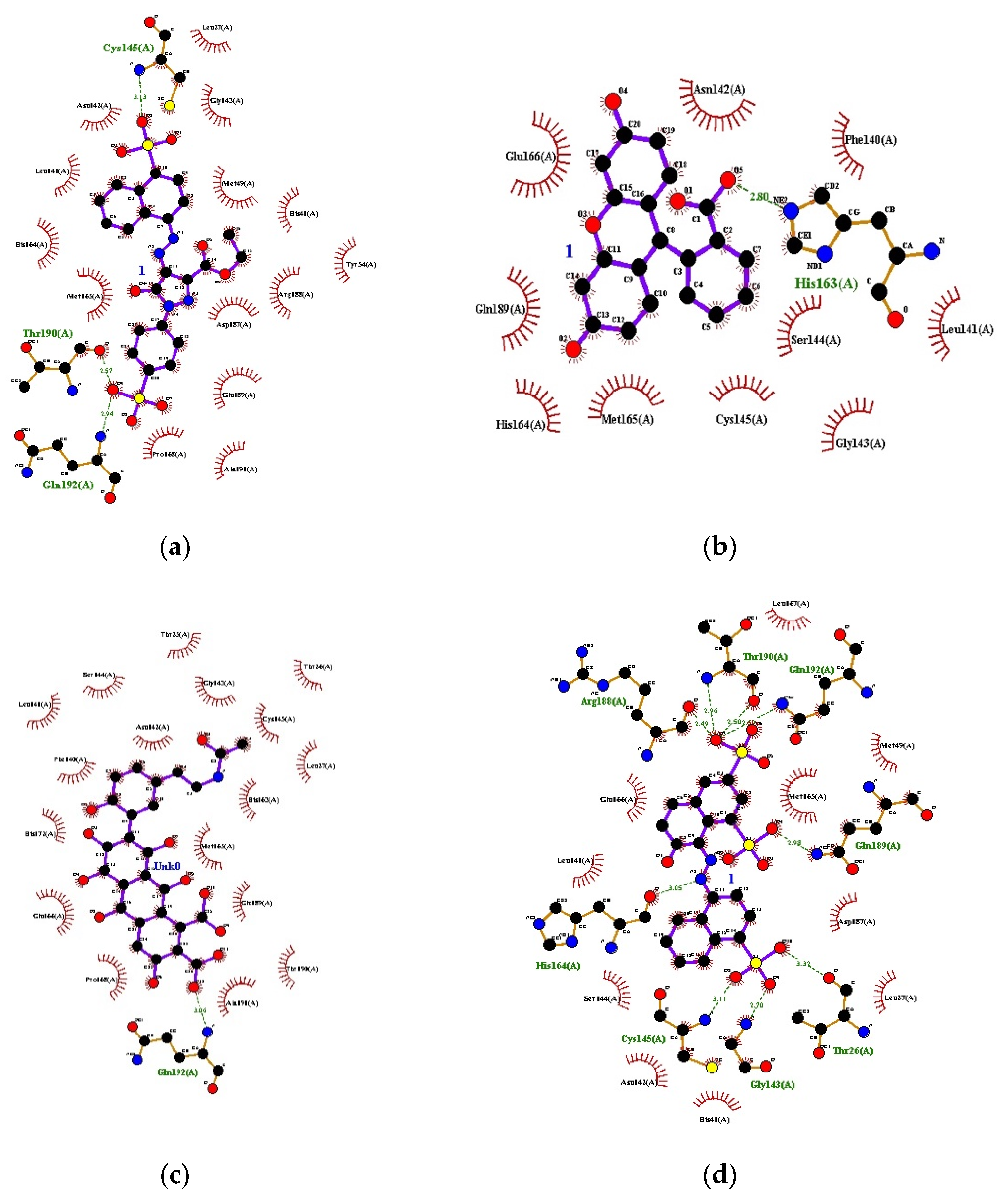
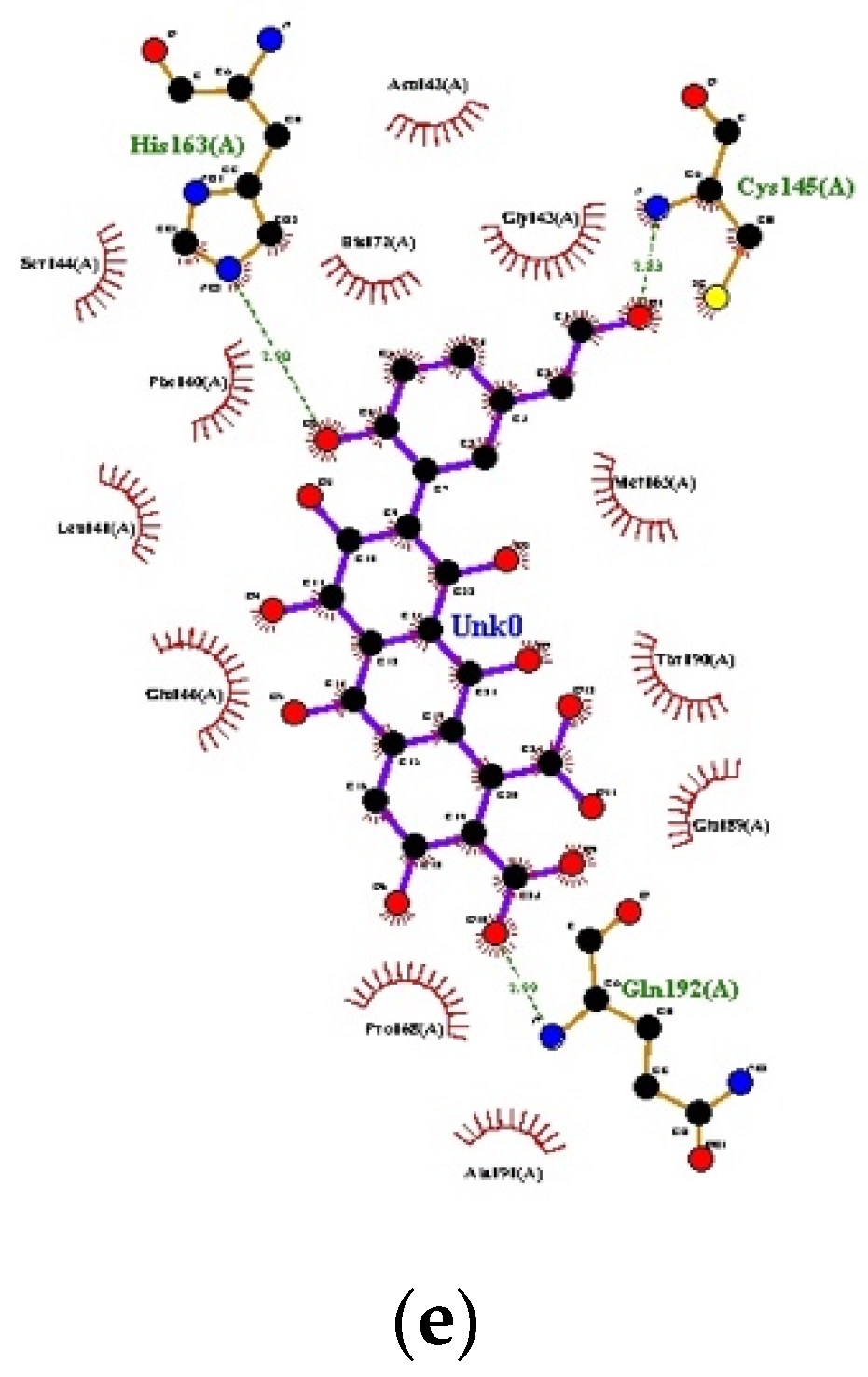

{kind=link}
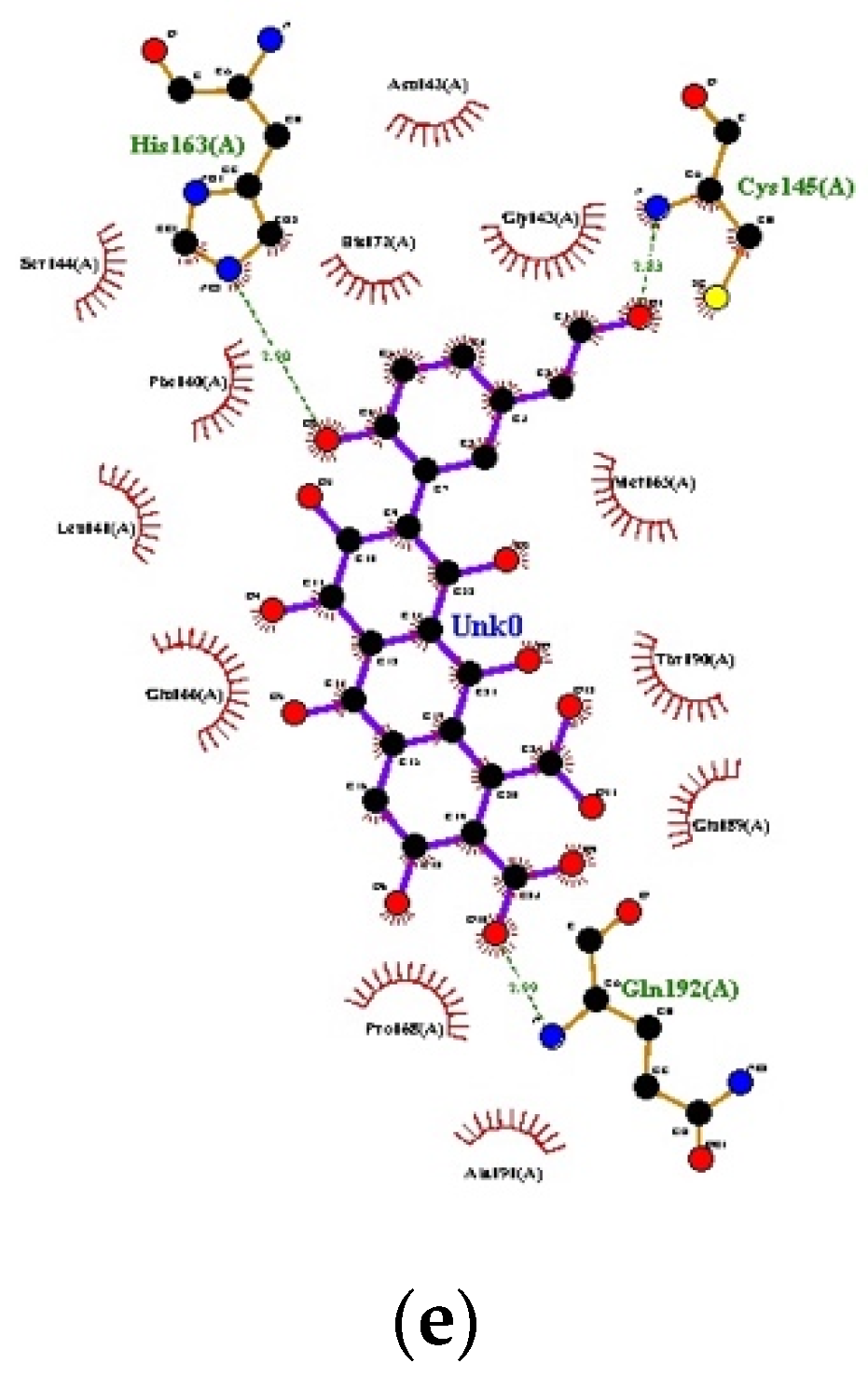{kind=link}
| S. No. | Ligands | 1st Run | 2nd Run | 3rd Run | |||
|---|---|---|---|---|---|---|---|
| Binding Energy | Inhibition Constant | Binding Energy | Inhibition Constant | Binding Energy | Inhibition Constant | ||
| 1 | DG01 | −10.35 | 26.12 nM | −9.99 | 47.43 nM | −9.91 | 54.73 nM |
| 2 | DG02 | −9.52 | 104.45 nM | −9.07 | 225.6 nM | −8.99 | 259.33 nM |
| 3 | DG03 | −9.43 | 121.71 nM | −9.29 | 154.77 nM | −9.28 | 158.05 nM |
| 4 | DG04 | −9.1 | 214.18 nM | −8.98 | 261.41 nM | −8.66 | 447.14 nM |
| 5 | DG05 | −9.00 | 251.81 nM | −8.89 | 305.47 | −8.87 | 314.38 nM |
| 6 | DG06 | −8.86 | 322.93 nM | −8.63 | 472.32 nM | −8.63 | 475.09 nM |
| 7 | DG07 | −8.53 | 555.76 nM | −8.53 | 561.87 nM | −8.52 | 571.48 nM |
| 8 | DG08 | −7.97 | 1.44 μM | −7.6 | 2.67 uM | −7.11 | 6.1 uM |
| 9 | DG09 | −7.86 | 1.73 μM | −7.72 | 2.2 uM | −7.63 | 2.54 uM |
| 10 | DG10 | −7.81 | 1.87 μM | −7.81 | 1.87 uM | −7.80 | 1.92 uM |
| 11 | DG11 | −7.42 | 3.63 μM | −7.33 | 4.24 uM | −7.28 | 4.6 uM |
| 12 | DG12 | −7.35 | 4.12 μM | −6.33 | 22.87 uM | −6.30 | 24.27 uM |
| 13 | DG13 | −7.34 | 4.14 μM | −7.28 | 4.62 uM | −7.32 | 4.32 uM |
| 14 | DG14 | −6.14 | 31.82 μM | −6.13 | 31.97 uM | −6.12 | 32.46 uM |
| 15 | DG15 | −6.24 | 26.75 μM | −4.79 | 307.68 uM | −5.78 | 58.44 uM |
| Compounds | DG01 | DG02 | DG03 | DG04 | DG05 | DG06 | DG07 | DG08 | DG09 | DG10 | DG11 | DG12 | DG13 | DG14 | DG15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Properties | |||||||||||||||
| MW | 546.53 | 538.53 | 835.89 | 537.43 | 496.38 | 458.46 | 273.29 | 561.69 | 539.4 | 314.25 | 468.42 | 408.41 | 422.39 | 495.39 | 538.41 |
| HBA | 11 | 11 | 5 | 12 | 12 | 9 | 3 | 7 | 14 | 7 | 12 | 9 | 8 | 12 | 13 |
| HBD | 3 | 4 | 2 | 8 | 8 | 3 | 0 | 3 | 9 | 4 | 3 | 3 | 4 | 8 | 7 |
| MR | 138.93 | 123.99 | 139.61 | 131.61 | 120.15 | 113.81 | 79.7 | 149.36 | 128.28 | 77.74 | 109.69 | 96.31 | 101.04 | 121.7 | 129.89 |
| TPSA | 208.86 | 229.71 | 75.99 | 238.99 | 230.12 | 170.45 | 47.03 | −1.14 | 273.21 | 132.13 | 220.19 | 170.45 | 183.7 | 235.91 | 236.19 |
| LOG Po/w | 1.54 | 1.37 | 5.23 | −1.25 | −1.25 | 2.8 | 2.05 | 2.94 | −4 | 0 | 0.32 | 2.02 | −0.18 | −0.71 | −0.31 |
| Solubility (mg/mL) | 1.13 × 10−2 | 6.97 × 10−2 | 4.44 × 10−7 | 6.15 × 10−3 | 7.05 × 10−3 | 4.58 × 10−3 | 6.63 × 10−3 | 4.22 × 10−6 | 3.51 × 10−1 | 4.22 × 10−2 | 5.74 × 10−1 | 5.59 × 10−2 | 1.63 × 10−1 | 5.74 × 10−4 | 2.90 × 10−3 |
| G.I absorption | Low | Low | High | Low | Low | Low | High | Low | Low | High | Low | Low | Low | Low | Low |
| BBB Permeant | No | No | No | No | No | No | Yes | No | No | No | No | No | No | No | No |
| CYP1A2 | No | No | Yes | No | Yes | No | Yes | No | No | No | No | No | No | Yes | Yes |
| CYP2D6 | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No |
| Veber | No | Yes | Yes | No | No | No | Yes | No | No | Yes | No | No | No | No | No |
| Lipinski | No | No | No | No | No | Yes | Yes | No | No | Yes | No | Yes | Yes | No | No |
| Bioavailability Score | 0.11 | 0.11 | 0.17 | 0.11 | 0.11 | 0.11 | 0.55 | 0.11 | 0.11 | 0.56 | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 |
| Compounds | Toxicity Class |
|---|---|
| DG01 | High Class III |
| DG02 | Low Class I |
| DG03 | High Class III |
| DG04 | High Class III |
| DG05 | High Class III |
| DG06 | Low Class I |
| DG07 | High Class III |
| DG08 | Low Class I |
| DG09 | High Class III |
| DG10 | High Class III |
| DG11 | Low Class I |
| DG12 | Low Class I |
| DG13 | Low Class I |
| DG14 | High Class III |
| DG15 | High Class III |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagur, P.; Seijas, J.A.A.; Mishra, A.; Ghosh, M. Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19. Chem. Proc. 2021, 3, 111. https://doi.org/10.3390/ecsoc-24-08353
Dagur P, Seijas JAA, Mishra A, Ghosh M. Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19. Chemistry Proceedings. 2021; 3(1):111. https://doi.org/10.3390/ecsoc-24-08353
Chicago/Turabian StyleDagur, Pankaj, Julio A. A. Seijas, Amarnath Mishra, and Manik Ghosh. 2021. "Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19" Chemistry Proceedings 3, no. 1: 111. https://doi.org/10.3390/ecsoc-24-08353
APA StyleDagur, P., Seijas, J. A. A., Mishra, A., & Ghosh, M. (2021). Molecular Docking Studies on Various Food Grade Dyes as a Potential Inhibitor of COVID-19. Chemistry Proceedings, 3(1), 111. https://doi.org/10.3390/ecsoc-24-08353