
Synthesis, ADME, and In Silico Molecular Docking Study of Novel N-Substituted β-Carboline Analogs as a Potential Anticancer Agent †


Abstract
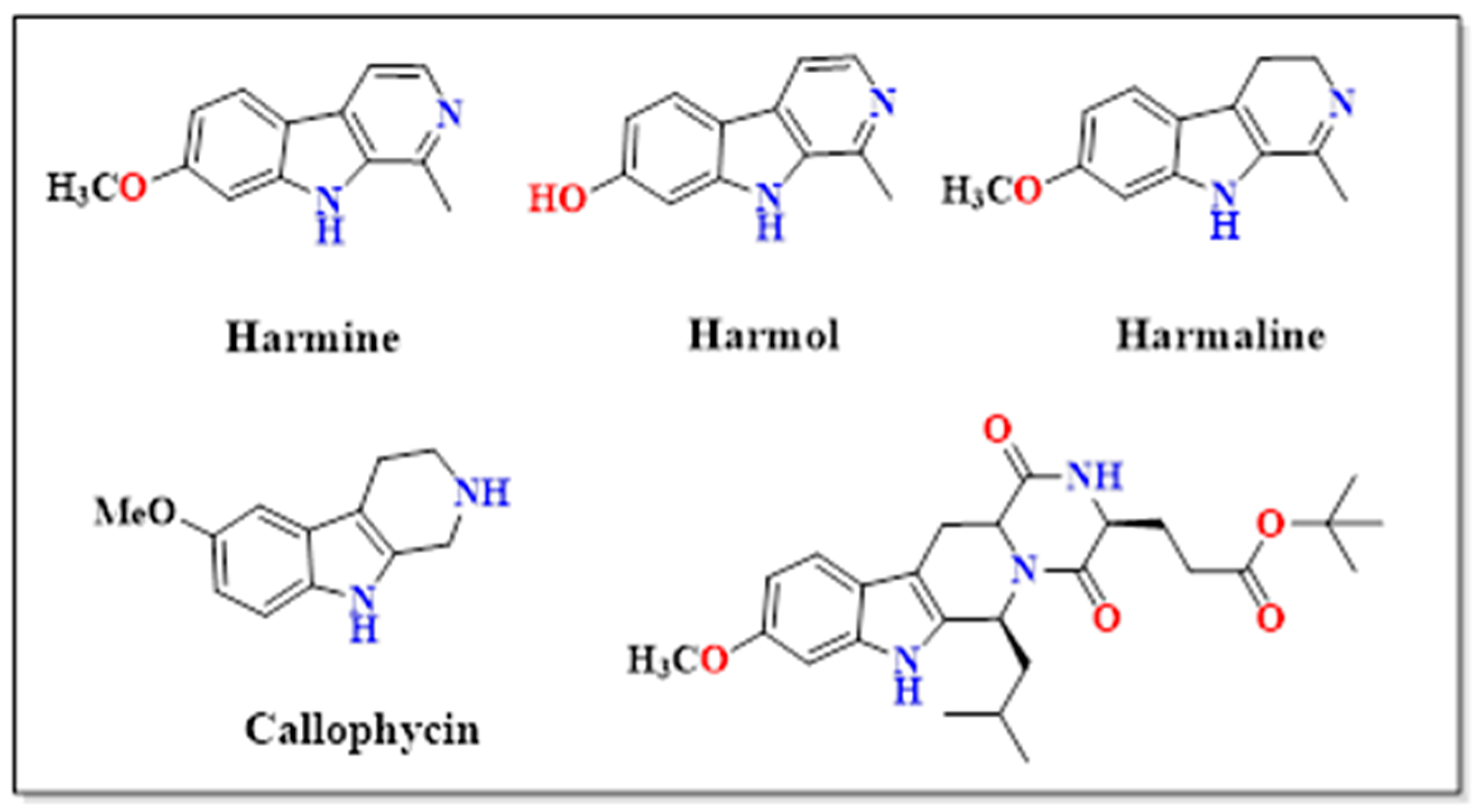
1. Introduction
2. Results and Discussion
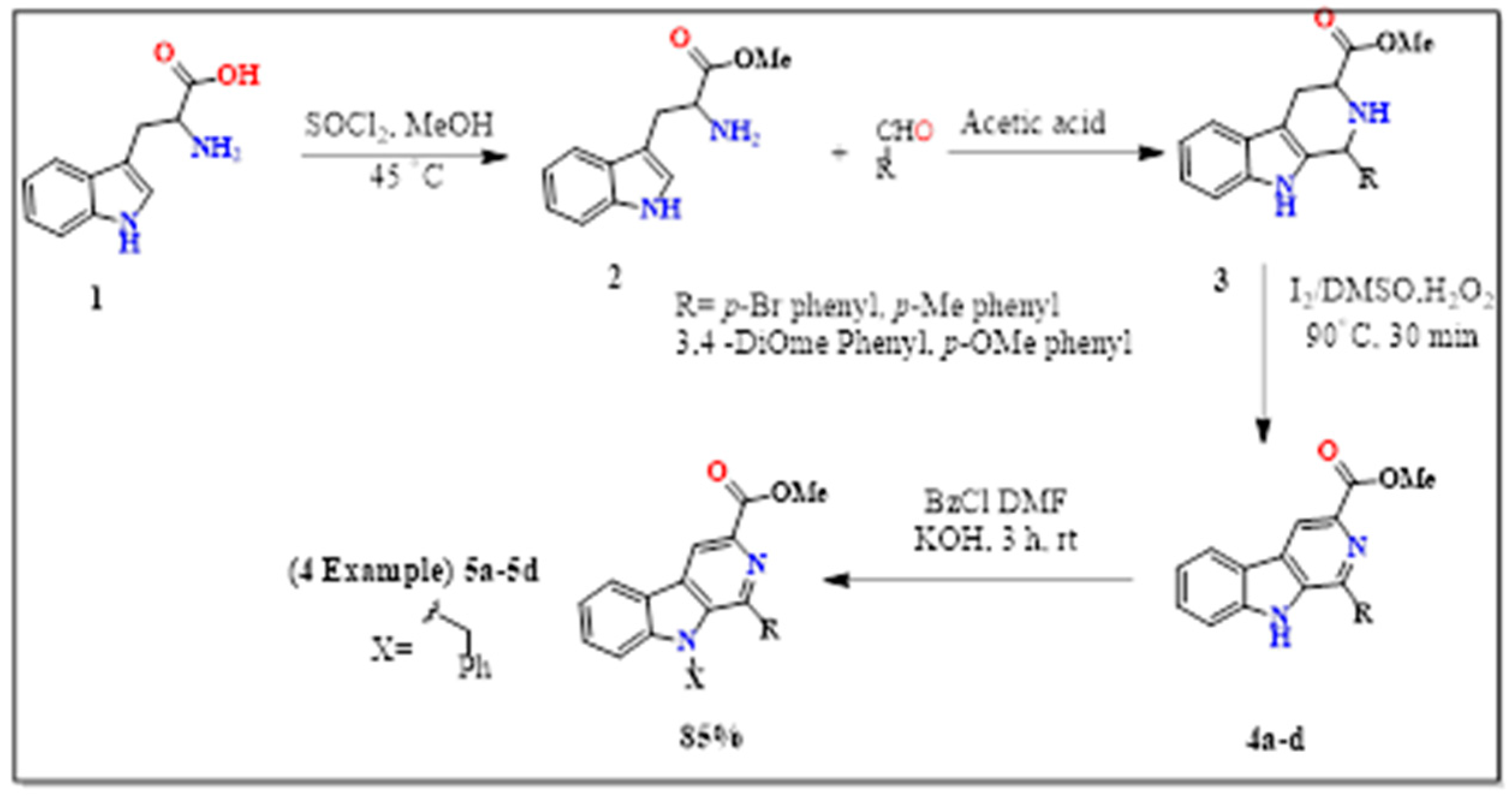
Synthesis of Methyl 9-(2,4-Dichlorobenzyl)-1-(p-Tolyl)-9H-Pyrido [3,4-b]Indole-3-Carboxylate
3. Experimental Section
3.1. Preparation of N-9-Alkyl-β-Carboline Methyl Ester
3.2. Methyl 1-(4-Methylphenyl)-9-(2,4-Dichlorobenzyl)-9H-Pyrido [3,4-b]Indole-3-Carboxylate
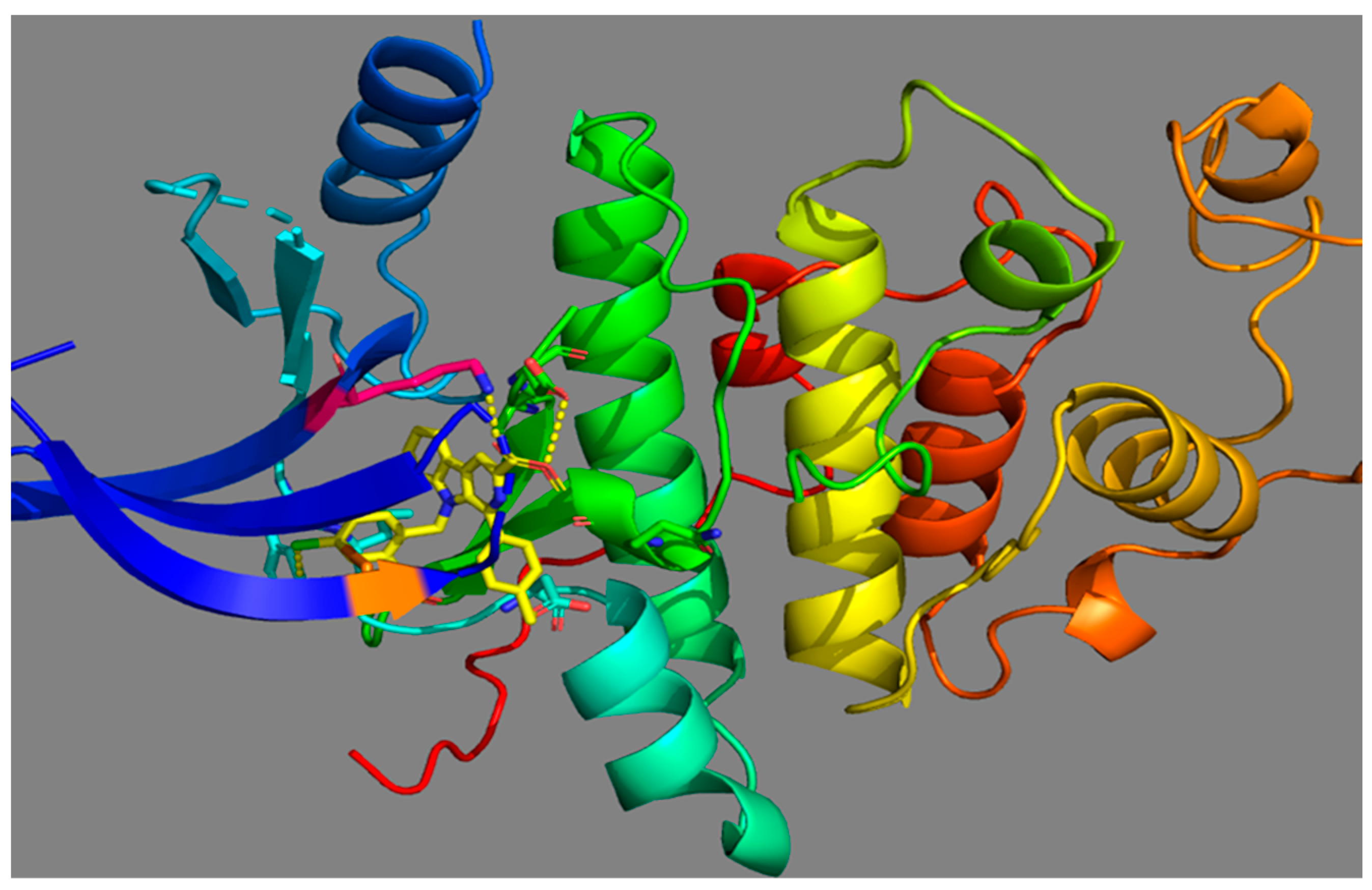
4. Molecular Docking
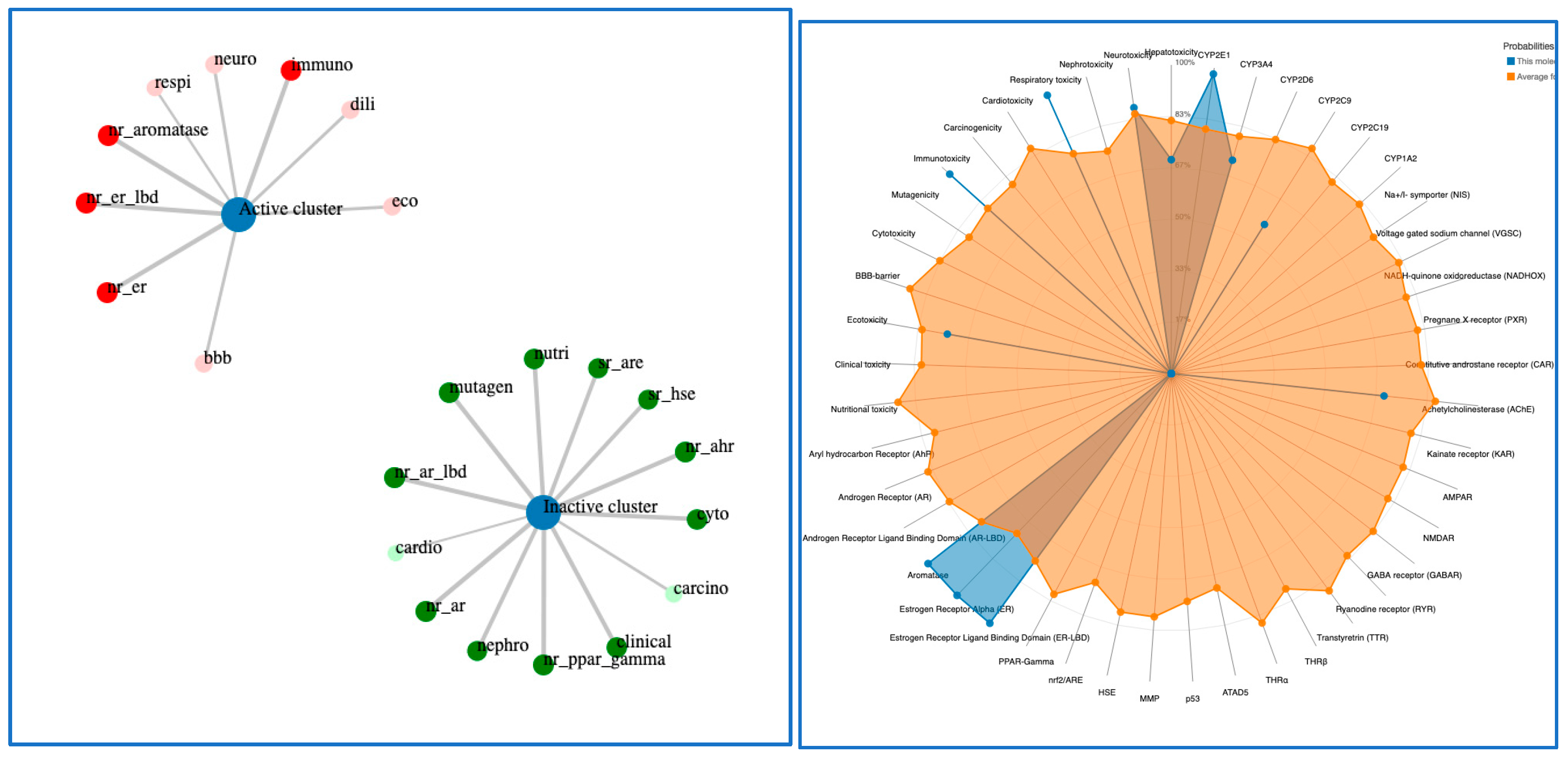
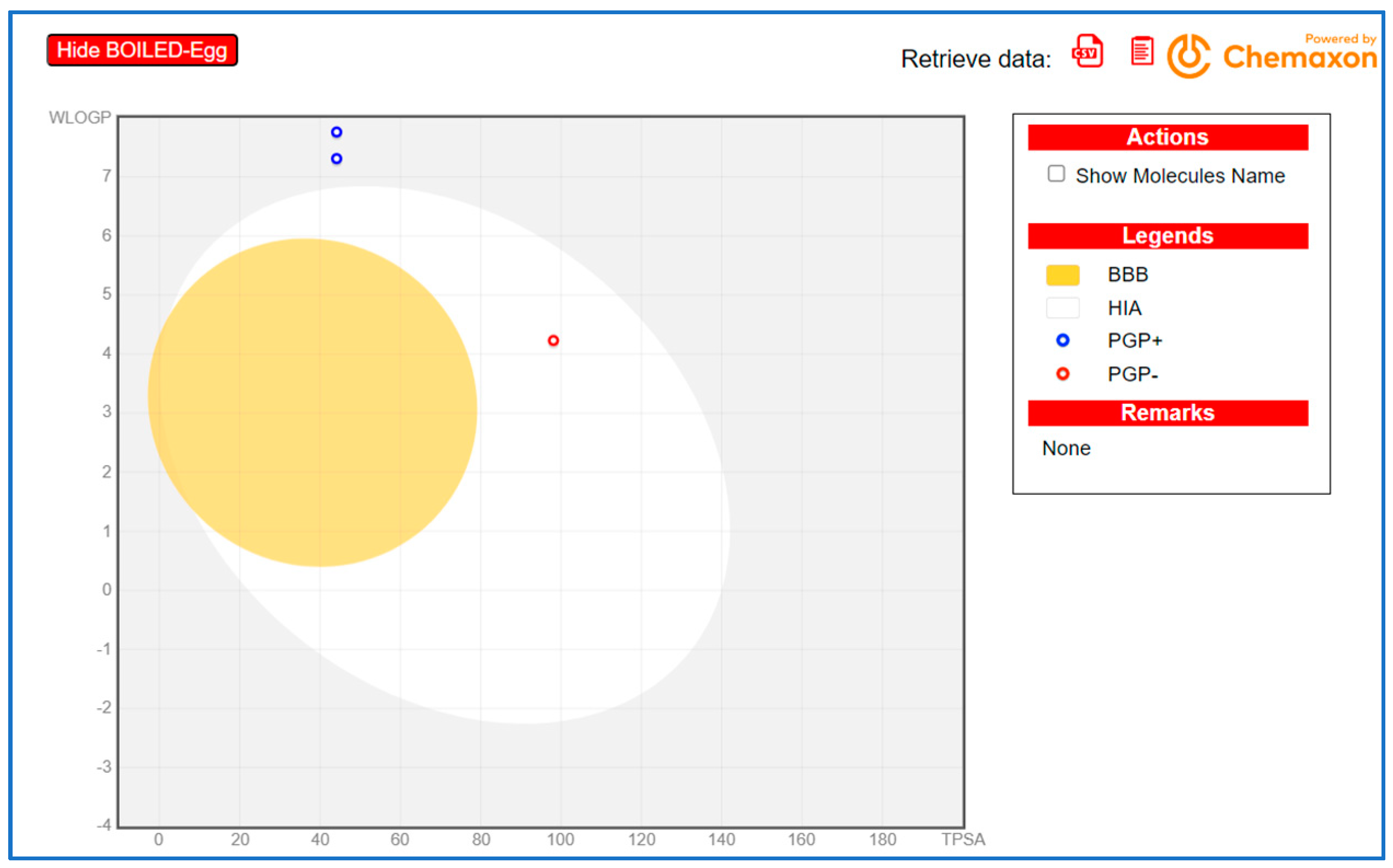
5. ADME Study and Toxicity
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sathishkumar, K.; Chaturvedi, M.; Das, P.; Stephen, S.; Mathur, P. Cancer incidence estimates for 2022 & projection for 2025: Result from National Cancer Registry Programme, India. Indian J. Med. Res. 2022, 156, 598–607. [Google Scholar] [PubMed]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Kinnel, B.; Singh, S.K.; Oprea-Ilies, G.; Singh, R. Targeted Therapy and Mechanisms of Drug Resistance in Breast Cancer. Cancers 2023, 15, 1320. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Ajith, A.K.; Subramani, S.; Manickam, A.H.; Ramasamy, S. Chemotherapeutic resistance genes of breast cancer patients-an overview. Adv. Pharm. Bull. 2022, 12, 649. [Google Scholar] [CrossRef]
- Kluth, M. Molecular In Vitro Analysis of the Human ABC Transporter MDR3. Doctoral Dissertation, Heinrich-Heine-Universität Düsseldorf, Düsseldorf, Germany, 2015. [Google Scholar]
- Gaikwad, S.; Kováčiková, L.; Pawar, P.; Gaikwad, M.; Boháč, A.; Dawane, B. An updates: Oxidative aromatization of THβC to β-carbolines and their application for the β-carboline alkaloids synthesis. Tetrahedron 2024, 155, 133903. [Google Scholar] [CrossRef]
- Manda, S.; Khan, S.I.; Jain, S.K.; Mohammed, S.; Tekwani, B.L.; Khan, I.A.; Vishwakarma, R.A.; Bharate, S.B. Synthesis, antileishmanial and antitrypanosomal activities of N-substituted tetrahydro-β-carbolines. Bioorganic Med. Chem. Lett. 2014, 24, 3247–3250. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Zhao, M.; Zhang, X.; Wang, Y.; Li, L.; Zheng, M.; Peng, S. A class of oral N-[(1S, 3S)-1-methyl-1, 2, 3, 4-tetrahydro-β-carboline-3-carbonyl]-N′-(amino-acid-acyl) hydrazine: Discovery, synthesis, in vitro anti-platelet aggregation/in vivo anti-thrombotic evaluation and 3D QSAR analysis. Eur. J. Med. Chem. 2011, 46, 3237–3249. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, G.; Cui, G.; Wang, W.X.; Zhao, M.; Wang, C.; Zhang, Z.; Peng, S. A new class of anti-thrombosis hexahydropyrazino-[1′, 2′: 1, 6] pyrido-[3, 4-b]-indole-1, 4-dions: Design, synthesis, log K determination, and QSAR analysis. Bioorganic Med. Chem. 2007, 15, 5672–5693. [Google Scholar] [CrossRef]
- Banerjee, R.; Kumar, M.; Gaurav, I.; Thakur, S.; Thakur, A.; Singh, K.; Karak, S.; Das, R.; Chhabra, M. In-silico Prediction of the Beta-carboline Alkaloids Harmine and Harmaline as Potent Drug Candidates for the Treatment of Parkinson’s disease. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2021, 20, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Rüben, K.; Wurzlbauer, A.; Walte, A.; Sippl, W.; Bracher, F.; Becker, W. Selectivity profiling and biological activity of novel β-carbolines as potent and selective DYRK1 kinase inhibitors. PLoS ONE 2015, 10, e0132453. [Google Scholar] [CrossRef]
- Gaikwad, S.V.; Nadimetla, D.N.; Kobaisi, M.A.; Devkate, M.; Joshi, R.; Shinde, R.G.; Gaikwad, M.V.; Nikalje, M.D.; Bhosale, S.V.; Lokhande, P.D. Iodine-DMSO-Catalyzed Chemoselective Biomimetic Aromatization of Tetrahydro-β-carbolines-3-carboxylic Acid: Mechanism Study with DFT-Calculation. Chem. Sel. 2019, 4, 10054–10059. [Google Scholar] [CrossRef]
- Gaikwad, S.V.; Gaikwad, M.V.; Lokhande, P.D. Iodine-DMSO catalyzed chemoselective oxidative aromatization and deallylation, nondeallylation of aryl allyl ether of tetrahydro-β-carboline. J. Heterocycl. Chem. 2021, 58, 1408–1414. [Google Scholar] [CrossRef]
- Gaikwad, S.; Kamble, D.; Lokhande, P. Iodine-catalyzed chemoselective dehydrogenation and aromatization of tetrahydro-β-carbolines: A short synthesis of Kumujian-C, Eudistomin-U, Norharmane, Harmane Harmalan and Isoeudistomine-M. Tetrahedron Lett. 2018, 59, 2387–2392. [Google Scholar] [CrossRef]
- Gaikwad, M.V.; Gaikwad, S.V.; Kamble, R.D. Mild and efficient ammonium chloride catalyzed Greener synthesis of tetrahydro-β-carboline. Curr. Res. Green Sustain. Chem. 2022, 5, 100268. [Google Scholar] [CrossRef]
- Gaikwad, S.; González, C.M.; Vilariño, D.; Lasanta, G.; Villaverde, C.; Mouriño, A.; Verlinden, L.; Verstuyf, A.; Peluso-Iltis, C.; Rochel, N.; et al. Lithocholic acid-based design of noncalcemic vitamin D receptor agonists. Bioorganic Chem. 2021, 111, 104878. [Google Scholar] [CrossRef] [PubMed]
- Madej, T.; Lanczycki, C.J.; Zhang, D.; Thiessen, P.A.; Geer, R.C.; Marchler-Bauer, A.; Bryant, S.H. MMDB and VAST+: Tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014, 42, D297–D303. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, M.; Gaikwad, S.; Kamble, R. Synthesis of novel series of 1-(6-hydroxy-4-(1H-indol-3-yl)-3, 6-dimethyl-4, 5, 6, 7-tetrahydro-1H-indazol-5-yl) ethan-1-oneas evaluations of their antimicrobial activity with insilco docking study. J. Med. Chem. Sci. 2022, 5, 239–248. [Google Scholar]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr/No | Product | %Yield | Product | %Yield |
|---|---|---|---|---|
| 1 | 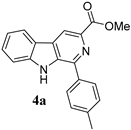 | 90% | 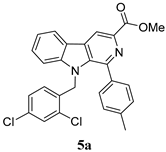 | 71% |
| 2 | 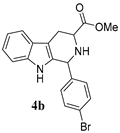 | 91% |  | 68% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaikwad, S. Synthesis, ADME, and In Silico Molecular Docking Study of Novel N-Substituted β-Carboline Analogs as a Potential Anticancer Agent. Chem. Proc. 2024, 16, 76. https://doi.org/10.3390/ecsoc-28-20166
Gaikwad S. Synthesis, ADME, and In Silico Molecular Docking Study of Novel N-Substituted β-Carboline Analogs as a Potential Anticancer Agent. Chemistry Proceedings. 2024; 16(1):76. https://doi.org/10.3390/ecsoc-28-20166
Chicago/Turabian StyleGaikwad, Sunil. 2024. "Synthesis, ADME, and In Silico Molecular Docking Study of Novel N-Substituted β-Carboline Analogs as a Potential Anticancer Agent" Chemistry Proceedings 16, no. 1: 76. https://doi.org/10.3390/ecsoc-28-20166
APA StyleGaikwad, S. (2024). Synthesis, ADME, and In Silico Molecular Docking Study of Novel N-Substituted β-Carboline Analogs as a Potential Anticancer Agent. Chemistry Proceedings, 16(1), 76. https://doi.org/10.3390/ecsoc-28-20166