
Theoretical Study of Intermolecular Interactions in Benzopyrans Substituted with Polyhaloalkyl Groups †



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
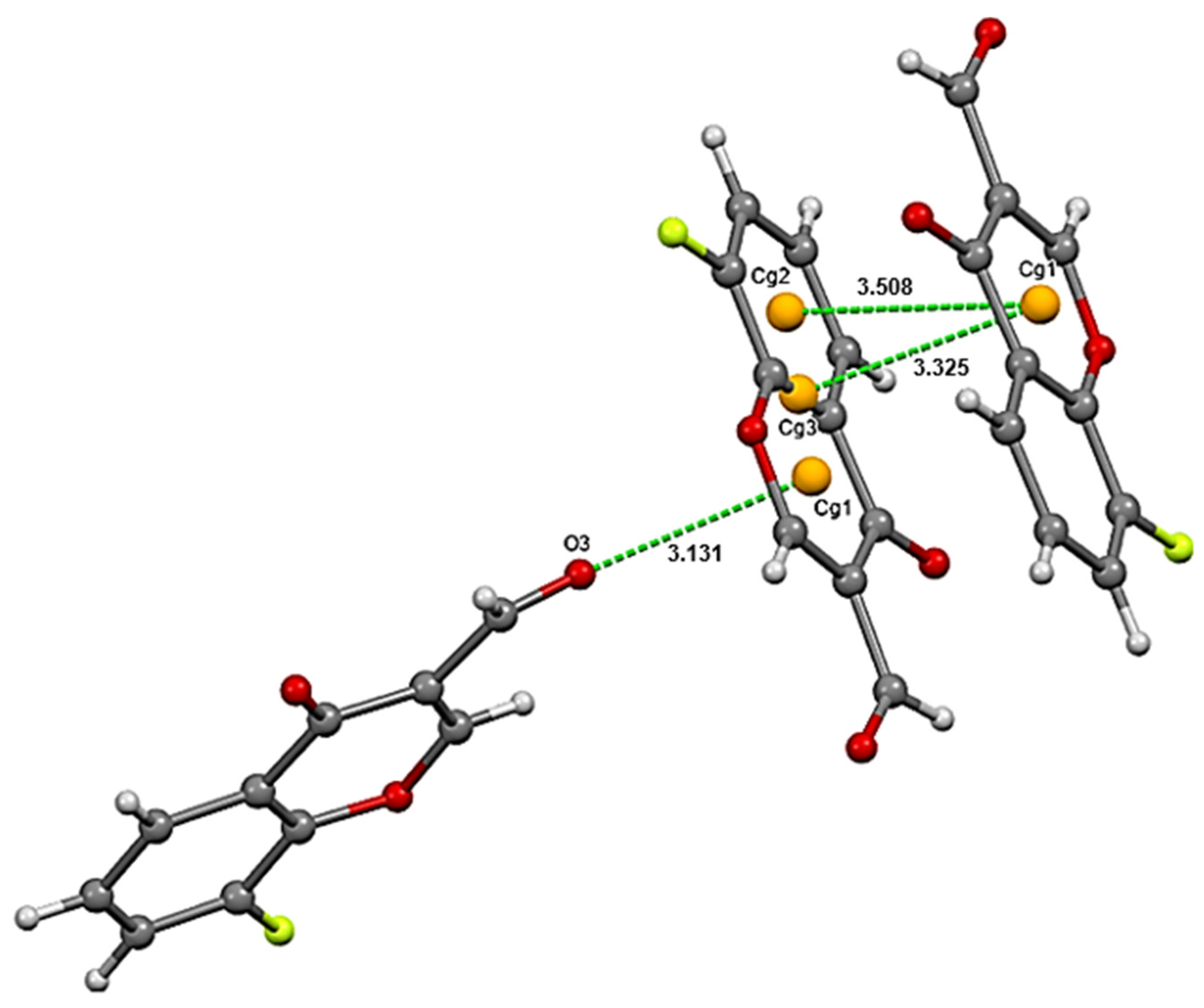
3.1. Crystalline Structure, Supramolecular Features and Intermolecular Interactions
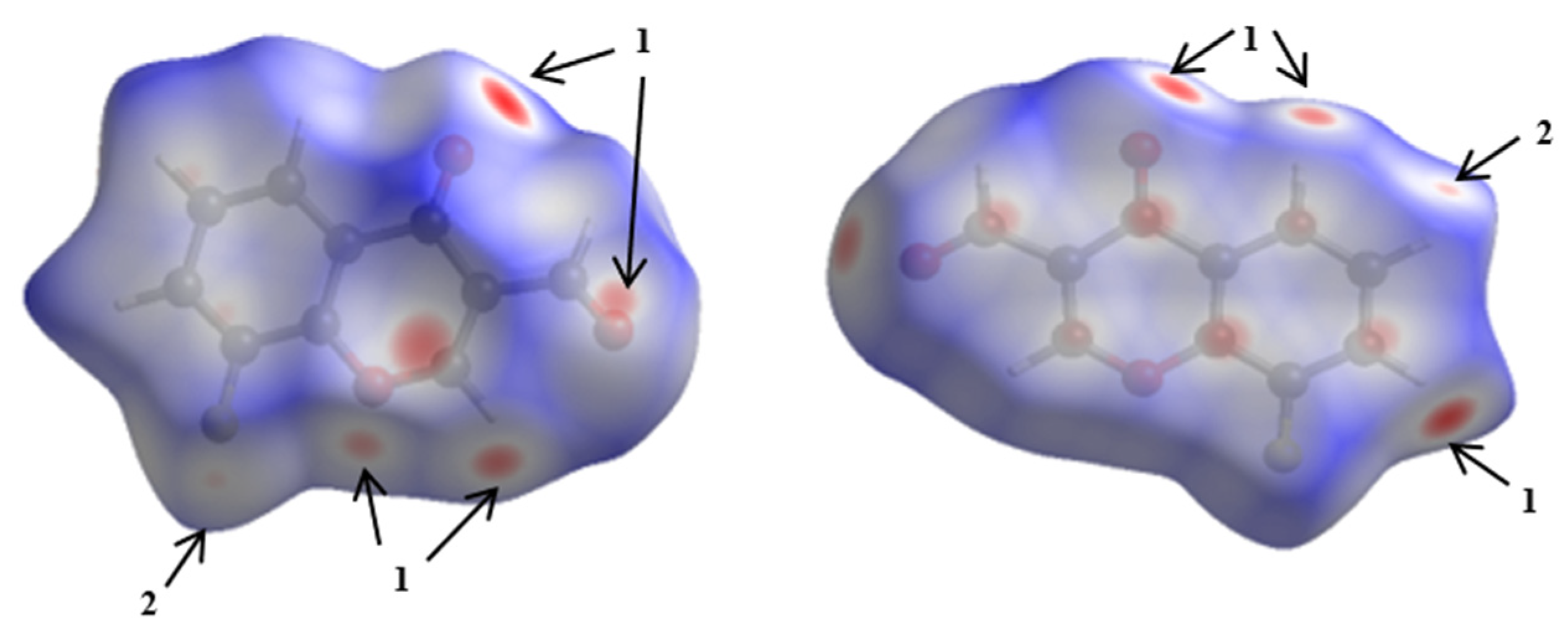
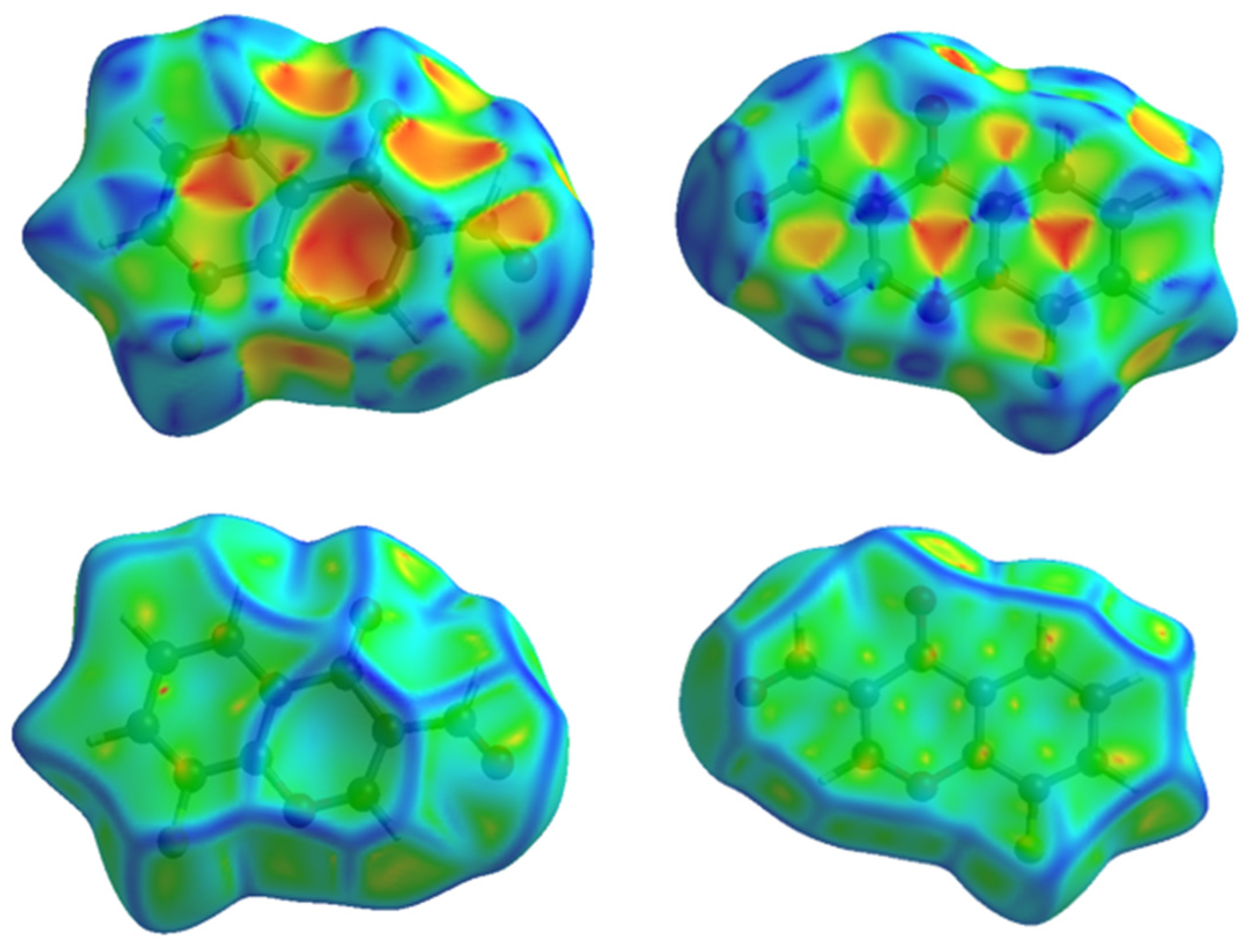
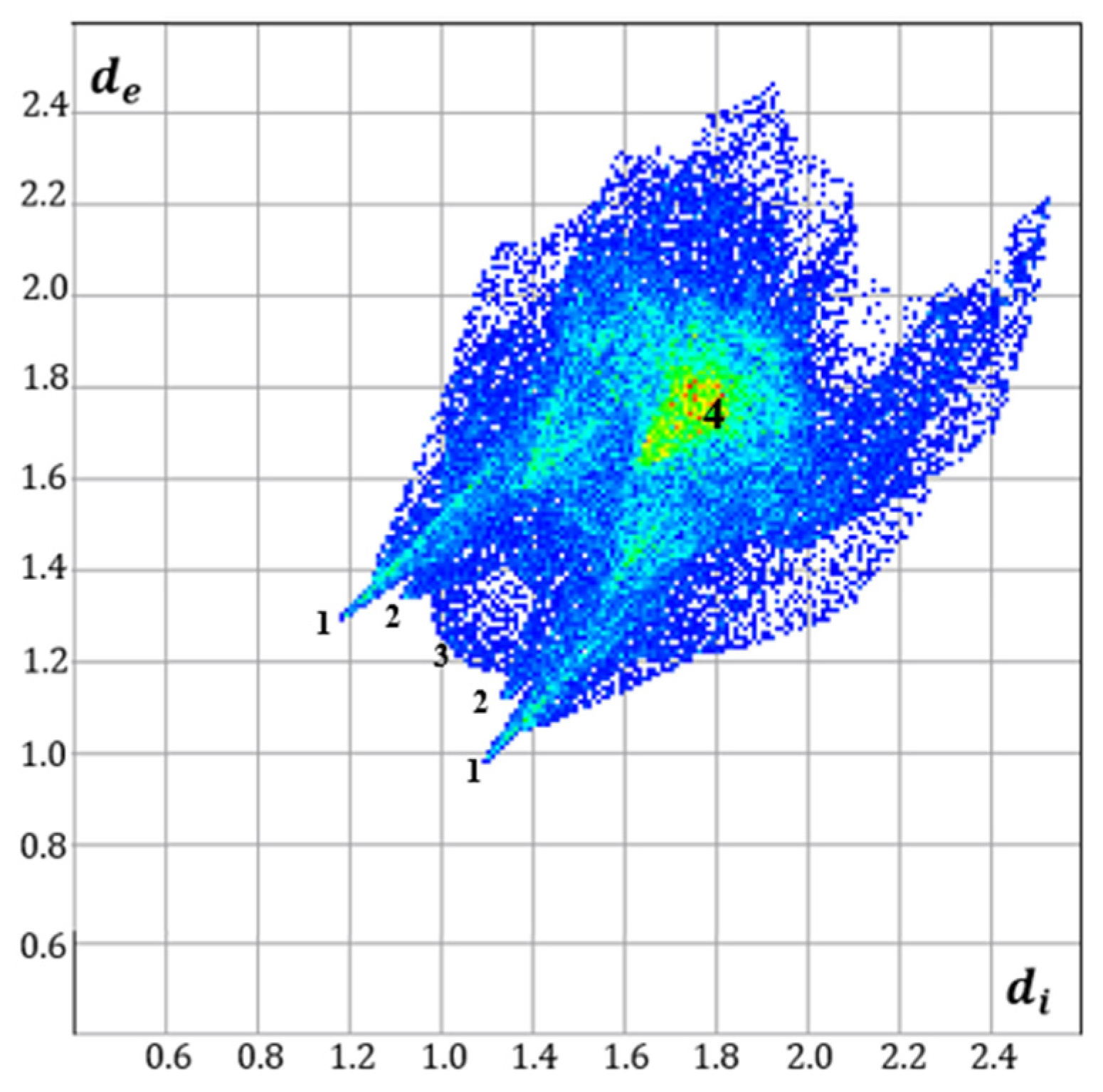
3.2. Hirshfeld Surface Analyses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaspar, A.; Garrido, E.M.P.J.; Borges, F.; Garrido, J.M.P.J. Biological and Medicinal Properties of Natural Chromones and Chromanones. ACS Omega 2024, 9, 21706–21726. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, N.U.A.; Irfan, M.; Hassan, S.U.; Saleem, U. Current Strategies in Development of New Chromone Derivatives with Diversified Pharmacological Activities: A Review. Pharm. Chem. J. 2020, 54, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Sosnovskikh, V.Y. Synthesis and Reactions of Halogen-Containing Chromones. Russ. Chem. Rev. 2003, 72, 489–516. [Google Scholar] [CrossRef]
- Sosnovskikh, V.Y.; Usachev, B.I. 2-Polyfluoroalkylchromones. 5. Nitration and Chlorination of 2-Trifluoromethylchromones. Russ. Chem. Bull. 2000, 49, 2074–2076. [Google Scholar] [CrossRef]
- Sosnovskikh, V.Y.; Korotaev, V.Y.; Chizhov, D.L.; Kutyashev, I.B.; Yachevskii, D.S.; Kazheva, O.N.; Dyachenko, O.A.; Charushin, V.N. Reaction of Polyhaloalkyl-Substituted Chromones, Pyrones, and Furanones with Salicylaldehydes as a Direct Route to Fused 2H-Chromenes. J. Org. Chem. 2006, 71, 4538–4543. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Jiang, Y.; Guo, F.; Chen, L.; Xu, L.; Zhang, W.; Liu, B. The Antitumor Activity of Naturally Occurring Chromones: A Review. Fitoterapia 2019, 135, 114–129. [Google Scholar] [CrossRef]
- Pastuszko, A.; Majchrzak, K.; Czyz, M.; Kupcewicz, B.; Budzisz, E. The Synthesis, Lipophilicity and Cytotoxic Effects of New Ruthenium(II) Arene Complexes with Chromone Derivatives. J. Inorg. Biochem. 2016, 159, 133–141. [Google Scholar] [CrossRef]
- Zhu, Y.; Yao, X.; Long, J.; Li, R.; Liu, Y.; Yang, Z.; Zheng, X. Fluorine-Containing Chrysin Derivatives: Synthesis and Biological Activity. Nat. Prod. Commun. 2019, 14, 1934578X19878921. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Adán, E.; Hernández-Ortega, S.; Toscano, R.A.; Germán-Acacio, J.M.; Sánchez-Pacheco, A.D.; Hernández-Vergara, M.; Barquera, J.E.; Valdés-Martínez, J. Competition of Hydrogen Bonds, Halogen Bonds, and π–π Interactions in Crystal Structures. Exploring the Effect of One Atom Substitution. Cryst. Growth Des. 2024, 24, 1888–1897. [Google Scholar] [CrossRef]
- Narváez, O.E.G.; Bonilla, V.P.M.; Zurita, D.A.; Alcívar, L.C.D.; Heredia-Moya, J.; Ulic, S.E.; Jios, J.L.; Piro, O.E.; Echeverría, G.A.; Langer, P. Synthesis, Experimental and Theoretical Study of Novel 2-Haloalkyl (-CF2H, -CCl2H, -CF2CF3)-, 3-Bromo and Bromomethyl Substituted Chromones. J. Fluor. Chem. 2021, 242, 109717. [Google Scholar] [CrossRef]
- Alcívar León, C.D.; Piro, O.E.; Echeverria, G.A.; Ulic, S.E.; Jios, J.L. Synthesis and Conformational Theoretical Study of Novel Derivatives of 2-(Trifluoromethyl)Chromone: 3-Cyanomethyl and 3-Aminomethyl 2-(Trifluoromethyl)Chromones. J. Argent. Chem. Soc. 2014, 101, 4960. [Google Scholar]
- Alcívar León, C.D.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L. A Detailed Experimental and Theoretical Study of Two Novel Substituted Trifluoromethylchromones. The Influence of the Bulky Bromine Atom on the Crystal Packing. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 136, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Alcívar León, C.D.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L.; Burgos Paci, M.; Argüello, G.A. The Role of Halogen C-X1⋯X2-C Contact on the Preferred Conformation of 2-Perhalomethylchromones in Solid State. Chem. Phys. 2016, 472, 142–155. [Google Scholar] [CrossRef]
- Alcívar León, C.D.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L.; Pereañez, J.A.; Henao Castañeda, I.C.; Pérez, H. The Role of Non-Covalent Interactions in Some 2-Trifluoromethylchromones in the Solid State. New J. Chem. 2017, 41, 14659–14674. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haro-Saltos, L.A.; Bonilla-Valladares, P.M.; Alcívar-León, C.D. Theoretical Study of Intermolecular Interactions in Benzopyrans Substituted with Polyhaloalkyl Groups. Chem. Proc. 2024, 16, 32. https://doi.org/10.3390/ecsoc-28-20209
Haro-Saltos LA, Bonilla-Valladares PM, Alcívar-León CD. Theoretical Study of Intermolecular Interactions in Benzopyrans Substituted with Polyhaloalkyl Groups. Chemistry Proceedings. 2024; 16(1):32. https://doi.org/10.3390/ecsoc-28-20209
Chicago/Turabian StyleHaro-Saltos, Lissette A., Pablo M. Bonilla-Valladares, and Christian D. Alcívar-León. 2024. "Theoretical Study of Intermolecular Interactions in Benzopyrans Substituted with Polyhaloalkyl Groups" Chemistry Proceedings 16, no. 1: 32. https://doi.org/10.3390/ecsoc-28-20209
APA StyleHaro-Saltos, L. A., Bonilla-Valladares, P. M., & Alcívar-León, C. D. (2024). Theoretical Study of Intermolecular Interactions in Benzopyrans Substituted with Polyhaloalkyl Groups. Chemistry Proceedings, 16(1), 32. https://doi.org/10.3390/ecsoc-28-20209