
A Refined Model of the CFTR Membrane Transporter as a Tool to Revert Misbehavior †

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
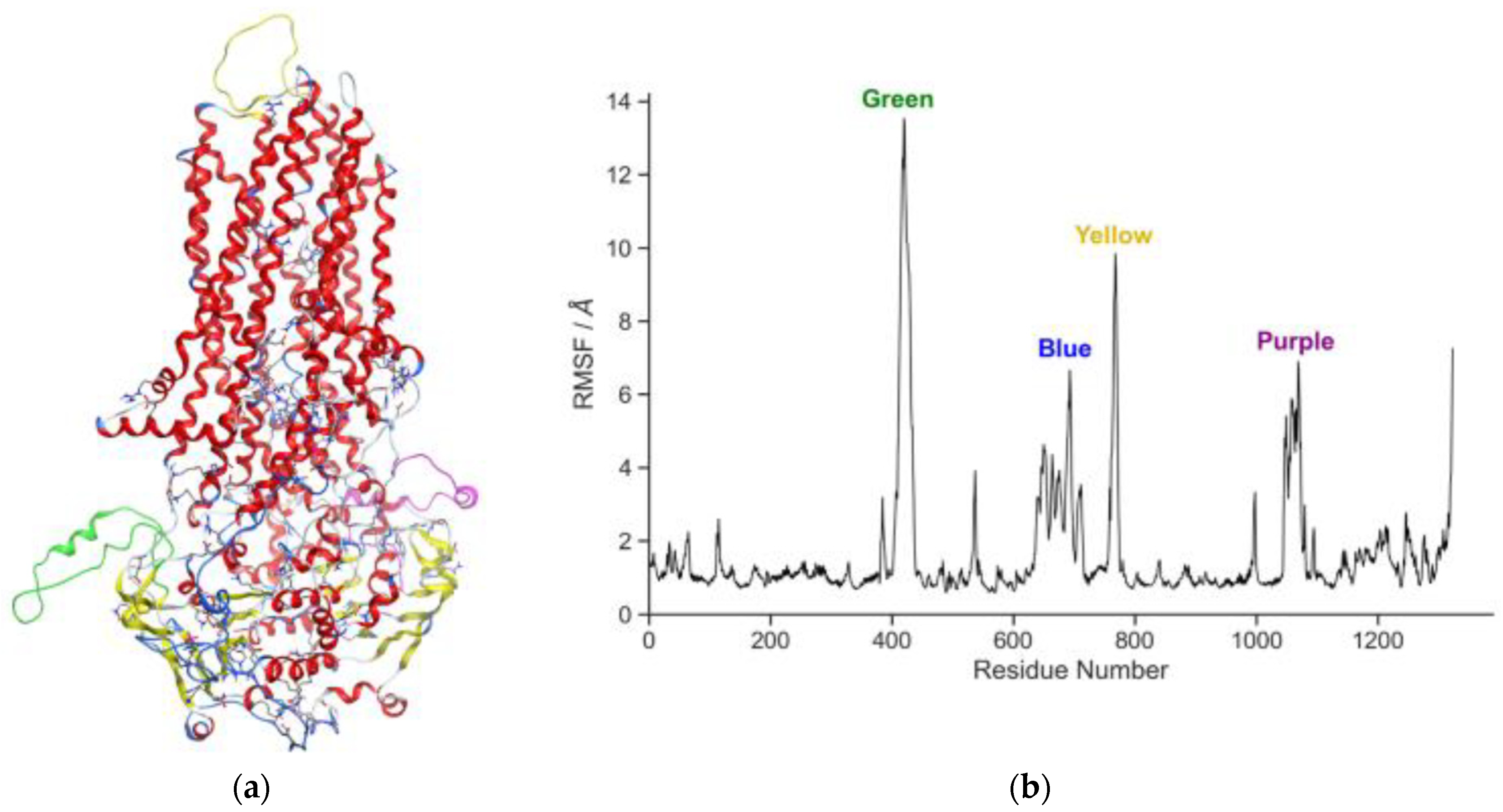
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, C.; Higgins, M.; Liu, L.; Volkova, N.; Zolin, A.; Naehrlich, L.; Andreas, P.; Elise, L.; Duška, T.-D.; Pavel, D.; et al. Effectiveness of Lumacaftor/Ivacaftor Initiation in Children with Cystic Fibrosis Aged 2 through 5 Years on Disease Progression: Interim Results from an Ongoing Registry-Based Study. J. Cyst. Fibros. 2024, 23, 436–442. [Google Scholar] [CrossRef]
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-S.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric Folding Correction of F508del and Rare CFTR Mutants by Elexacaftor-Tezacaftor-Ivacaftor (Trikafta) Combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Dastoor, P.; Muiler, C.; Garrison, A.; Egan, M.; Carlos Dos Reis, D.; Santos, A.; Ameen, N.A. Localization and Function of Humanized F508del-CFTR in Mouse Intestine Following Activation of Serum Glucocorticoid Kinase 1 and Trikafta. Eur. J. Pharmacol. 2024, 978, 176771. [Google Scholar] [CrossRef]
- Yeh, J.; Yu, Y.; Hwang, T. Structural Mechanisms for Defective CFTR Gating Caused by the Q1412X Mutation, a Severe Class VI Pathogenic Mutation in Cystic Fibrosis. J. Physiol. 2019, 597, 543–560. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef]
- Alam, A.; Locher, K.P. Structure and Mechanism of Human ABC Transporters. Annu. Rev. Biophys. 2023, 52, 275–300. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular Structure of the ATP-Bound, Phosphorylated Human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef]
- Levring, J.; Terry, D.S.; Kilic, Z.; Fitzgerald, G.; Blanchard, S.C.; Chen, J. CFTR Function, Pathology and Pharmacology at Single-Molecule Resolution. Nature 2023, 616, 606–614. [Google Scholar] [CrossRef]
- Ostedgaard, L.S.; Baldursson, O.; Welsh, M.J. Regulation of the Cystic Fibrosis Transmembrane Conductance Regulator Cl− Channel by Its R Domain. J. Biol. Chem. 2001, 276, 7689–7692. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural Identification of a Hotspot on CFTR for Potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Insights on P-Glycoprotein’s Efflux Mechanism Obtained by Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Bonito, C.A.; Cordeiro, M.N.D.S.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Structure-Function Relationships in ABCG2: Insights from Molecular Dynamics Simulations and Molecular Docking Studies. Sci Rep 2017, 7, 15534. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Assessing the Stabilization of P-Glycoprotein’s Nucleotide-Binding Domains by the Linker, Using Molecular Dynamics. Mol. Inform. 2013, 32, 529–540. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2019.01 Chemical Computing Group ULC. 910-1010 Sherbrooke St. W., Montreal, QC H3A 2R7, 2019. Available online: https://www.chemcomp.com/en/index.htm (accessed on 26 September 2024).
- Bernhofer, M.; Dallago, C.; Karl, T.; Satagopam, V.; Heinzinger, M.; Littmann, M.; Olenyi, T.; Qiu, J.; Schütze, K.; Yachdav, G.; et al. PredictProtein—Predicting Protein Structure and Function for 29 Years. Nucleic Acids Res. 2021, 49, W535–W540. [Google Scholar] [CrossRef]
- Bozoky, Z.; Krzeminski, M.; Chong, P.A.; Forman-Kay, J.D. Structural Changes of CFTR R Region upon Phosphorylation: A Plastic Platform for Intramolecular and Intermolecular Interactions. FEBS J. 2013, 280, 4407–4416. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of Proteins in Membranes Database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Strain Fluctuations and Elastic Constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Corradi, V.; Gu, R.-X.; Vergani, P.; Tieleman, D.P. Structure of Transmembrane Helix 8 and Possible Membrane Defects in CFTR. Biophys. J. 2018, 114, 1751–1754. [Google Scholar] [CrossRef]
- Farkas, B.; Tordai, H.; Padányi, R.; Tordai, A.; Gera, J.; Paragi, G.; Hegedűs, T. Discovering the Chloride Pathway in the CFTR Channel. Cell. Mol. Life Sci. 2020, 77, 765–778. [Google Scholar] [CrossRef]
- Zeng, Z.W.; Linsdell, P.; Pomès, R. Molecular Dynamics Study of Cl− Permeation through Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Cell. Mol. Life Sci. 2023, 80, 51. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzano, P.M.S.; González-Durruthy, M.; Ferreira, R.J.; Bonito, C.A.; Amaral, M.D.; dos Santos, D.J.V.A. A Refined Model of the CFTR Membrane Transporter as a Tool to Revert Misbehavior. Chem. Proc. 2024, 16, 109. https://doi.org/10.3390/ecsoc-28-20247
Suzano PMS, González-Durruthy M, Ferreira RJ, Bonito CA, Amaral MD, dos Santos DJVA. A Refined Model of the CFTR Membrane Transporter as a Tool to Revert Misbehavior. Chemistry Proceedings. 2024; 16(1):109. https://doi.org/10.3390/ecsoc-28-20247
Chicago/Turabian StyleSuzano, Pedro M. S., Michael González-Durruthy, Ricardo J. Ferreira, Cátia A. Bonito, Margarida D. Amaral, and Daniel J. V. A. dos Santos. 2024. "A Refined Model of the CFTR Membrane Transporter as a Tool to Revert Misbehavior" Chemistry Proceedings 16, no. 1: 109. https://doi.org/10.3390/ecsoc-28-20247
APA StyleSuzano, P. M. S., González-Durruthy, M., Ferreira, R. J., Bonito, C. A., Amaral, M. D., & dos Santos, D. J. V. A. (2024). A Refined Model of the CFTR Membrane Transporter as a Tool to Revert Misbehavior. Chemistry Proceedings, 16(1), 109. https://doi.org/10.3390/ecsoc-28-20247