
Molecular Dynamics Simulations on the Mesophilic Enzyme Vibrio Cholerae Endonuclease I: Salt Effect Study †

Abstract
:1. Introduction
2. Materials and Methods
3. Results
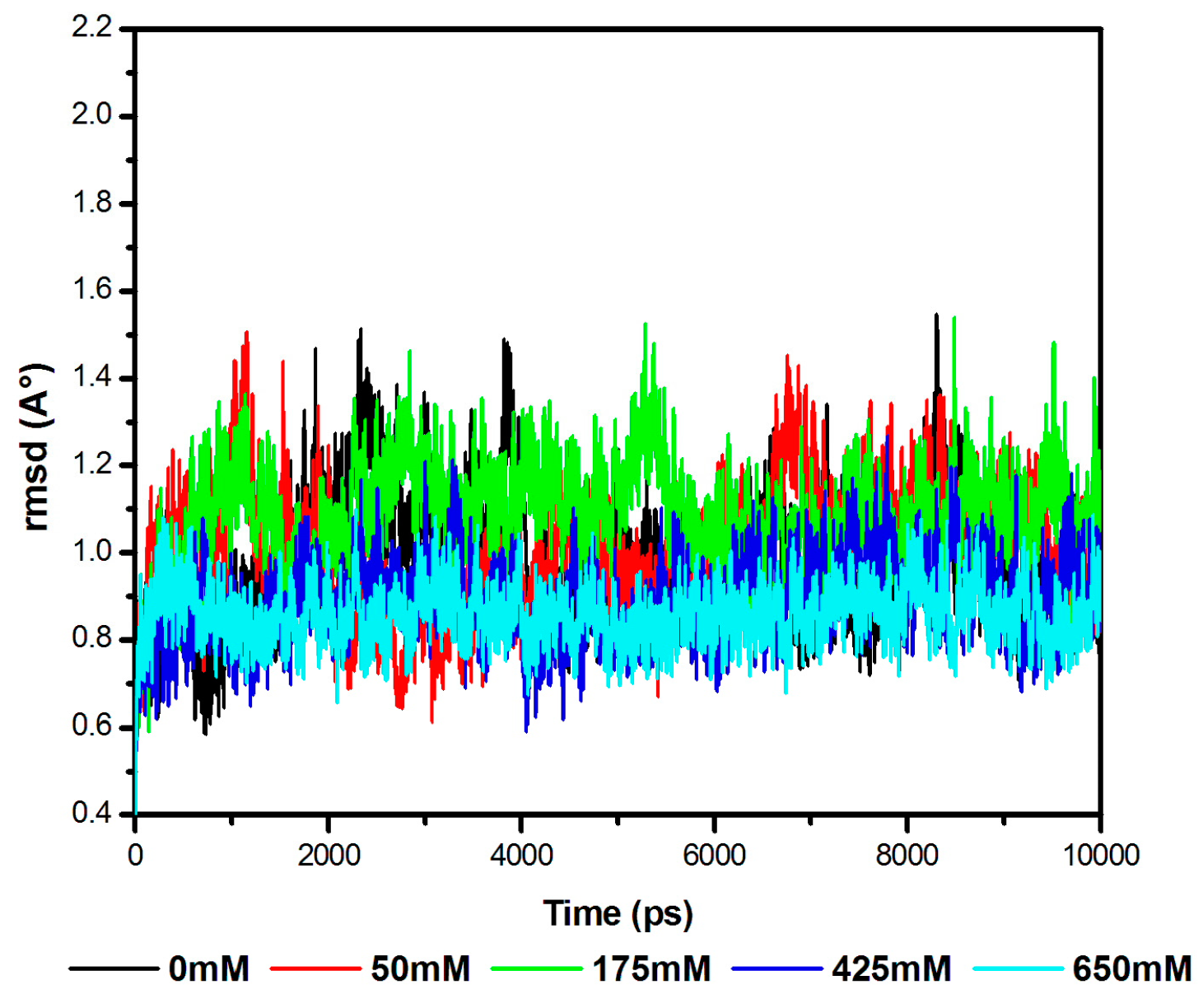
3.1. Stability of the Model
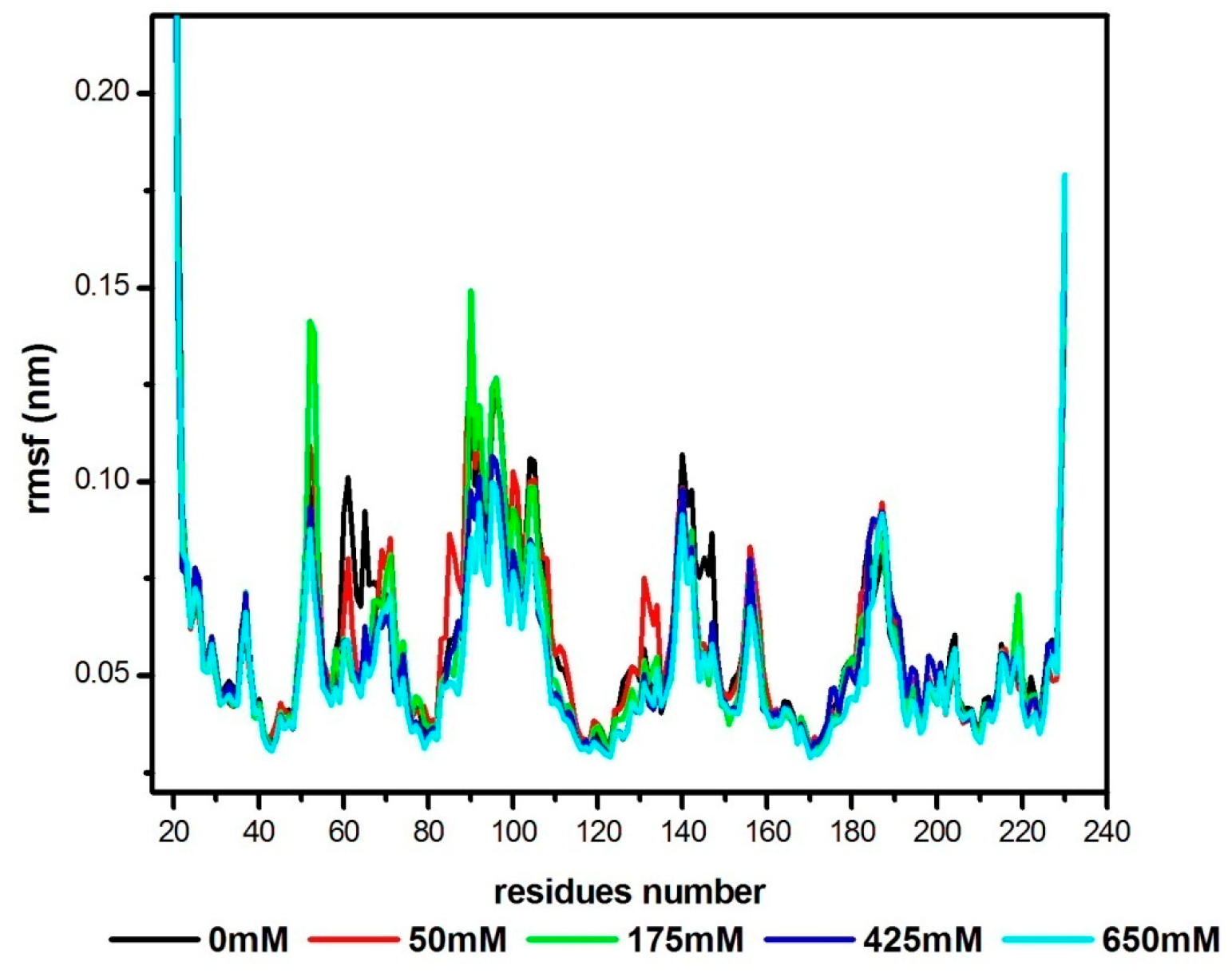
3.2. Structural Flexibility
3.3. Radial Distribution Functions
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaper, J.B.; Morris, J.G., Jr.; Levine, M.L. Cholera. Clin. Microbiol. Rev. 1995, 8, 48–86. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.V.; Matte, M.H.; Matte, G.R.; Jiang, S.; Sabeena, F.; Shukla, B.N.; Sanyal, S.C.; Huq, A.; Colwell, R.R. Molecular analysis of Vibrio cholerae O1, O139, non-O1, and non O139 strains: Clonal Relationships between clinical and environmental isolates. Appl. Environ. Microbiol. 2001, 67, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Focareta, T.; Manning, P.A. Extracellular proteins of Vibrio cholerae: Molecular cloning, nucleotide sequence and characterization of the deoxyribonuclease (DNase) together with its periplasmic localization on Esterichia coli K-12. Gene 1987, 53, 31–40. [Google Scholar] [CrossRef]
- Altermark, B.; Smalås, A.O.; Willassen, N.P.; Helland, R. The structure of Vibrio cholerae extracellular endonuclease I reveals the presence of a buried chloride ion. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1387–1391. [Google Scholar] [CrossRef]
- Altermark, B.; Willassen, N.P.; Smalås, A.O.; Moe, E. Comparative studies of endonuclease I from cold-adapted Vibrio salmonicida and mesophilic Vibrio cholerae. FEBS J. 2007, 274, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Chang, S.Y.; Chen, S.L.; Chuang, S.M. Cloning and expression in Escherichia coli of the gene encoding an extracellular deoxyribonuclease (DNase) from Aeromonas hydrophila. Gene 1992, 122, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.I.; Lo, S.K.; Shao, C.P.; Tsai, H.W.; Hor, L.I. Cloning and characterization of a periplasmic nuclease of Vibrio vulnificus and its role in preventing uptake of foreign DNA. Appl. Environ. Microbiol. 2001, 67, 82–88. [Google Scholar] [CrossRef]
- Salikhova, Z.Z.; Sokolova, R.B.; Ponomareva, A.Z.; Iusupova, D.V. Endonuclease from Proteus mirabilis. Prikl. Biokhim. Mikrobiol. 2001, 37, 43–47. [Google Scholar]
- Altermark, B.; Helland, R.; Moe, E.; Willassen, N.P.; Smalas, A.O. Structural adaptation of endonuclease I from the cold-adapted and halophilic bacterium Vibrio salmonicida. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 368–376. [Google Scholar] [CrossRef]
- MacKerell, J.A.D.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetic in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comp. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van der Spoel, D.; Lindahl, E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comp. Chem. 2005, 26, 1701–1719. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Dinola, A.; Hakk, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N-log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pederson, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8592. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Benrezkallah, D.; Dauchez, M.; Krallafa, A.M. Molecular dynamics of the salt dependence of a cold adapted enzyme: Endonuclease I. J. Biomol. Struct. Dyn. 2015, 33, 2511–2521. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VcEndA_0 mM | VcEndA_50 mM | VcEndA_175 mM | VcEndA_425 mM | VcEndA_650 mM | |
|---|---|---|---|---|---|
| Cl− | 6 | 15 | 37 | 76 | 122 |
| Na+ | 0 | 9 | 31 | 82 | 116 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benrezkallah, D. Molecular Dynamics Simulations on the Mesophilic Enzyme Vibrio Cholerae Endonuclease I: Salt Effect Study. Chem. Proc. 2023, 14, 40. https://doi.org/10.3390/ecsoc-27-16165
Benrezkallah D. Molecular Dynamics Simulations on the Mesophilic Enzyme Vibrio Cholerae Endonuclease I: Salt Effect Study. Chemistry Proceedings. 2023; 14(1):40. https://doi.org/10.3390/ecsoc-27-16165
Chicago/Turabian StyleBenrezkallah, Djamila. 2023. "Molecular Dynamics Simulations on the Mesophilic Enzyme Vibrio Cholerae Endonuclease I: Salt Effect Study" Chemistry Proceedings 14, no. 1: 40. https://doi.org/10.3390/ecsoc-27-16165
APA StyleBenrezkallah, D. (2023). Molecular Dynamics Simulations on the Mesophilic Enzyme Vibrio Cholerae Endonuclease I: Salt Effect Study. Chemistry Proceedings, 14(1), 40. https://doi.org/10.3390/ecsoc-27-16165