
Designing and In Silico Evaluation of Some Non-Nucleoside MbtA Inhibitors: On Track to Tackle Tuberculosis †



Abstract
1. Background
2. Materials and Methods Employed
2.1. Hardwares and Softwares Used
2.2. Molecular Docking Simulations
2.2.1. Preparation of Protein
2.2.2. Preparation of Ligands
2.2.3. Molecular Docking Studies
2.3. Predictive Absorption, Distribution, Metabolism, and Excretion (ADME)
2.4. Prediction of Toxicity
2.5. Molecular Dynamics Simulations
3. Results and Discussions
3.1. Molecular Docking Simulations
Interaction Analysis of GV08
3.2. Predictive Absorption, Distribution, Metabolism, and Excretion (ADME)
3.2.1. Drug-Likeness, Alerts, Lead-Likeness, and Synthetic Accessibility
3.2.2. Analysis of Pharmacokinetics Compliance through In Silico Evaluation
3.3. Prediction of Toxicity
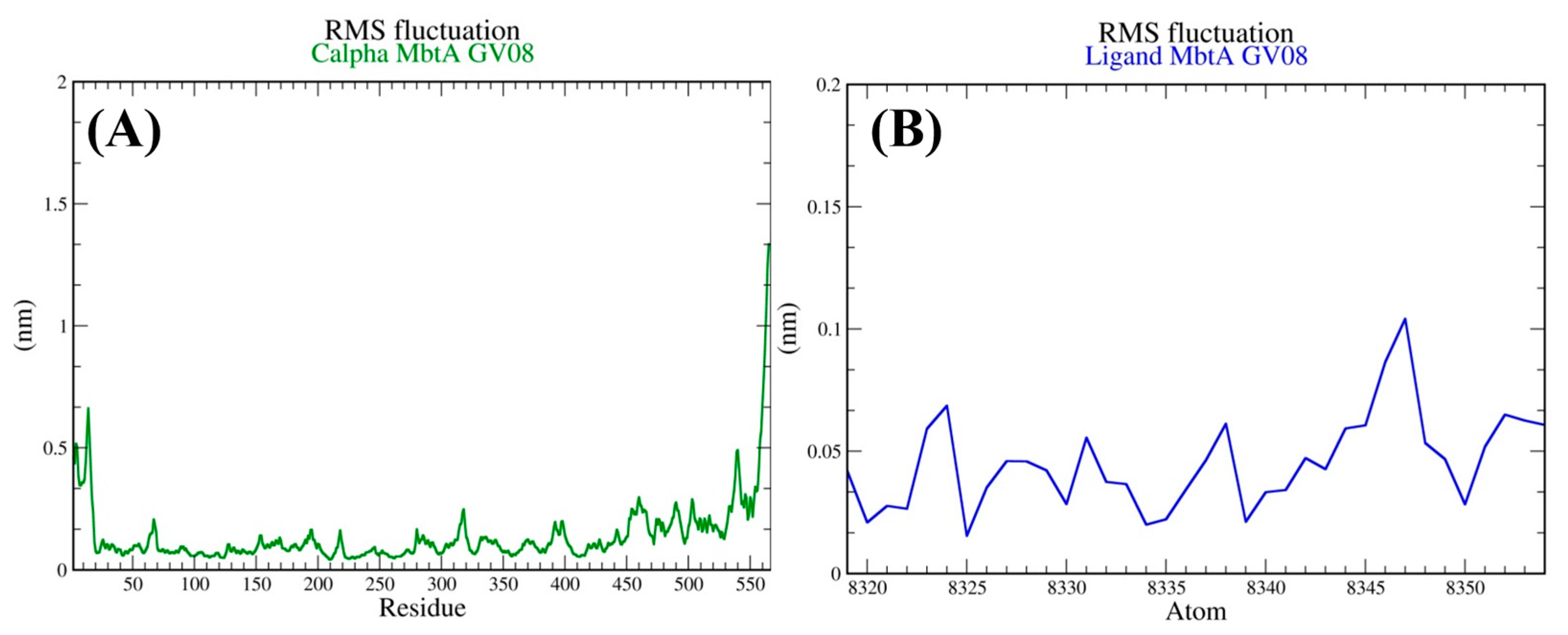
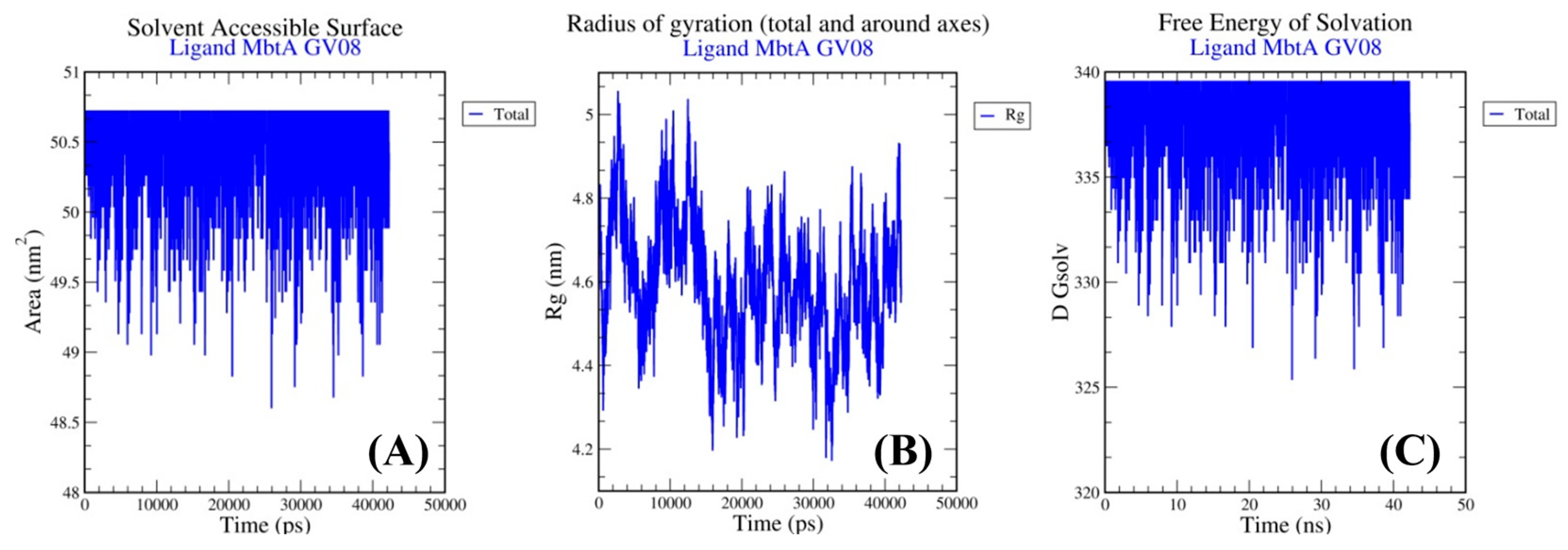
3.4. Molecular Dynamics Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loddenkemper, R.; Murray, J.F.; Gradmann, C.; Hopewell, P.C.; Kato-Maeda, M. History of tuberculosis. Tuberculosis 2018, 8–27. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- World Health Organization. Tuberculosis. 2022. Available online: https://www.who.int/health-topics/tuberculosis#tab=tab_1 (accessed on 17 February 2022).
- Nakajima, H. Tuberculosis: A global emergency. World Health 1993, 46, 3. [Google Scholar]
- Bruchfeld, J.; Correia-Neves, M.; Källenius, G. Tuberculosis and HIV Coinfection: Table 1. Cold Spring Harb. Perspect. Med. 2015, 5, a017871. [Google Scholar] [CrossRef] [PubMed]
- Shyam, M.; Shilkar, D.; Verma, H.; Dev, A.; Sinha, B.N.; Brucoli, F.; Bhakta, S.; Jayaprakash, V. The Mycobactin Biosynthesis Pathway: A Prospective Therapeutic Target in the Battle against Tuberculosis. J. Med. Chem. 2020, 64, 71–100. [Google Scholar] [CrossRef]
- Stirrett, K.L.; Ferreras, J.; Jayaprakash, V.; Sinha, B.N.; Ren, T.; Quadri, L.E. Small molecules with structural similarities to siderophores as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett. 2008, 18, 2662–2668. [Google Scholar] [CrossRef] [PubMed]
- Ferreras, J.A.; Gupta, A.; Amin, N.D.; Basu, A.; Sinha, B.N.; Worgall, S.; Jayaprakash, V.; Quadri, L.E.N. Chemical scaffolds with structural similarities to siderophores of non-ribosomal peptide–polyketide origin as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett. 2011, 21, 6533–6537. [Google Scholar] [CrossRef] [PubMed]
- Shyam, M.; Verma, H.; Bhattacharje, G.; Mukherjee, P.; Singh, S.; Kamilya, S.; Jalani, P.; Das, S.; Dasgupta, A.; Mondal, A.; et al. Mycobactin Analogues with Excellent Pharmacokinetic Profile Demonstrate Potent Antitubercular Specific Activity and Exceptional Efflux Pump Inhibition. J. Med. Chem. 2022, 65, 234–256. [Google Scholar] [CrossRef]
- Dassault Systèmes. BIOVIA Discovery Studio Visualizer; V16.1.0.15350; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.M.D.; Shakil, S.; Haneef, M. A simple click by click protocol to perform docking: Autodock 4.2 made easy for non-bioinformaticians. EXCLI J. 2013, 12, 830–857. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open-source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| S. No. | Code | R | R1 |
|---|---|---|---|
| 01 | GV01 |  | 2-CH3 |
| 02 | GV02 | | 3-CH3 |
| 03 | GV03 | | 4-CH3 |
| 04 | GV04 | | 2-OCH3 |
| 05 | GV05 | | 3-OCH3 |
| 06 | GV06 | | 4-OCH3 |
| 07 | GV07 | | 2-Cl |
| 08 | GV08 | | 3-Cl |
| 09 | GV09 | | 4-Cl |
| 10 | GV10 | | 2-OH |
| 11 | GV11 | | 3-OH |
| 12 | GV12 | | 4-OH |
| S.No. | Coding | Docking Score (kcal/mol) | Inhibition Constant (Ki) |
|---|---|---|---|
| 01 | GV01 | −8.19 | 996.73 nM |
| 02 | GV02 | −8.53 | 563.3 nM |
| 03 | GV03 | −8.59 | 508.51 nM |
| 04 | GV04 | −8.26 | 878.26 nM |
| 05 | GV05 | −7.97 | 1.45 µM |
| 06 | GV06 | −7.88 | 1.67 µM |
| 07 | GV07 | −8.54 | 553.44 nM |
| 08 | GV08 | −8.80 | 352.58 nM |
| 09 | GV09 | −8.61 | 499.91 nM |
| 10 | GV10 | −7.96 | 1.47 µM |
| 11 | GV11 | −7.88 | 1.67 µM |
| 12 | GV12 | −7.70 | 2.29 µM |
| S.No. | Coding | H-Bond Interacting Residues |
|---|---|---|
| 1. | GV08 | Glu357, Ala356, Thr462, Gly460 |
| 2. | GV09 | Glu357, Ala356, Thr462, Gly460, Gly214 |
| 3. | GV03 | Glu357, Ala356, Thr462, Gly460 |
| 4. | GV07 | Gly330, Thr462, Gly460 |
| Sl No. | Compound Code | Drug-Likeness Rules | Alerts | Lead-Likeness | Synthetic Accessibility | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipinski (Pfizer) | Ghose (Amgen) | Veber (GSK) | Egan (Pharmacia) | Muege (Bayer) | Bioavailability Score | PAINS | Brenk | ||||
| 1. | GV08 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 1 | Yes | 3.43 |
| 2. | GV09 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 1 | Yes | 3.43 |
| 3. | GV03 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 1 | Yes | 3.54 |
| 4. | GV07 | Yes | Yes | Yes | Yes | Yes | 0.55 | 1 | 1 | Yes | 3.51 |
| GV08 | GV09 | GV03 | GV07 | |||
|---|---|---|---|---|---|---|
| A D M E T P R O F I L I N G | Physiochemical parameters | Formula | C16H14ClN3OS | C16H14ClN3OS | C17H17N3OS | C16H14ClN3OS |
| Molecular weight | 331.82 g/mol | 331.82 g/mol | 311.40 g/mol | 331.82 g/mol | ||
| Mol. refractivity | 99.56 | 99.56 | 99.51 | 99.56 | ||
| TPSA | 93.94 Å2 | 93.94 Å2 | 93.94 Å2 | 93.94 Å2 | ||
| Lipophilicity | ILOGP | 2.43 | 2.41 | 2.40 | 2.14 | |
| SILICOS-IT | 3.90 | 3.90 | 3.77 | 3.90 | ||
| Water solubility | Log S (ESOL), class | −3.99 Soluble | −3.99 Soluble | −3.70 Soluble | −3.99 Soluble | |
| Log S (Ali), class | −4.64 Moderately soluble | −4.64 Moderately soluble | −4.37 Moderately soluble | −4.64 Moderately soluble | ||
| SILICOS-IT, class | −4.69 Moderately soluble | −4.69 Moderately soluble | −4.47 Moderately soluble | −4.69 Moderately soluble | ||
| Pharmacokinetics | GI absorption | High | High | High | High | |
| BBB permeant | No | No | No | No | ||
| Log Kp (skin perm.) | −6.19 cm/s | −6.19 cm/s | −6.25 cm/s | −6.19 cm/s | ||
| CYP1A2 | Yes | Yes | No | Yes | ||
| CYP2C19 | Yes | Yes | Yes | Yes | ||
| CYP2C9 | Yes | Yes | Yes | Yes | ||
| CYP2D6 | No | No | No | No | ||
| CYP3A4 | No | No | No | No | ||
| Name of Model | Unit | GV08 | GV09 | GV03 | GV07 |
|---|---|---|---|---|---|
| AMES toxicity | Yes/No | No | No | No | No |
| Max. tolerated dose (human) | Log mg/kg/day | 0.053 | 0.085 | 0.101 | 0.087 |
| hERG I inhibitor | Yes/No | No | No | No | No |
| hERG II inhibitor | Yes/No | No | No | No | No |
| Oral rat chronic toxicity (LD50) | Mol/kg | 2.47 | 2.46 | 2.393 | 2.461 |
| Oral rat chronic toxicity | Log mg/kg_bw/day | 1.115 | 1.167 | 1.313 | 1.096 |
| Hepatotoxicity | Yes/No | No | No | Yes | No |
| Skin sensitization | Yes/No | No | No | No | No |
| T. Pyriformis toxicity | Log ug/L | 2.113 | 2.1 | 2.037 | 2.127 |
| Minnow toxicity | Log mM | 0.629 | 0.882 | 1.1 | 0.893 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakshit, G.; Jayaprakash, V. Designing and In Silico Evaluation of Some Non-Nucleoside MbtA Inhibitors: On Track to Tackle Tuberculosis. Chem. Proc. 2022, 12, 78. https://doi.org/10.3390/ecsoc-26-13688
Rakshit G, Jayaprakash V. Designing and In Silico Evaluation of Some Non-Nucleoside MbtA Inhibitors: On Track to Tackle Tuberculosis. Chemistry Proceedings. 2022; 12(1):78. https://doi.org/10.3390/ecsoc-26-13688
Chicago/Turabian StyleRakshit, Gourav, and Venkatesan Jayaprakash. 2022. "Designing and In Silico Evaluation of Some Non-Nucleoside MbtA Inhibitors: On Track to Tackle Tuberculosis" Chemistry Proceedings 12, no. 1: 78. https://doi.org/10.3390/ecsoc-26-13688
APA StyleRakshit, G., & Jayaprakash, V. (2022). Designing and In Silico Evaluation of Some Non-Nucleoside MbtA Inhibitors: On Track to Tackle Tuberculosis. Chemistry Proceedings, 12(1), 78. https://doi.org/10.3390/ecsoc-26-13688