
Computational Approaches for Structure-Based Molecular Characterization and Functional Annotation of the Fusion Protein of Nipah henipavirus †




Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Sequence Retrieval
2.2. Identification of the Physicochemical Properties
2.3. Secondary Structure Identification and Assessment of the Selected Protein
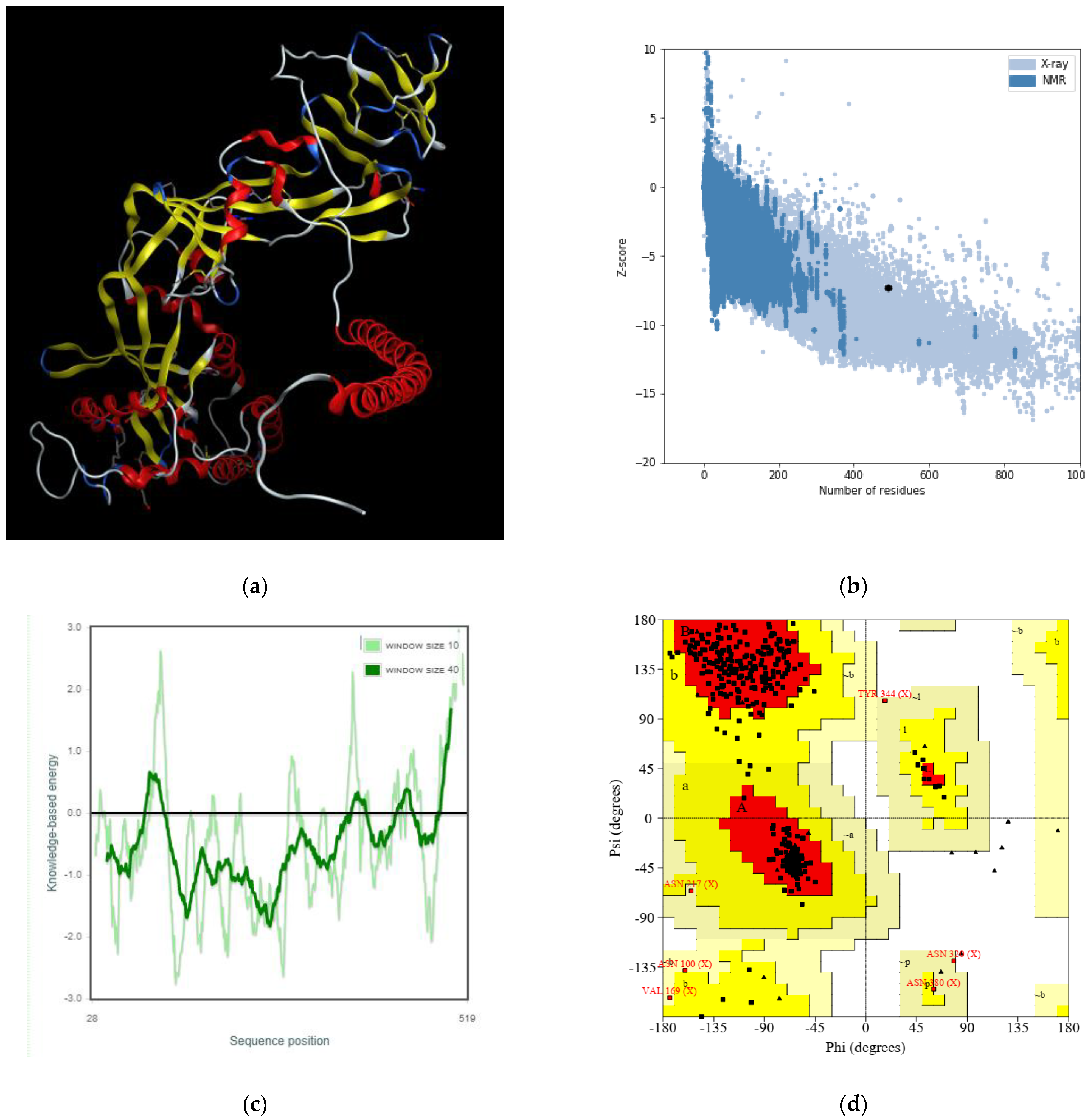
2.4. Determination and Validation of the Three-Dimensional Protein Structure
3. Results and Discussion
3.1. Sequence Retrieval of the Selected Protein
3.2. Physicochemical Parameters Determination of the Selected Protein
3.3. Identification and Validation of the Predicted Secondary Structure of the Selected Protein
3.4. The Three-Dimensional Protein Structure Anticipation and Assessment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goh, K.J.; Tan, C.T.; Chew, N.K.; Tan, P.S.K.; Kamarulzaman, A.; Sarji, S.A.; Wong, K.T.; Abdullah, B.J.J.; Chua, K.B.; Lam, S.K. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N. Engl. J. Med. 2000, 342, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Chakraborty, A. Chakraborty, Nipah virus outbreaks in Bangladesh: A deadly infectious disease. WHO South East Asia J. Public Health 2012, 1, 208–212. [Google Scholar] [CrossRef]
- Chadha, M.S.; Comer, J.A.; Lowe, L.; Rota, P.A.; Rollin, P.; Bellini, W.J.; Ksiazek, T.G.; Mishra, A.C. Nipah virus-associated encephalitis outbreak, Siliguri, India. Emerg. Infect. Dis. 2006, 12, 235–240. [Google Scholar]
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef]
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: A comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.H.; Field, H.E.; Luby, S.; Pulliam, J.R.; Daszak, P. Nipah virus: Impact, origins, and causes of emergence. Curr. Infect. Dis. Rep. 2006, 8, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Gupta, N.; Kodan, P.; Mittal, A.; Ray, Y.; Nischal, N.; Soneja, M.; Biswas, A.; Wig, N. Nipah virus disease: A rare and intractable disease. Intractable Rare Dis. Res. 2019, 8, 1–8. [Google Scholar] [CrossRef]
- Luby, S.P.; Rahman, M.; Hossain, M.J.; Blum, L.S.; Husain, M.M.; Gurley, E.; Khan, R.; Ahmed, B.N.; Rahman, S.; Nahar, N.; et al. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis. 2006, 12, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, G.; Chandni, R.; Mourya, D.T.; Singh, S.K.; Sadanandan, R.; Sudan, P.; Bhargava, B. Outbreak Investigation of Nipah Virus Disease in Kerala, India, 2018. J. Infect. Dis. 2019, 219, 1867–1878. [Google Scholar] [CrossRef]
- Pillai, V.S.; Krishna, G.; Veettil, M.V. Nipah Virus: Past Outbreaks and Future Containment. Viruses 2020, 12, 465. [Google Scholar] [CrossRef]
- Aguilar, H.C.; Iorio, R.M. Henipavirus membrane fusion and viral entry. Curr. Top. Microbiol. Immunol. 2012, 359, 79–94. [Google Scholar] [PubMed]
- Ang, B.S.; Lim, T.C.; Wang, L. Nipah Virus Infection. J. Clin. Microbiol. 2018, 56, e01875-17. [Google Scholar] [CrossRef] [PubMed]
- Paul, L. Nipah virus in Kerala: A deadly Zoonosis. Clin. Microbiol. Infect. 2018, 24, 1113–1114. [Google Scholar] [CrossRef] [PubMed]
- Hickey, C.A.; Broder, C.C. The mechanism of henipavirus fusion: Examining the relationships between the attachment and fusion glycoproteins. Virol. Sin. 2009, 24, 110–120. [Google Scholar] [CrossRef]
- Lee, B.; Ataman, Z.A. Modes of paramyxovirus fusion: A Henipavirus perspective. Trends Microbiol. 2011, 19, 89–399. [Google Scholar] [CrossRef]
- Steffen, D.L.; Xu, K.; Nikolov, D.B.; Broder, C.C. Henipavirus mediated membrane fusion, virus entry and targeted therapeutics. Viruses 2012, 4, 280–308. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Wang, Z. Research Progress in Enveloped Glycoproteins and the Membrane-fusion Mechanism of Nipah Virus. Bing Du Xue Bao 2016, 32, 361–368. [Google Scholar] [PubMed]
- Mathieu, C.; Horvat, B. Henipavirus pathogenesis and antiviral approaches. Expert Rev. Anti Infect. Ther. 2015, 13, 343–354. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; E Bolton, E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef]
- Schneider, M.; Tognolli, M.; Bairoch, A. The Swiss-Prot protein knowledgebase and ExPASy: Providing the plant community with high quality proteomic data and tools. Plant Physiol. Biochem. 2004, 42, 1013–1021. [Google Scholar] [CrossRef]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Moffat, L.; Jones, D.T. Increasing the accuracy of single sequence prediction methods using a deep semi-supervised learning framework. Bioinformatics 2021, 37, 3744–3751. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.P.; Daniels, R.D.; Wong, L.; Gatta, A.T.; Gerondopoulos, A.; Barr, F. Discovery of new Longin and Roadblock domains that form platforms for small GTPases in Ragulator and TRAPP-II. Small GTPases 2013, 4, 62–69. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35 (Suppl. S2), W407–W410. [Google Scholar] [CrossRef]
- Tran, T.L.N.; Miranda, A.F.; Mouradov, A.; Adhikari, B. Physicochemical Characteristics of Protein Isolated from Thraustochytrid Oilcake. Foods 2020, 9, 779. [Google Scholar] [CrossRef]
- Dey, D.; Biswas, P.; Paul, P.; Mahmud, S.; Ema, T.I.; Khan, A.A.; Ahmed, S.Z.; Hasan, M.M.; Saikat, A.S.M.; Fatema, B.; et al. Natural flavonoids effectively block the CD81 receptor of hepatocytes and inhibit HCV infection: A computational drug development approach. Mol. Divers. 2022. [Google Scholar] [CrossRef]
- Lu, Z.X.; He, J.F.; Zhang, Y.C.; Bing, D.J. Composition, physicochemical properties of pea protein and its application in functional foods. Crit. Rev. Food Sci. Nutr. 2020, 60, 2593–2605. [Google Scholar] [CrossRef] [PubMed]
- Karadag, M.; Arslan, M.; Kaleli, N.E.; Kalyoncu, S. Physicochemical determinants of antibody-protein interactions. Adv. Protein. Chem. Struct Biol. 2020, 121, 85–114. [Google Scholar]
- Khan, R.A.; Hossain, R.; Siyadatpanah, A.; Al-Khafaji, K.; Khalipha, A.B.R.; Dey, D.; Asha, U.H.; Biswas, P.; Saikat, A.S.M.; Chenari, H.A.; et al. Diterpenes/Diterpenoids and Their Derivatives as Potential Bioactive Leads against Dengue Virus: A Computational and Network Pharmacology Study. Molecules 2021, 26, 6821. [Google Scholar] [CrossRef]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef]
- Sleep, D. Albumin and its application in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 793–812. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chem. Proc. 2022, 8, 5. [Google Scholar]
- Zaman, R.; Islam, R.A.; Ibnat, N.; Othman, I.; Zaini, A.; Lee, C.Y.; Chowdhury, E.H. Current strategies in extending half-lives of therapeutic proteins. J. Control. Release 2019, 301, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Louw, A. GR Dimerization and the Impact of GR Dimerization on GR Protein Stability and Half-Life. Front. Immunol. 2019, 10, 1693. [Google Scholar] [CrossRef]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino. Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Podust, V.N.; Balan, S.; Sim, B.-C.; Coyle, M.P.; Ernst, U.; Peters, R.T.; Schellenberger, V. Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J. Control. Release 2016, 240, 52–66. [Google Scholar] [CrossRef]
- Rajib, H.; Islam; Torequl, M.; Pranta, R.; Divya, J.; Mohammad, S.A.S.; Lutfun, N.; Das, T.A.; Satyajit, S.; Abdulmajid, A.S.; et al. Amentoflavone, New Hope against SARS-CoV-2: An Outlook through its Scientific Records and an in silico Study. Pharmacogn. Res. 2021, 13, 149–157. [Google Scholar]
- Niu, X.; Li, N.; Chen, D.; Wang, Z. Interconnection between the protein solubility and amino acid and dipeptide compositions. Protein Pept. Lett. 2013, 20, 88–95. [Google Scholar] [CrossRef]
- Huang, H.-L.; Charoenkwan, P.; Kao, T.-F.; Lee, H.-C.; Chang, F.-L.; Huang, W.-L.; Ho, S.-J.; Shu, L.-S.; Chen, W.-L. Prediction and analysis of protein solubility using a novel scoring card method with dipeptide composition. BMC Bioinform. 2012, 13 (Suppl. S17), S3. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M.; Uddin, E.; Ahmad, T.; Mahmud, S.; Imran, A.S.; Ahmed, S.; Alyami, S.A.; Moni, M.A. Structural and Functional Elucidation of IF-3 Protein of Chloroflexus aurantiacus Involved in Protein Biosynthesis: An In Silico Approach. BioMed Res. Int. 2021, 2021, 9050026. [Google Scholar] [CrossRef] [PubMed]
- Yagasaki, M.; Hashimoto, S. Synthesis and application of dipeptides; current status and perspectives. Appl. Microbiol. Biotechnol. 2008, 81, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Guruprasad, K.; Reddy, B.V.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M.; Islam, R.; Mahmud, S.; Imran, M.A.S.; Alam, M.S.; Masud, M.H.; Uddin, M.E. Structural and Functional Annotation of Uncharacterized Protein NCGM946K2_146 of Mycobacterium Tuberculosis: An In-Silico Approach. Proceedings 2020, 66, 13. [Google Scholar]
- Gamage, D.G.; Gunaratne, A.; Periyannan, G.; Russell, T.G. Applicability of Instability Index for In vitro Protein Stability Prediction. Protein Pept. Lett. 2019, 26, 339–347. [Google Scholar] [CrossRef]
- Panda, S.; Chandra, G. Physicochemical characterization and functional analysis of some snake venom toxin proteins and related non-toxin proteins of other chordates. Bioinformation 2012, 8, 891–896. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [PubMed]
- Enany, S. Structural and functional analysis of hypothetical and conserved proteins of Clostridium tetani. J. Infect. Public Health 2014, 7, 296–307. [Google Scholar] [CrossRef]
- Chang, Y.K.; Yang, J.R. Analysis and prediction of highly effective antiviral peptides based on random forests. PLoS ONE 2013, 8, e70166. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Karnik, S.; Barai, R.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010, 38, D774–80. [Google Scholar] [CrossRef]
- Hossain, R.; Quispe, C.; Saikat, A.S.M.; Jain, D.; Habib, A.; Janmeda, P.; Islam, M.T.; Radha, U.; Daştan, S.D.; Kumar, M.; et al. Biosynthesis of Secondary Metabolites Based on the Regulation of MicroRNAs. Biomed Res. Int. 2022, 2022, 9349897. [Google Scholar] [CrossRef]
- Gromiha, M.M. Chapter 1—Proteins. In Protein Bioinformatics; Gromiha, M.M., Ed.; Academic Press: Singapore, 2010; pp. 1–27. [Google Scholar]
- Wardah, W.; Khan, M.; Sharma, A.; Rashid, M. Protein secondary structure prediction using neural networks and deep learning: A review. Comput. Biol. Chem. 2019, 81, 1–8. [Google Scholar] [CrossRef]
- Koch, I.; Schäfer, T. Protein super-secondary structure and quaternary structure topology: Theoretical description and application. Curr. Opin. Struct. Biol. 2018, 50, 134–143. [Google Scholar] [CrossRef]
- Jisna, A.V.; Jayaraj, P.B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein J. 2021, 40, 522–544. [Google Scholar] [CrossRef]
- Hou, J.; Wu, T.; Cao, R.; Cheng, J. Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13. Proteins 2019, 87, 1165–1178. [Google Scholar] [CrossRef]
- Tamburrini, K.C.; Pesce, G.; Nilsson, J.; Gondelaud, F.; Kajava, A.V.; Berrin, J.-G.; Longhi, S. Predicting Protein Conformational Disorder and Disordered Binding Sites. Methods Mol. Biol. 2022, 2449, 95–147. [Google Scholar] [PubMed]
- Reinert, E.Z.; Horne, W.S. Protein backbone engineering as a strategy to advance foldamers toward the frontier of protein-like tertiary structure. Org. Biomol. Chem. 2014, 12, 8796–8802. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Cao, W.; Saad, G.; Shoji, M.; Terada, T. Comparative analysis of membrane protein structure databases. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1077–1091. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Individualities | Protein Information |
|---|---|
| Locus | QBQ56722 |
| Amino acid | 546 aa |
| Accession | QBQ56722 |
| Version | QBQ56722.1 |
| GenBank ID | QBQ56722.1 |
| Source | Nipah henipavirus |
| Organism | Nipah henipavirus |
| FASTA sequence | >QBQ56722.1 fusion protein [Nipah henipavirus] MAVILNKRYYSNLLLLILMISECSVGILHYEKLSKIGLVKGITRKYKIKSNPLTKDIVIKMIPNVSNMSQCTGSVMENYKTRLNGILTPIKGALEIYKNNTHDLVGDVRLAGVIMAGVAIGIATAAQITAGVALYEAMKNADNINKLKSSIESTNEAVVKLQETAEKTVYVLTALQDYINTNLVPTIDKISCKQTELSLDLALSKYLSDLLFVFGPNLQDPVSNSMTIQAISQAFGGNYETLLRTLGYATEDFDDLLESDSITGQIIYVDLSGYYIIVRVYFPILTEIQQAYIQELLPVSFNNDNSEWISIVPNFILVRNTLISNIEIGFCLITKRSVICNQDYATPMTNNMRECLTGSTEKCPRELVVSSHVPRFALSNGVLFANCISVTCQCQTTGRAISQSGEQTLLMIDNTTCPTAVLGNVIISLGKYLGSVNYNSEGIAIGPPVFTDKVDISSQISSMNQSLQQSKDYIKEAQRLLDTVNPSLISMLSMIILYVLSIASLCIGLITFISFIIVEKKRNTYSRLEDRRVRPTSSGDLYYIGT |
| Parameters | Values |
|---|---|
| Molecular weight | 60,280.90 Da |
| Theoretical pI | 6.08 (6.30 *) |
| Total number of positively charged residues (Arg + Lys) | 46 |
| Total number of negatively charged residues (Asp + Glu) | 48 |
| Total number of atoms | 8584 |
| Estimated half-life | (a) 30 h (mammalian reticulocytes, in vitro) (b) >20 h (yeast, in vivo) (c) >10 h (Escherichia coli, in vivo) |
| Instability index (II) | 38.05 |
| Aliphatic index | 112.27 |
| Grand average of hydropathicity (GRAVY) | 0.177 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saikat, A.S.M.; Das, R.C.; Das, M.C. Computational Approaches for Structure-Based Molecular Characterization and Functional Annotation of the Fusion Protein of Nipah henipavirus. Chem. Proc. 2022, 12, 32. https://doi.org/10.3390/ecsoc-26-13530
Saikat ASM, Das RC, Das MC. Computational Approaches for Structure-Based Molecular Characterization and Functional Annotation of the Fusion Protein of Nipah henipavirus. Chemistry Proceedings. 2022; 12(1):32. https://doi.org/10.3390/ecsoc-26-13530
Chicago/Turabian StyleSaikat, Abu Saim Mohammad, Ranjit Chandra Das, and Madhab Chandra Das. 2022. "Computational Approaches for Structure-Based Molecular Characterization and Functional Annotation of the Fusion Protein of Nipah henipavirus" Chemistry Proceedings 12, no. 1: 32. https://doi.org/10.3390/ecsoc-26-13530
APA StyleSaikat, A. S. M., Das, R. C., & Das, M. C. (2022). Computational Approaches for Structure-Based Molecular Characterization and Functional Annotation of the Fusion Protein of Nipah henipavirus. Chemistry Proceedings, 12(1), 32. https://doi.org/10.3390/ecsoc-26-13530