
Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes


Abstract
1. Introduction
2. Background of Autoimmune T1D
2.1. Immune Cells Involved in the Pathophysiology of Autoimmune Type 1 Diabetes
2.2. Immunotherapy-Based Approaches to Treating Autoimmune T1D
2.3. There Are a Few Obstacles to Immunotherapy-Based Treatment for Autoimmune T1D Cure
3. Conclusions and Future Perspectives
- What factors/agents cause the initial destruction of pancreatic β-cells in T1D patients?
- ○
- Genetic or epigenetic modifications;
- ○
- Viral infection;
- ○
- Gut microbial flora;
- ○
- Environmental or diet and nutrition;
- ○
- Aging or developmental changes destroy β-cells.
- Once pancreatic cells are destroyed, how do immune cells select their cognitive epitope; why do these epitopes express or present more? What factor or cells cause them?
- Self-epitope/antigen enters circulation—how/why do CD4/CD8 T cells begin recognizing self-antigen, indicating that TCR rearrangement occurred previously with the corresponding epitope? What causes the immune tolerance system to fail to circulate these immune cells?
- Once in the circulation, β-cells antigen/markers, specific T cells are generated; how do these T cells begin attacking remaining β-cell masses who give them command? Is there any expression of pancreas-specific signals or chemokine receptors on these cells? How they infiltrated the pancreatic islet to destroy-cells.
- What causes Treg cells to become inactive?
- How can we prevent an unwelcome immune attack on pancreatic β-cells? The best strategy is to hide and attack.
- Hide: Modify pancreatic β-cell-specific antigen recognition/presentation to preserve β-cell mass.
- Attack: Attacking the rebel immune cells is another option; we can generate antigen-specific Treg or cytotoxic CD4/CD8 T cells to kill the rebel immune cells.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Firestein, G.S.; Budd, R.C.; Harris, E.D.; McInnes, I.B.; Sergent, J.S. Kelley’s Textbook of Rheumatology, 8th ed.; WB Saunders: Philadephia, PA, USA, 2008. [Google Scholar]
- Goldman, L.; Ausiello, D.A. Cecil Medicine; Saunders Elsevier: Philadelphia, PA, USA, 2008. [Google Scholar]
- Herold, K.C.; Vignali, D.A.A.; Cooke, A.; Bluestone, J.A. Type 1 diabetes: Translating mechanistic observations into effective clinical outcomes. Nat. Rev. Immunol. 2013, 13, 243–256. [Google Scholar] [CrossRef]
- Tisch, R.; McDevitt, H. Insulin-dependent diabetes mellitus. Cell 1996, 85, 291–297. [Google Scholar] [CrossRef]
- Anderson, M.S.; Bluestone, J.A. THE NOD MOUSE: A Model of Immune Dysregulation. Annu. Rev. Immunol. 2005, 23, 447–485. [Google Scholar] [CrossRef] [PubMed]
- Burn, P. Type 1 diabetes. Nat. Rev. Drug Discov. 2010, 9, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Głowińska-Olszewska, B.; Szabłowski, M.; Panas, P.; Żołądek, K.; Jamiołkowska-Sztabkowska, M.; Milewska, A.J.; Kadłubiska, A.; Polkowska, A.; Łuczyński, W.; Bossowski, A. Increasing Co-occurrence of Additional Autoimmune Disorders at Diabetes Type 1 Onset Among Children and Adolescents Diagnosed in Years 2010–2018—Single-Center Study. Front. Endocrinol. 2020, 11, 476. [Google Scholar] [CrossRef]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef]
- Van Belle, T.L.; Coppieters, K.T.; Von Herrath, M.G. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef]
- Atkinson, M.A. The Pathogenesis and Natural History of Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007641. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef]
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- Pepper, A.R.; Bruni, A.; Shapiro, A.J. Clinical islet transplantation: Is the future finally now? Curr. Opin. Organ Transplant. 2018, 23, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Bassi, R.; Fiorina, P. Impact of Islet Transplantation on Diabetes Complications and Quality of Life. Curr. Diabetes Rep. 2011, 11, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, Å. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Harjutsalo, V.; Podar, T.; Tuomilehto, J. Cumulative Incidence of Type 1 Diabetes in 10,168 Siblings of Finnish Young-Onset Type 1 Diabetic Patients. Diabetes 2005, 54, 563–569. [Google Scholar] [CrossRef]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for Islet Autoimmunity among Monozygotic Twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Ali, M.A.; Liu, Y.-F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9, eaaf7779. [Google Scholar] [CrossRef]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Skyler, J.S.; Pugliese, A. Immunotherapy Trials for Type 1 Diabetes: The Contribution of George Eisenbarth. Diabetes Technol. Ther. 2013, 15, S2-13–S2-30. [Google Scholar] [CrossRef] [PubMed]
- Feutren, G.; Assan, R.; Karsenty, G.; Du Rostu, H.; Sirmai, J.; Papoz, L.; Vialettes, B.; Vexiau, P.; Rodier, M.; Lallemand, A.; et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset: Results of a multicentre double-blind trial. Lancet 1986, 328, 119–124. [Google Scholar] [CrossRef]
- Sigal, N.H.; Dumont, F.J.; Cyclosporin, A. FK-506, and rapamycin: Pharmacologic probes of lymphocyte signal transduction. Annu. Rev. Immunol. 1992, 10, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Canadian-European Randomized Control Trial Group. Cyclosporin-induced remission of IDDM after early intervention: Association of 1 yr of cyclosporin treatment with enhanced insulin secretion. Diabetes 1988, 37, 1574–1582. [Google Scholar] [CrossRef]
- Füchtenbusch, M.; Kredel, K.; Bonifacio, E.; Schnell, O.; Ziegler, A.G. Exposure to exogenous insulin promotes IgG1 and the T-helper 2-associated IgG4 responses to insulin but not to other islet autoantigens. Diabetes 2000, 49, 918–925. [Google Scholar] [CrossRef]
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor–CD3 complex. Nature 2019, 573, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Long, S.A.; Thorpe, J.; DeBerg, H.A.; Gersuk, V.; Eddy, J.A.; Harris, K.M.; Ehlers, M.; Herold, K.C.; Nepom, G.T.; Linsley, P.S. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci. Immunol. 2016, 1, eaai7793. [Google Scholar] [CrossRef]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef]
- Dolgin, E. Anti-CD3 drug keeps diabetes at bay. Nat. Biotechnol. 2019, 37, 1099–1101. [Google Scholar] [CrossRef]
- Wang, X.; Ni, L.; Chang, D.; Lu, H.; Jiang, Y.; Kim, B.-S.; Wang, A.; Liu, X.; Zhong, B.; Yang, X.; et al. Cyclic AMP-Responsive Element-Binding Protein (CREB) is Critical in Autoimmunity by Promoting Th17 but Inhibiting Treg Cell Differentiation. EBioMedicine 2017, 25, 165–174. [Google Scholar] [CrossRef]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-Hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, L.; Sheng, X.; Chen, W.; Tang, H.; Sheng, H.; Xi, B.; Zang, Y.Q. T-cell vaccination leads to suppression of intrapancreatic Th17 cells through Stat3-mediated RORγt inhibition in autoimmune diabetes. Cell Res. 2011, 21, 1358–1369. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stifter, K.; Schuster, C.; Schlosser, M.; Boehm, B.O.; Schirmbeck, R. Exploring the induction of preproinsulin-specific Foxp3+ CD4+ Treg cells that inhibit CD8+ T cell-mediated autoimmune diabetes by DNA vaccination. Sci. Rep. 2016, 6, 29419. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Robey, E.A.; Hsieh, C.-S. Central CD4+ T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol. 2019, 19, 7–18. [Google Scholar] [CrossRef]
- Kitashima, D.Y.; Kobayashi, T.; Woodring, T.; Idouchi, K.; Döbel, T.; Voisin, B.; Adachi, T.; Ouchi, T.; Takahashi, H.; Nishifuji, K.; et al. Langerhans Cells Prevent Autoimmunity via Expansion of Keratinocyte Antigen-Specific Regulatory T Cells. EBioMedicine 2018, 27, 293–303. [Google Scholar] [CrossRef]
- Boardman, D.; Levings, M.K. Cancer immunotherapies repurposed for use in autoimmunity. Nat. Biomed. Eng. 2019, 3, 259–263. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Tack, C.J.; Kleijwegt, F.S.; Van Riel, P.L.C.M.; Roep, B.O. Development of type 1 diabetes in a patient treated with anti-TNF-α therapy for active rheumatoid arthritis. Diabetologia 2009, 52, 1442–1444. [Google Scholar] [CrossRef]
- Mastrandrea, L.; Yu, J.; Behrens, T.; Buchlis, J.; Albini, C.; Fourtner, S.; Quattrin, T. Etanercept Treatment in Children With New-Onset Type 1 Diabetes: Pilot Randomized, Placebo-Controlled, Double-Blind Study: Response to Peters. Diabetes Care 2009, 32, e154. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.L. TNF, TNF inducers, and TNFR2 agonists: A new path to type 1 diabetes treatment. Diabetes/Metab. Res. Rev. 2018, 34, e2941. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.A.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moneim, A.; Bakery, H.H.; Allam, G. The potential pathogenic role of IL-17/Th17 cells in both type 1 and type 2 diabetes mellitus. Biomed. Pharmacother. 2018, 101, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Nambam, B.; Haller, M.J. Updates on Immune Therapies in Type 1 Diabetes. Eur. Endocrinol. 2016, 12, 89–95. [Google Scholar] [CrossRef]
- Cabrera, S.M.; Wang, X.; Chen, Y.-G.; Jia, S.; Kaldunski, M.; Greenbaum, C.J.; Mandrup-Poulsen, T.; Hessner, M.J.; the Type 1 Diabetes TrialNet Canakinumab Study Group; the AIDA Study Group. Interleukin-1 antagonism moderates the inflammatory state associated with Type 1 diabetes during clinical trials conducted at disease onset. Eur. J. Immunol. 2015, 46, 1030–1046. [Google Scholar] [CrossRef]
- Van Asseldonk, E.J.; Van Poppel, P.C.; Ballak, D.B.; Stienstra, R.; Netea, M.G.; Tack, C.J. One week treatment with the IL-1 receptor antagonist anakinra leads to a sustained improvement in insulin sensitivity in insulin resistant patients with type 1 diabetes mellitus. Clin. Immunol. 2015, 160, 155–162. [Google Scholar] [CrossRef]
- Mariño, E.; Silveira, P.A.; Stolp, J.; Grey, S.T. B cell-directed therapies in type 1 diabetes. Trends Immunol. 2011, 32, 287–294. [Google Scholar] [CrossRef]
- Pescovitz, M.D.; Greenbaum, C.J.; Bundy, B.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; Moran, A.; Raskin, P.; et al. B-Lymphocyte Depletion With Rituximab and β-Cell Function: Two-Year Results. Diabetes Care 2014, 37, 453–459. [Google Scholar] [CrossRef]
- Wu, L.; Dakic, A. Development of dendritic cell system. Cell. Mol. Immunol. 2004, 1, 112–118. [Google Scholar]
- Shang, N.; Figini, M.; Shangguan, J.; Wang, B.; Sun, C.; Pan, L.; Ma, Q.; Zhang, Z. Dendritic cells based immunotherapy. Am. J. Cancer Res. 2017, 7, 2091. [Google Scholar]
- Marin-Gallen, S.; Clemente-Casares, X.; Planas, R.; Pujol-Autonell, I.; Carrascal, J.; Carrillo, J.; Ampudia, R.; Verdaguer, J.; Pujol-Borrell, R.; Borràs, F.E.; et al. Dendritic cells pulsed with antigen-specific apoptotic bodies prevent experimental type 1 diabetes. Clin. Exp. Immunol. 2009, 160, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Creusot, R.J.; Giannoukakis, N.; Trucco, M.; Clare-Salzler, M.J.; Fathman, C.G. It’s Time to Bring Dendritic Cell Therapy to Type 1 Diabetes. Diabetes 2013, 63, 20–30. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (Safety) Study of Autologous Tolerogenic Dendritic Cells in Type 1 Diabetic Patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Regnell, S.E.; Lernmark, Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Qaisar, N.; Jurczyk, A.; Wang, J.P. Potential role of type I interferon in the pathogenic process leading to type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Harrison, L.C.; von Herrath, M.G. Trials in type 1 diabetes: Antigen-specific therapies. Clin. Immunol. 2013, 149, 345–355. [Google Scholar] [CrossRef][Green Version]
- Luo, X.; Herold, K.C.; Miller, S.D. Immunotherapy of Type 1 Diabetes: Where Are We and Where Should We Be Going? Immunity 2010, 32, 488–499. [Google Scholar] [CrossRef]
- Walker, L.S.; Abbas, A.K. The enemy within: Keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2002, 2, 11–19. [Google Scholar] [CrossRef]
- Grebinoski, S.; Vignali, D.A. Inhibitory receptor agonists: The future of autoimmune disease therapeutics? Curr. Opin. Immunol. 2020, 67, 1–9. [Google Scholar] [CrossRef]
- Tang, Q.; Henriksen, K.J.; Bi, M.; Finger, E.B.; Szot, G.; Ye, J.; Masteller, E.L.; McDevitt, H.; Bonyhadi, M.; Bluestone, J.A. In Vitro–expanded Antigen-specific Regulatory T Cells Suppress Autoimmune Diabetes. J. Exp. Med. 2004, 199, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Reya, T. Illuminating immune privilege—A role for regulatory T cells in preventing rejection. N. Engl. J. Med. 2011, 365, 956–957. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.F.; Di Ianni, M.; Ruggeri, L.; Falzetti, F.; Carotti, A.; Terenzi, A.; Pierini, A.; Massei, M.S.; Amico, L.; Urbani, E.; et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood 2014, 124, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Hull, C.M.; Nickolay, L.E.; Estorninho, M.; Richardson, M.W.; Riley, J.L.; Peakman, M.; Maher, J.; Tree, T.I. Generation of human islet-specific regulatory T cells by TCR gene transfer. J. Autoimmun. 2017, 79, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Safari, F.; Farajnia, S.; Arya, M.; Zarredar, H.; Nasrolahi, A. CRISPR and personalized Treg therapy: New insights into the treatment of rheumatoid arthritis. Immunopharmacol. Immunotoxicol. 2018, 40, 201–211. [Google Scholar] [CrossRef]
- Aronson, R.; Gottlieb, P.A.; Christiansen, J.S.; Donner, T.W.; Bosi, E.; Bode, B.W.; Pozzilli, P.; the DEFEND Investigator Group. Low-Dose Otelixizumab Anti-CD3 Monoclonal Antibody DEFEND-1 Study: Results of the Randomized Phase III Study in Recent-Onset Human Type 1 Diabetes. Diabetes Care 2014, 37, 2746–2754. [Google Scholar] [CrossRef]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J.; Bode, B.; Aronoff, S.; Holand, C.; Carlin, D.; et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef]
- Hagopian, W.; Ferry, R.J.; Sherry, N.; Carlin, D.; Bonvini, E.; Johnson, S.; Stein, K.; Koenig, S.; Daifotis, A.; Herold, K.; et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: Two-year results from the randomized, placebo-controlled Protégé trial. Diabetes 2013, 62, 3901–3908. [Google Scholar] [CrossRef]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: Metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-Lymphocyte Depletion, and Preservation of Beta-Cell Function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Costimulation Modulation With Abatacept in Patients With Recent-Onset Type 1 Diabetes: Follow-up 1 Year After Cessation of Treatment. Diabetes Care 2014, 37, 1069–1075. [Google Scholar] [CrossRef]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef]
- Rigby, M.R.; Harris, K.M.; Pinckney, A.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Griffin, K.J.; Tsalikian, E.; et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J. Clin. Investig. 2015, 125, 3285–3296. [Google Scholar] [CrossRef]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- Moran, A.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; Raskin, P.; et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: Two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 2013, 381, 1905–1915. [Google Scholar] [CrossRef]
- Sumpter, K.M.; Adhikari, S.; Grishman, E.K.; White, P.C. Preliminary studies related to anti-interleukin-1β therapy in children with newly diagnosed type 1 diabetes. Pediatr. Diabetes 2011, 12, 656–667. [Google Scholar] [CrossRef]
- Bonifacio, E.; Ziegler, A.G.; Klingensmith, G.; Schober, E.; Bingley, P.J.; Rottenkolber, M.; Theil, A.; Eugster, A.; Puff, R.; Peplow, C.; et al. Effects of high-dose oral insulin on immune responses in children at high risk for type 1 diabetes: The Pre-POINT randomized clinical trial. JAMA 2015, 313, 1541–1549. [Google Scholar] [CrossRef]
- Voltarelli, J.C.; Couri, C.E.B.; Stracieri, A.B.P.L.; Oliveira, M.C.; Moraes, D.A.; Pieroni, F.; Coutinho, M.; Malmegrim, K.C.R.; Foss-Freitas, M.C.; Simões, B.P.; et al. Autologous Nonmyeloablative Hematopoietic Stem Cell Transplantation in Newly Diagnosed Type 1 Diabetes Mellitus. JAMA J. Am. Med. Assoc. 2007, 297, 1568–1576. [Google Scholar] [CrossRef]
- D’Addio, F.; Vasquez, A.V.; Ben Nasr, M.; Franek, E.; Zhu, D.; Li, L.; Ning, G.; Snarski, E.; Fiorina, P. Autologous Nonmyeloablative Hematopoietic Stem Cell Transplantation in New-Onset Type 1 Diabetes: A Multicenter Analysis. Diabetes 2014, 63, 3041–3046. [Google Scholar] [CrossRef]
- Haller, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Michels, A.; Rosenthal, S.M.; Shuster, J.J.; Zou, B.; Brusko, T.M.; Hulme, M.A.; Wasserfall, C.; et al. Anti-thymocyte globulin/G-CSF treatment preserves β cell function in patients with established type 1 diabetes. J. Clin. Investig. 2015, 125, 448–455. [Google Scholar] [CrossRef]
- Michels, A.W.; Landry, L.G.; McDaniel, K.A.; Yu, L.; Campbell-Thompson, M.; Kwok, W.W.; Jones, K.L.; Gottlieb, P.A.; Kappler, J.W.; Tang, Q.; et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes 2016, 66, 722–734. [Google Scholar] [CrossRef]
- Wong, F.S.; Karttunen, J.; Dumont, C.; Wen, L.; Visintin, I.; Pilip, I.M.; Shastri, N.; Pamer, E.G.; Janeway, C.A. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat. Med. 1999, 5, 1026–1031. [Google Scholar] [CrossRef]
- Chen, J.; Grieshaber, S.; Mathews, C.E. Methods to Assess Beta Cell Death Mediated by Cytotoxic T Lymphocytes. J. Vis. Exp. 2011, 2011, e2724. [Google Scholar] [CrossRef]
- Krishnamurthy, B.; Dudek, N.L.; McKenzie, M.D.; Purcell, A.; Brooks, A.; Gellert, S.; Colman, P.G.; Harrison, L.C.; Lew, A.; Thomas, H.E.; et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J. Clin. Investig. 2006, 116, 3258–3265. [Google Scholar] [CrossRef]
- Nakayama, M.; Abiru, N.; Moriyama, H.; Babaya, N.; Liu, E.; Miao, D.; Yu, L.; Wegmann, D.R.; Hutton, J.C.; Elliott, J.F.; et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005, 435, 220–223. [Google Scholar] [CrossRef]
- Prasad, S.; Kohm, A.P.; McMahon, J.S.; Luo, X.; Miller, S.D. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading. J. Autoimmun. 2012, 39, 347–353. [Google Scholar] [CrossRef]
- Zhang, L.; Nakayama, M.; Eisenbarth, G.S. Insulin as an autoantigen in NOD/human diabetes. Curr. Opin. Immunol. 2008, 20, 111–118. [Google Scholar] [CrossRef]
- Kracht, M.J.L.; Van Lummel, M.; Nikolic, T.; Joosten, A.M.; Laban, S.; van der Slik, A.; van Veelen, P.; Carlotti, F.; De Koning, F.C.E.J.P.; Hoeben, R.; et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat. Med. 2017, 23, 501–507. [Google Scholar] [CrossRef]
- Jin, N.; Wang, Y.; Crawford, F.; White, J.; Marrack, P.; Dai, S.; Kappler, J.W. N-terminal additions to the WE14 peptide of chromogranin A create strong autoantigen agonists in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2015, 112, 13318–13323. [Google Scholar] [CrossRef]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Armstrong, M.; Powell, R.L.; Reisdorph, N.; Kumar, N.; et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351, 711–714. [Google Scholar] [CrossRef]
- Wiles, T.A.; Powell, R.; Michel, C.R.; Beard, K.S.; Hohenstein, A.; Bradley, B.; Reisdorph, N.; Haskins, K.; Delong, T. Identification of Hybrid Insulin Peptides (HIPs) in Mouse and Human Islets by Mass Spectrometry. J. Proteome Res. 2019, 18, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.L.; Rihanek, M.; Hohenstein, A.C.; Nakayama, M.; Michels, A.; Gottlieb, P.A.; Haskins, K.; Delong, T. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes 2019, 68, 1830–1840. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.L.; Clare-Salzler, M.; Tian, J.; Forsthuber, T.; Ting, G.S.P.; Robinson, P.; Atkinson, M.A.; Sercarz, E.E.; Tobin, A.J.; Lehmann, P.V. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature 1993, 366, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Kaufman, D.L.; Newman, D.; Tobin, A.J.; Maclaren, N.K. Islet cell cytoplasmic autoantibody reactivity to glutamate decarboxylase in insulin-dependent diabetes. J. Clin. Investig. 1993, 91, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Schloot, N.C.; Daniel, D.; Norbury-Glaser, M.; Wegmann, D.R. Peripheral T cell Clones from NOD Mice Specific for GAD65 Peptides: Lack of Islet Responsiveness or Diabetogenicity. J. Autoimmun. 1996, 9, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Videbæk, N.; Harach, S.; Phillips, J.; Hutchings, P.; Ozegbe, P.; Michelsen, B.K.; Cooke, A. An islet-homing NOD CD8+cytotoxic T cell clone recognizes GAD65and causes insulitis. J. Autoimmun. 2003, 20, 97–109. [Google Scholar] [CrossRef]
- Wenzlau, J.M.; Walter, M.; Gardner, T.J.; Frisch, L.M.; Yu, L.; Eisenbarth, G.S.; Ziegler, A.-G.; Davidson, H.W.; Hutton, J.C. Kinetics of the Post-Onset Decline in Zinc Transporter 8 Autoantibodies in Type 1 Diabetic Human Subjects. J. Clin. Endocrinol. Metab. 2010, 95, 4712–4719. [Google Scholar] [CrossRef]
- Dang, M.; Rockell, J.; Wagner, R.; Wenzlau, J.M.; Yu, L.; Hutton, J.C.; Gottlieb, P.A.; Davidson, H.W. Human Type 1 Diabetes Is Associated with T Cell Autoimmunity to Zinc Transporter. J. Immunol. 2011, 186, 6056–6063. [Google Scholar] [CrossRef]
- Nayak, D.; Calderon, B.; Vomund, A.N.; Unanue, E.R. ZnT8-Reactive T Cells Are Weakly Pathogenic in NOD Mice but Can Participate in Diabetes Under Inflammatory Conditions. Diabetes 2014, 63, 3438–3448. [Google Scholar] [CrossRef]
- Xu, X.; Gu, Y.; Bian, L.; Shi, Y.; Cai, Y.; Chen, Y.; Chen, H.; Qian, L.; Wu, X.; Xu, K.; et al. Characterization of immune response to novel HLA-A2-restricted epitopes from zinc transporter 8 in type 1 diabetes. Vaccine 2015, 34, 854–862. [Google Scholar] [CrossRef]
- Émmanuelle, É.; Kratzer, R.; Arnoux, J.-B.; Barilleau, E.; Hamel, Y.; Marchi, C.; Beltrand, J.; Michaud, B.; Chatenoud, L.; Robert, J.-J.; et al. ZnT8 Is a Major CD8+ T Cell–Recognized Autoantigen in Pediatric Type 1 Diabetes. Diabetes 2012, 61, 1779–1784. [Google Scholar] [CrossRef]
- Wenzlau, J.M.; Frisch, L.M.; Hutton, J.C.; Davidson, H.W. Mapping of conformational autoantibody epitopes in ZNT8. Diabetes/Metab. Res. Rev. 2011, 27, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Kubosaki, A.; Miura, J.; Notkins, A.L. IA-2 is not required for the development of diabetes in NOD mice. Diabetologia 2004, 47, 149–150. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ouyang, Q.; Standifer, N.E.; Qin, H.; Gottlieb, P.; Verchere, C.B.; Nepom, G.T.; Tan, R.; Panagiotopoulos, C. Recognition of HLA class I-restricted beta-cell epitopes in type 1 diabetes. Diabetes 2006, 55, 3068–3074. [Google Scholar] [CrossRef]
- Zhao, Z.; Miao, D.; Michels, A.; Steck, A.; Dong, F.; Rewers, M.; Yu, L. A multiplex assay combining insulin, GAD, IA-2 and transglutaminase autoantibodies to facilitate screening for pre-type 1 diabetes and celiac disease. J. Immunol. Methods 2016, 430, 28–32. [Google Scholar] [CrossRef]
- Kawasaki, E.; Hutton, J.C.; Eisenbarth, G.S. Molecular Cloning and Characterization of the Human Transmembrane Protein Tyrosine Phosphatase Homologue, Phogrin, an Autoantigen of Type 1 Diabetes. Biochem. Biophys. Res. Commun. 1996, 227, 440–447. [Google Scholar] [CrossRef]
- Kawasaki, E.; Eisenbarth, G.S.; Wasmeier, C.; Hutton, J.C. Autoantibodies to protein tyrosine phosphatase-like proteins in type I diabetes: Overlapping specificities to phogrin and ICA512/IA-2. Diabetes 1996, 45, 1344–1349. [Google Scholar] [CrossRef]
- Kelemen, K.; Crawford, M.L.; Gill, R.G.; Hutton, J.C.; Wegmann, D. Cellular immune response to phogrin in the NOD mouse: Cloned T-cells cause destruction of islet transplants. Diabetes 1999, 48, 1529–1534. [Google Scholar] [CrossRef]
- Kelemen, K.; Gottlieb, P.A.; Putnam, A.L.; Davidson, H.W.; Wegmann, D.R.; Hutton, J.C. HLA-DQ8-associated T cell responses to the diabetes autoantigen phogrin (IA-2 beta) in human prediabetes. J. Immunol. 2004, 172, 3955–3962. [Google Scholar] [CrossRef]
- Karges, W.; Hammond-McKibben, D.; Gaedigk, R.; Shibuya, N.; Cheung, R.; Dosch, H.M. Loss of self-tolerance to ICA69 in nonobese diabetic mice. Diabetes 1997, 46, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Astsaturov, I.; Gaedigk, R.; Hammond-McKibben, D.; Pilon, M.; Song, A.; Kubiak, V.; Karges, W.; Arpaia, E.; McKerlie, C.; et al. ICA69null Nonobese Diabetic Mice Develop Diabetes, but Resist Disease Acceleration by Cyclophosphamide. J. Immunol. 2002, 168, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Tsui, H.; Lau, A.; Song, A.; Li, X.; Cheung, R.K.; Sampson, A.; Afifiyan, F.; Elford, A.; Jackowski, G.; et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat. Med. 2003, 9, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Bergerot, I.; Elliott, J.F.; Harrison, L.C.; Abiru, N.; Eisenbarth, G.S.; Delovitch, T.L. Evidence that a peptide spanning the B-C junction of proinsulin is an early Autoantigen epitope in the pathogenesis of type 1 diabetes. J. Immunol. 2001, 167, 4926–4935. [Google Scholar] [CrossRef]
- Spitzenberger, F.; Pietropaolo, S.; Verkade, P.; Habermann, B.; Lacas-Gervais, S.; Mziaut, H.; Pietropaolo, M.; Solimena, M. Islet Cell Autoantigen of 69 kDa Is an Arfaptin-related Protein Associated with the Golgi Complex of Insulinoma INS-1 Cells. J. Biol. Chem. 2003, 278, 26166–26173. [Google Scholar] [CrossRef]
- Stadinski, B.D.; Delong, T.; Reisdorph, N.; Reisdorph, R.; Powell, R.L.; Armstrong, M.; Piganelli, J.D.; Barbour, G.; Bradley, B.; Crawford, F.; et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat. Immunol. 2010, 11, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.A.; Delong, T.; Baker, R.L.; Fitzgerald-Miller, L.; Wagner, R.; Cook, G.; Rewers, M.R.; Michels, A.; Haskins, K. Chromogranin A is a T cell antigen in human type 1 diabetes. J. Autoimmun. 2014, 50, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Nikoopour, E.; Krougly, O.; Lee-Chan, E.; Haeryfar, S.M.; Singh, B. Detection of vasostatin-1-specific CD8+ T cells in non-obese diabetic mice that contribute to diabetes pathogenesis. Clin. Exp. Immunol. 2016, 185, 292–300. [Google Scholar] [CrossRef]
- Delong, T.; Baker, R.L.; Reisdorph, N.; Reisdorph, R.; Powell, R.L.; Armstrong, M.; Barbour, G.; Bradley, B.; Haskins, K. Islet Amyloid Polypeptide Is a Target Antigen for Diabetogenic CD4+ T Cells. Diabetes 2011, 60, 2325–2330. [Google Scholar] [CrossRef]
- Baker, R.L.; Delong, T.; Barbour, G.; Bradley, B.; Nakayama, M.; Haskins, K. Cutting edge: CD4 T cells reactive to an islet amyloid polypeptide peptide accumulate in the pancreas and contribute to disease pathogenesis in nonobese diabetic mice. J. Immunol. 2013, 191, 3990–3994. [Google Scholar] [CrossRef]
- Viret, C.; Mahiddine, K.; Baker, R.L.; Haskins, K.; Guerder, S. The T Cell Repertoire–Diversifying Enzyme TSSP Contributes to Thymic Selection of Diabetogenic CD4 T Cell Specificities Reactive to ChgA and IAPP Autoantigens. J. Immunol. 2015, 195, 1964–1973. [Google Scholar] [CrossRef]
- Wiles, T.A.; Delong, T.; Baker, R.; Bradley, B.; Barbour, G.; Powell, R.L.; Reisdorph, N.; Haskins, K. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J. Autoimmun. 2017, 78, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Serra, P.; Amrani, A.; Yamanouchi, J.; Marée, A.F.M.; Edelstein-Keshet, L.; Santamaria, P. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: High efficiency of a low-affinity peptide. Nat. Med. 2005, 11, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Danke, N.A.; Berger, D.; Reichstetter, S.; Reijonen, H.; Greenbaum, C.; Pihoker, C.; James, E.A.; Kwok, W.W. Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein-Reactive CD4+ T Cells in Human Subjects. J. Immunol. 2006, 176, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-J.; Chee, J.; Sutherland, R.M.; Thomas, H.E.; Zhan, Y.; Krishnamurthy, B.; Kay, T.W.H.; Lew, A.M. Functional cytotoxic T lymphocytes against IGRP 206-214 predict diabetes in the non-obese diabetic mouse. Immunol. Cell Biol. 2014, 92, 640–644. [Google Scholar] [CrossRef] [PubMed]




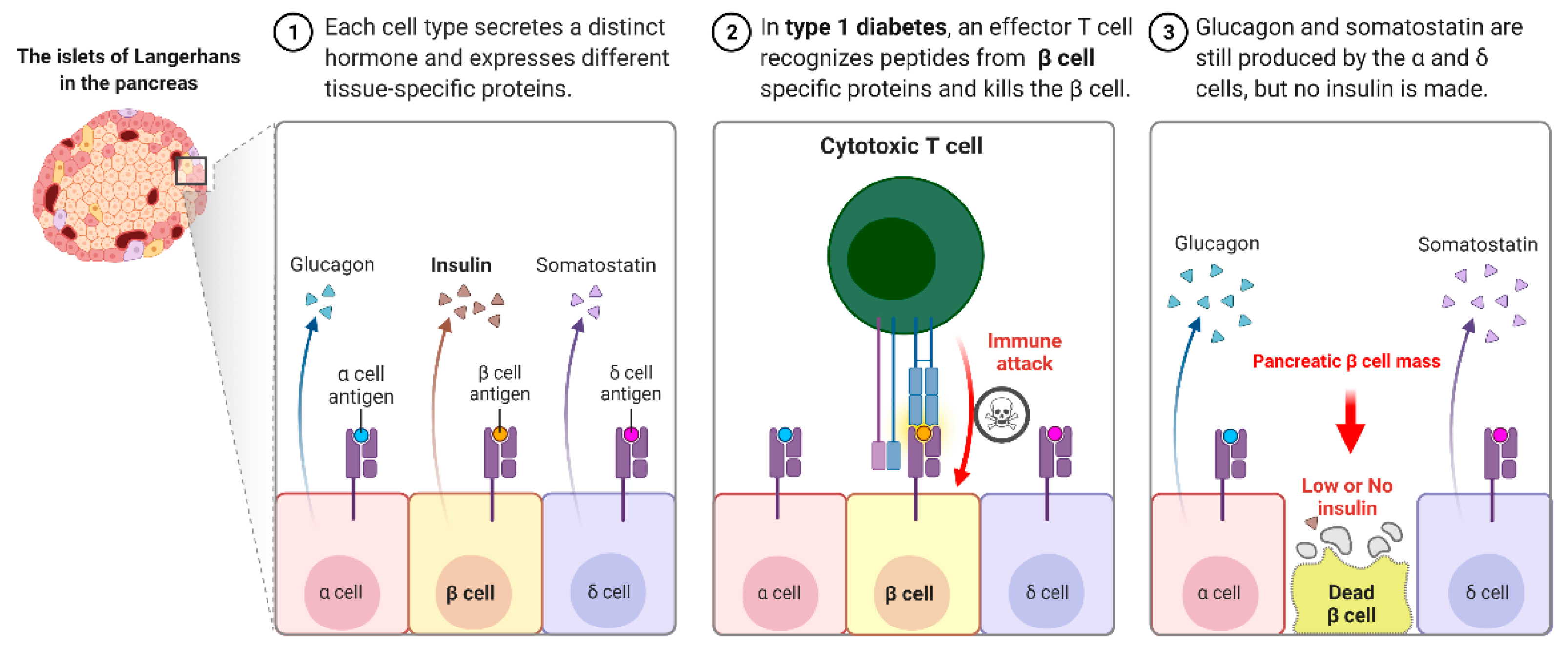{kind=link}
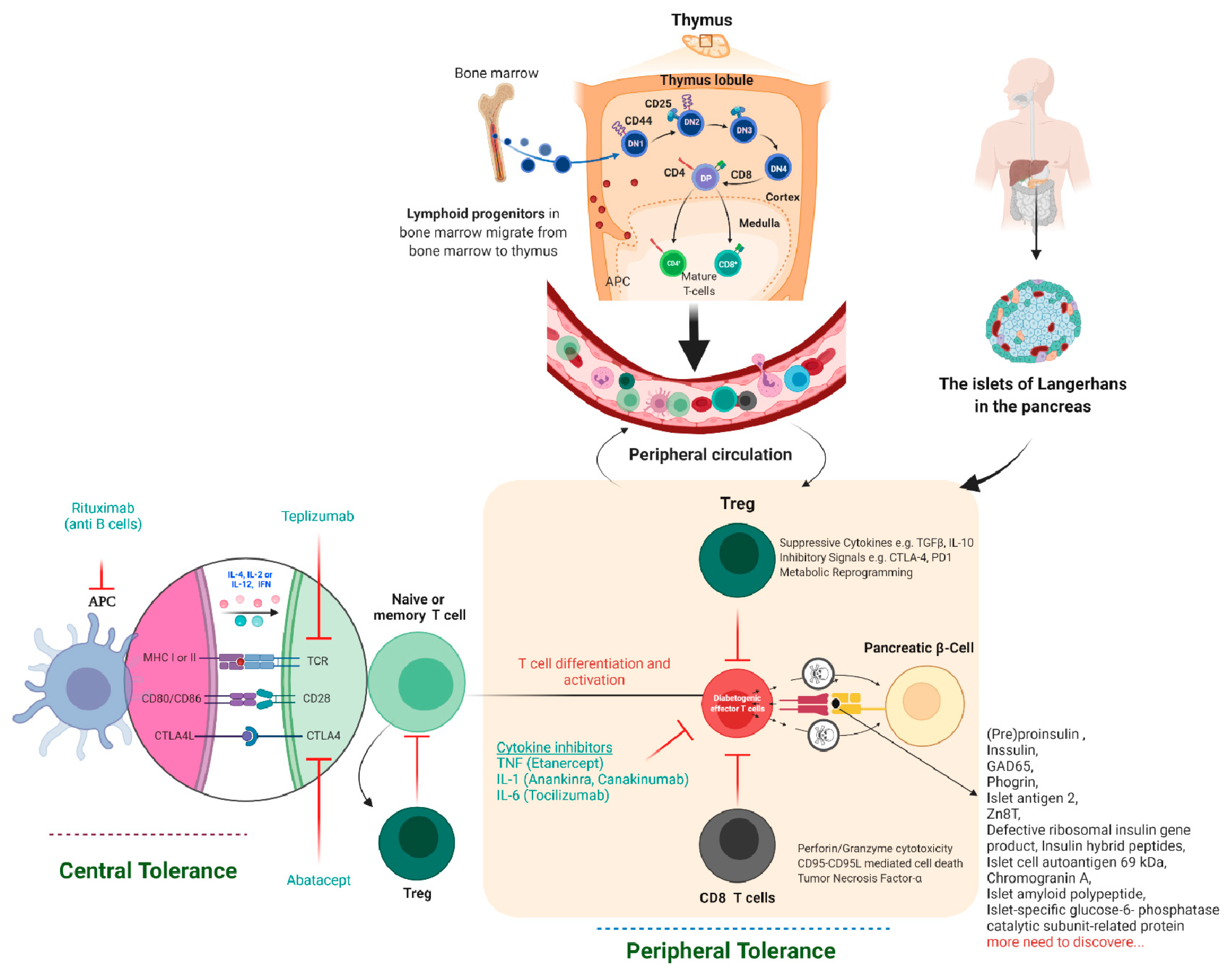{kind=link}
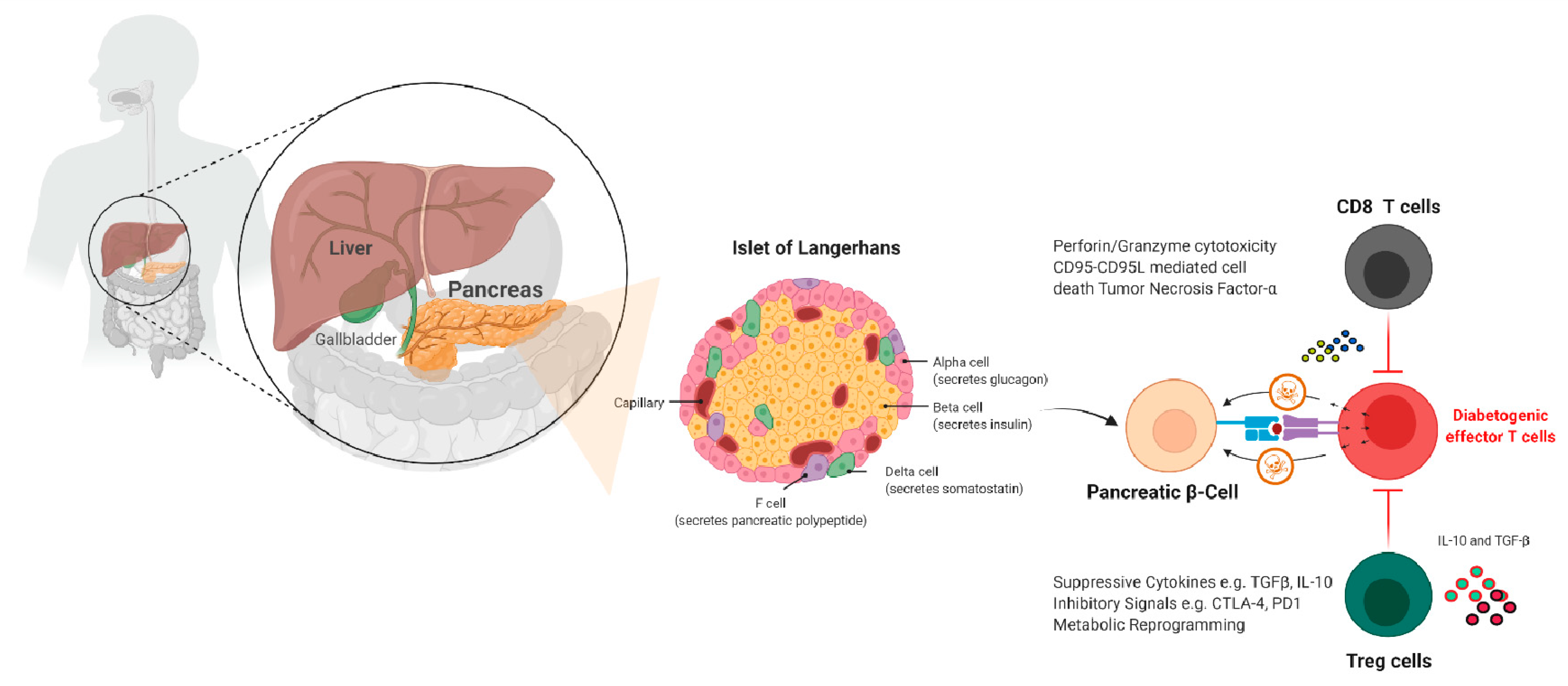{kind=link}
| Therapeutic Agents | Study/Authors and Intervention | Outcome | Citations |
|---|---|---|---|
T cell-based:
| DEFEND-1, 2 (Otelexizumab) | There was no EBV in the therapy group, but there was no statistically significant difference in 2-h MMTT AUC C-peptide at 12 months. | [28] |
| Protégé (Teplizumab) | At 1 year, there was no significant difference in HbA1c1 < 6.5 percent or insulin dose < 0.5 U/kg per day: At year 2, AUC C-peptide in the high dose group was considerably greater than in the placebo group. | [29,30] | |
| AbATE (Teplizumab) | The treatment group’s baseline adjusted AUC C-peptide reduced at year 2 was considerably lower. | [31] | |
| B cell-based: The monoclonal anti-CD20 antibody, which blocks the B cell function | Rituximab | HbA1c lowers as the rate of C peptide declines and insulin levels decrease. | [32,33] |
| Co-stimulation blockade | TrialNet CTLA4-Ig (abatacept); CTLA-4-IgG1 chimeric protein acts as a decoy receptor for CD80/86 and blocks CD28-CD80/86 induced co-stimulation of T-cells | Significantly higher stimulated C-peptide 2-h AUC in the treated group at the end of treatment and 1-year post-treatment | [34,35] |
| TIDAL (alafacept); Alafacept: chimeric protein (2 LFA-3 molecule-IgG1) binds to CD2 and blocks T-cell-stimulation | Significantly higher stimulated AUC C-peptide in the treatment group compared to placebo; insulin use lower in the treatment group | [36] | |
| Cytokine-based: IL-2 agonist | Aldesleukin; IL-2 maintains Treg population and function | A dose-dependent elevation of Treg cells in the treatment group compared to placebo | [37] |
| TNF antagonism | Etanercept | HbA1c decreases while endogenous insulin production increases. | [38] |
| IL-1 receptor blockade | Anakinra |
| [39,40] |
| IL-1beta antagonism | Canakinumab | There was no C peptide reaction | [39] |
| IL-1 receptor blockade IL-1beta antagonism | Anakinra/canakinumab | Immunomodulation/reverse relationship between inflammation and C peptide stimulation | [41] |
| Anti-IL-6 therapy | Tocilizumab in New-onset T1D (EXTEND) | Ongoing study | Clinical trial NCT02293837 |
| Antigen-based therapy: | Antigen-specific therapies may involve direct targeting of pathogenic T cells and/or boosting Tregs for bystander suppression | Tregs were shown to be more prevalent in those who got a larger dose of oral insulin (62.5 mg) | [42] |
| Treg-based: | Expansion of autologous Treg cells | A subset of adoptively transferred Treg is still in circulation (25% of peak) at year 1, with no significant adverse effects. C-peptide preservation in those receiving a lower dose | [20] |
| DC-based: | In T1D individuals who get their autologous DCs exhibited limited output. In this study, autologous DCs were given by infused via abdominal intradermal injections each 2 weeks apart | The autologous DC-based therapy was very well tolerated; no important differences were seen in glycemia | [37] |
| Combination therapy |
| C-peptide significantly increased at 30 months follow up; increased side effects | [43] |
| 32% were insulin-free at 4 years, maintenance of C-peptide, but with increased side effects | [44] | ||
| Mean AUC C-peptide at 12 months was significantly higher in the study group compared to the placebo group | Low-dose ATG + plus pegylated G-CSF [45] |
| T1D Autoantigens | Tissue Distribution | Source (NOD Mouse or T1D Patients) | Effector CD4 and/or CD8 T Cells | Citations |
|---|---|---|---|---|
| (Pre) proinsulin | β cells, thymus | Mouse and human | CD4 and CD8 | [77,78,79,80] |
| Insulin | Islet cells | Mouse and human | CD4 and CD8 | [78,81,82,83] |
| A defective ribosomal insulin gene product | Islet cells | Human | CD8 | [84] |
| Hybrid insulin peptides (HIPs) | Islet cells | Mouse and human | CD4 | [85,86,87,88] |
| Glutamic acid decarboxylase (GAD65) | Islet cells, adrenal gland, CNS, neurons, testis, ovary | Mouse and human | CD4 and CD8 | [89,90,91,92] |
| Zinc transporter 8 (ZnT8) | Pancreatic β cells | Mouse and human | CD4 and CD8 | [93,94,95,96,97,98] |
| Tyrosine phosphatase like autoantigen or insulinoma antigen-2 (IA-2; ICA512, PTPRN) | Islets | Human | CD4 and CD8 | [99,100,101] |
| IA-2β (Phogrin, PTPRN2) | Islets | Mouse and human | CD4 | [102,103,104,105] |
| Islet cell autoantigen of 69 kDa (ICA69) | Pancreas, heart, and brain | Human | CD4 | [106,107,108,109,110] |
| Chromogranin A | Neuroendocrine cells | Mouse and human | CD4 and CD8 | [111,112,113] |
| Islet amyloid polypeptide (ppIAPP) | Islets | Mouse and human | CD4 and CD8 | [114,115,116,117] |
| IGRP; islet-specific glucose-6-phosphatase catalytic subunit-related protein | Islets | Mouse and human | CD4 and CD8 | [80,118,119,120] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathod, S. Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes. Diabetology 2022, 3, 79-96. https://doi.org/10.3390/diabetology3010007
Rathod S. Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes. Diabetology. 2022; 3(1):79-96. https://doi.org/10.3390/diabetology3010007
Chicago/Turabian StyleRathod, Sanjay. 2022. "Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes" Diabetology 3, no. 1: 79-96. https://doi.org/10.3390/diabetology3010007
APA StyleRathod, S. (2022). Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes. Diabetology, 3(1), 79-96. https://doi.org/10.3390/diabetology3010007