Abstract
This work proposes a simple, fast and low-cost voltammetric method for the determination of trimethoprim at low concentrations in an analytical and real matrix (river water sample, bovine serum and synthetic urine). For this, a glassy carbon electrode was modified with Printex(6L) carbon and gold nanoparticles in a chitosan film crosslinked with epichlorohydrin. After that, the electrochemical measurement system contained a solution of phosphate buffer at pH 4.0 with commands for the square wave voltammetry technique. The results achieved showed a limit of detection equal to 12.4 nmol L−1 and a linear concentration range from 0.20 to 6.0 μmol L−1. The sensor selectivity was tested in the presence of various electroactive molecules, and the results showed that the detection of TMP in the presence of possible interferents was not masked. In addition, the applicability of the AuNPs–Printex(6L)–CTS:EPH/GCE sensor was also verified in synthetic samples of urine, bovine serum and river water through standard addition and recovery tests. Finally, the results of this analytical proposal portray a simple, fast and efficient method for the detection of TMP in different matrices.
1. Introduction
In recent years, the body of research related to the documentation of emerging contaminants in aqueous matrices has grown substantially [1]. This is due to the widespread use of different classes of pharmaceuticals, such as antibiotics [2]. Antibiotics are characterized as one of the most important categories of emerging contaminants due to their presence in different environmental compartments, making them a potential threat for the increase in bacterial resistance [3].
Thus, the arrival of this type of medication occurs through different routes in water bodies, but the greatest concern is related to the inappropriate disposal of medicines as well as to untreated sewage from industries, hospitals and swine farms that flow into rivers and lakes [2]. This attitude puts animals, food and water at risk of carrying and transmitting some form of disease in the long term [4]. Therefore, it is necessary to develop sensitive methods for detecting antibiotics in real environmental samples.
In this context, the synthetic antibiotic trimethoprim (TMP) stands out, which is marketed for the treatment of intestinal and urinary infections, chronic bronchitis, pneumonia and another respiratory disease [5]. It is used both in humans and in the veterinary clinic and acts by inhibiting the enzyme dihydrofolate reductase. In addition, TMP has antibacterial activity against many Gram-positive and Gram-negative bacteria, and is a drug commonly used as a synergist with other antibiotics aiming at greater effectiveness in the treatment of bacteria [6,7].
Considering its widespread use and the risks it poses to humans and the environment, it is necessary to develop increasingly sensitive, practical and low-cost analytical methods for the determination and/or monitoring of TMP in environmental and biological samples. In this sense, electrochemical sensors stand out for their chemical and physical characteristics, in addition to the possibility of modification, which in turn can further increase their sensitivity and/or selectivity [8,9,10].
Among the different types of nanomaterials used to manufacture electrochemical sensors for the detection of electroactive organic molecules, carbon nanomaterials are notable for their intrinsic properties, such as interaction with the analyte, altering the dielectric atmosphere due to the processes of oxidation/reduction on the surface of the carbonaceous material [11,12,13]. Carbon has structural polymorphism characteristics; excellent catalytic and electronic properties; and different morphologies and chemical nanoforms suitable for analytical detection studies, such as carbon black/Printex, graphene, graphene oxide, graphite, fullerenes, carbon nanotubes, diamond nanoparticles, carbon dots and nanofibers [14,15,16].
On the other hand, strategies to amplify the catalytic response of a carbon-based sensor are often detailed in full, but it is observed that the support of metallic nanoparticles in a carbonaceous structure provides a greater synergistic effect, contributing to the increase in the peak current when the metals are well incorporated into the carbon structure [17]. This phenomenon can be attributed not only to the properties related to the electrical conductivity, surface area and chemical stability of metallic nanoparticles, but also to the size and morphology of these metals [18]. Based on these characteristics, nanoparticles of transition metals such as Au, Ag, Pt, Pd and Ru are increasingly explored in this area of research due to their excellent properties [19,20].
Therefore, studies related to the detection of antibiotics with sensors of simple experimental architecture, capable of detecting low concentrations in analytical and real matrices (environmental and biological), are considered important. In this perspective, other research groups also consider a study of this magnitude necessary, such as the study recently carried out by Martins et al. [21], who manufactured a low-cost electrochemical sensor with reduced graphene nanoribbons on disposable platforms to detect sulfamethoxazole and trimethoprim in the sample results, achieving good analytical performance. In this same context, Yue et al. [5] proposed a system with vitreous carbon modified with graphene nanorods for the detection of trimethoprim in tap water, lake, urine and serum, achieving promising results. On the other hand, Sgobbi et al. [6] produced a miniaturized system in screen-printed electrodes for the detection of trimethoprim, using multi-walled carbon nanotubes decorated with Prussian blue nanocubes, obtaining promising results.
In this work, an electrochemical device based on Printex(6L) carbon modified with gold nanoparticles (AuNPs) is proposed for the determination of TMP in river water samples with high sensitivity and reproducibility. Therefore, it can serve as a strategy for monitoring and/or quantifying TMP in environmental and biological samples.
2. Materials and Methods
2.1. Reagents and Solutions
All reagents necessary to carry out this work were of analytical grade and purity greater than 99.0%. All aqueous solutions were prepared with deionized water with resistivity ≥ 18.0 MΩ cm obtained from a Milli-Q, Direct 8 system (Millipore, Burlington, VT, USA). The following items: C14H18N4O3 (TMP), chitosan (CTS), epichlorohydrin (EPH), urea, bovine serum, phosphate salts (KH2PO4; K2HPO4; Na3PO4) and HAuCl4.3H2O (gold (III) acid chloride trihydrate) were purchased from Sigma Aldrich (St. Louis, MO, USA). Na3C6H507 (sodium citrate) was purchased from Synth. Printex(6L) carbon was purchased from Degussa (Essen, North Rhine-Westphalia, Germany).
2.2. Apparatus
The electrochemical characterization of the films proposed in this work was obtained by an Autolab PGSTAT-30 potentiostat/galvanostat (Utrecht, The Netherlands) controlled by NOVA 2.1 software. The electrochemical measurement system contained a cell with a volumetric capacity of 10 mL with input for three electrodes: working electrode (glassy carbon modified with Printex(6L) carbon and gold nanoparticles, counter electrode (platinum plate) and reference electrode (Ag/AgCl/KCl (3.0 mol L−1)).
The morphological characterization of the materials that comprise the sensor was also performed by the scanning electron microscopy technique (FEI Magellan 400 L microscope–TermoFischer, Former FEI, Waltham, MA, USA) and transmission electron microscopy (FEI TECNAI F20 HRTEM, FEI Company, Eindhoven, The Netherlands).
2.3. Au Nanoparticles Synthesis
The synthesis of gold nanoparticles (AuNPs) was achieved as already reported in full [22]. An amount of 16.08 g of anhydrous sodium citrate (equivalent to 0.6 mol L−1) was weighed and dissolved in 100 mL of deionized water under heating to 100 °C. After the solution reached boiling point, approximately 40 mg of the gold salt was added, conditioned to magnetic stirring for 4 min. Then, the reaction mixture was transferred to an ice bath until the temperature equilibrated to 25 °C. At the end of the procedure, it was possible to visualize the color change from yellow to red, as is characteristic of the formation of AuNPs.
2.4. Preparation of the Modified Glassy Carbon Electrodes
The surfaces of the glassy carbon electrodes (GCE) were polished with 0.5 μm alumina microparticles on a polishing cloth. Then, the electrodes were submitted to ultrasonic cleaning steps followed by water, isopropyl alcohol and water, successively, for one minute each.
In parallel, two suspensions were prepared to modify the GCE surface. The first suspension contained 2.0 mg of Printex(6L) carbon, 200 μL of a 0.10% (v/v) CTS solution, 200 μL of a 0.10% (v/v) EPH solution and 1600 μL of deionized water. In the second suspension was 2.0 mg of Printex(6L) carbon, 200 μL of a 0.10% (v/v) CTS solution, 200 μL of a 0.10% (v/v) EPH solution, 1000 μL of AuNPs solution and 600 μL of deionized water. Both suspensions were left under ultrasonic agitation for 40 min to obtain a homogeneous dispersion. Finally, an 8 μL aliquot of the dispersions was dripped onto the surface of the GCE (3.0 mm diameter) and dried at room temperature for approximately 2 h to obtain the electrodes: Printex(6L)–CTS:EPH/GCE and AuNPs–Printex(6L)–CTS:EPH/GCE.
2.5. Samples Preparation
Synthetic urine was prepared with a mixture of salts and ultrapure water in the respective concentrations: 0.18 mmol L−1, 0.18 mmol L−1, 0.10 mmol L−1, 0.15 mmol L−1, 0.10 mmol L−1 and 0.20 mol L−1 of urea, ammonium chloride, calcium chloride, monobasic potassium phosphate, sodium chloride and potassium chloride, respectively [23]. Serum samples were used as purchased, without any type of pre-treatment.
Finally, water samples were collected from the Monjolinho River located in the city of São Carlos, in São Paulo, Brazil. Water samples were collected at 3 different points, filtered and stored in a single bottle in a refrigerator at 3 °C.
All samples were doped and evaluated for recovery rate using two standard TMP concentration levels (A = 0.40 μmol L−1 and B = 3.0 μmol L−1). For this, aliquots of 300 μL of each sample were used in an electrochemical cell containing 10 mL of supporting electrolyte.
3. Results
Initially, a study was carried out to evaluate the composition/proportion of the nanomaterials (Printex(6L) and AuNPs) used in the modification of the GCE. For this, in all cases the proportions of CTS (0.10% (v/v)) and EPH (0.10% (v/v)) were kept constant (0.10 mL/each). Thus, we first evaluated the proportion of Printex(6L) carbon in the ratio (nanomaterial:solution (mg/mL)) of 0.50:1.0, 1.0:1.0, 1.5:1.0 and 2.0:1.0 (mg/mL), the solution being composed of CTS, EPH and water. The highest peak current magnitude and film stability signals were obtained using a dispersion ratio of 1.0:1.0 (mg/mL). Subsequently, with the proportion of Printex(6L) carbon fixed (1.0 mg/mL), the amount of AuNPs used was evaluated. Thus, dispersions in the proportions (AuNPs:solution (mL/mL)) of 0.25:0.75; 0.50:0.50 and 0.75:0.25 (mL/mL) were evaluated. Therefore, the highest peak current magnitude (n = 3) for TMP was obtained using the proportion of 1.0 mg of Printex(6L) carbon, 0.50 mL of AuNPs, 0.10 mL of CTS, 0.10 mL of EPH and 0.30 mL of deionized water. Thus, this proportion was used for further studies.
3.1. Morphological Characterization
The morphological characterization of Printex(6L) carbon, Printex(6L) carbon modified with AuNPs and AuNPs was obtained by scanning electron microscopy (SEM) or transmission electron microscopy (TEM). It can be seen in Figure 1A that the Printex(6L) carbon has a homogeneous distribution in the shape of numerous spherical nanoparticles. This morphological profile has been previously reported in the literature, and it is justified that the shape of the material contributes to adding greater porosity to the electrode surface, which provides a better detection response in relation to the smoother surface of the GCE.
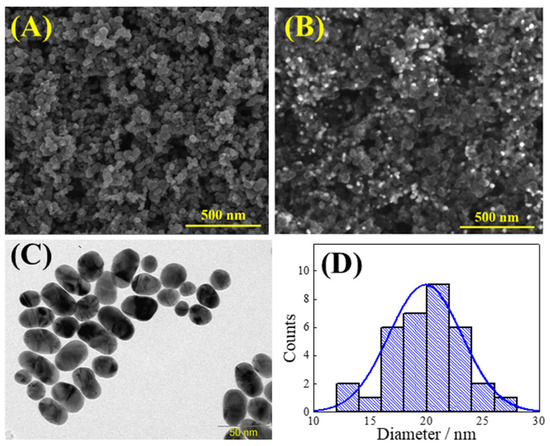
Figure 1.
SEM image of (A) Printex(6L) carbon and (B) AuNPs–Printex(6L). TEM images for (C) AuNPs and (D) histogram of AuNPs diameters.
Then, when the AuNPs were added together with the Printex(6L) carbon (Figure 1B), it was possible to observe, in BSE mode, small bright spots referring to the AuNPs that differ from the composition of the Printex(6L) carbon. Considering that gold nanoparticles can be geometrically confused with Printex(6L) carbon nanoparticles because they are spherical and very small, they were also evaluated by TEM (Figure 1C), whose average sphere size is 20 nm, as can be seen observed in the graph of Figure 1D.
3.2. Determination of Electroactive Area
The electroactive areas of the bare GCE, Printex(6L)–CTS:EPH/GCE and AuNPs–Printex(6L)–CTS:EPH/GCE were estimated by cyclic voltammetry in 0.10 mol L−1 of KCl solution in the presence of a 2.0 mmol L−1 [Fe(CN)6]3− (see Figure S1 in the Supplementary Materials), according to the Randles–Sevcik equation (1) [24]:
where Ip is the peak current (A); n is the number of electrons transferred; A is the electroactive area (cm2); D is the diffusion coefficient of [Fe(CN)6]3− in the 0.10 mol L−1 KCl solution (7.6 × 10−6 cm2 s−1); C the [Fe(CN)6]3− concentration (mol cm−3); and v is the potential scan rate (V s−1).
Ip = ±(2.69 × 105) n3/2 A D1/2 C v1/2
The averages of the obtained slopes of the Ipa and Ipc vs. v1/2 plots for the [Fe (CN)6]3− oxidation process were 5.11 × 10−5 A V−1/2 s1/2 for the GCE; 9.58 × 10−5 A V−1/2 s1/2 for the Printex(6L)–CTS:EPH/GCE; and 1.21 × 10−4 A V−1/2 s1/2 for the AuNPs–Printex(6L)–CTS:EPH/GCE. Then, the estimated electroactive areas were 0.045, 0.084 and 0.11 cm2, for GCE, Printex(6L)–CTS:EPH/GCE and AuNPs–Printex(6L)–CTS:EPH/GCE, respectively. It can be observed from the obtained data that the electrochemical response for the redox probe ([Fe(CN)6]3−) was clearly affected by modification on the surface of the electrode, where the presence of AuNPs and Printex(6L) on the electrode surface (AuNPs–Printex(6L)–CTS:EPH/GCE) resulted in an increase in the electroactive area by a factor of 2.4 times compared to the bare GCE, besides presenting a more reversible electrochemical behavior compared to the bare GCE and Printex(6L)–CTS:EPH/GCE.
3.3. Electrochemical Behavior of TMP
The electrochemical profile of the TMP molecule using the electrodes GCE, Printex(6L)–CTS:EPH/GCE and AuNPs–Printex(6L)–CTS:EPH/GCE was evaluated by cyclic voltammetry (n = 3), in which a 0.20 mol L−1 of phosphate buffer solution of pH 6.0 was used, using a potential range of 0.50–1.3 V with a sweep speed of 50 mV s−1, whose concentration of the analyte was 0.20 mmol L−1 (Figure 2).
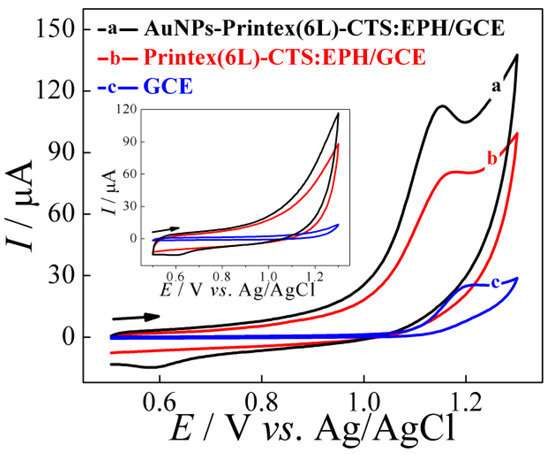
Figure 2.
Cyclic voltammograms obtained from the application of the following electrodes: a, AuNPs–Printex(6L)–CTS:EPH/GCE; b, Printex(6L)–CTS:EPH/GCE; and c, bare GCE in the presence (0.20 mmol L−1) and absence (insert) of TMP in 0.2 mol L−1 of phosphate buffer (pH 6.0). v = 50 mV s−1.
In the voltammetric profile for TMP, it is possible to observe that only a well-defined oxidation peak of the molecule occurs close to the potential of 1.2 V, which indicates an irreversible process without the presence of any reduction peak of the molecule. Furthermore, when only GCE was used in the analytical detection process, there was a relatively low peak current magnitude response (Ip = 24 ± 1 μA), which may indicate that GCE alone would not be ideal for achieving lower TMP concentration values than would be expected in the real world, such as samples from environmental or biological matrices.
In this context, when modifying the GCE with Printex(6L) carbon, an increase in the peak current magnitude of the analyte oxidation process can be observed (Ip = 66 ± 2 μA), as can be seen in Figure 2. This Printex(6L) carbon modification on the GCE surface contributes to a 2.8-fold increase in peak current magnitude for the TMP when compared to the bare GCE. Finally, when AuNPs were incorporated into the film (AuNPs–Printex(6L)–CTS:EPH), an even greater increase in peak current magnitude was observed (Ip = 96 ± 4 μA). Thus, the elevation of the monitored response increases 1.4 times higher than the Printex(6L)–CTS:EPH/GCE and 4.0 times higher than the bare GCE. This phenomenon is due to the synergistic effects between the carbonaceous (Printex(6L)) and metallic (AuNPs) structure, since in addition to raising the values of the measured property (current), it also contributes to the displacement of the oxidation potential. This contribution is positive because the less positive the oxidation potential, the better the response due to the possible presence of interfering reactions that occur at increasingly positive potentials, such as the oxidation reaction of water molecules. For the bare electrode, the Ep was observed close to 1.20 V. From the modifications carried out, it was possible to identify that for Printex(6L) there was a decrease to 1.16 V, and for the nanocomposite (AuNPs–Printex(6L)) the Ep was close to 1.14 V—that is, a decrease of approximately 0.04 V and 0.06 V, respectively.
3.4. Optimization of the Sensor
Initially, the influence of pH for 5.0 μmol L−1 of TMP was investigated by SWV, using only the AuNPs–Printex(6L)–CTS:EPH/GCE sensor in 0.20 mol L−1 of phosphate buffer solutions in the pH range of 3.0 to 7.0. As can be seen in Figure 3, the optimal pH was found considering the peak current magnitude was pH 4.0. Therefore, pH 4.0 was selected for the following studies.
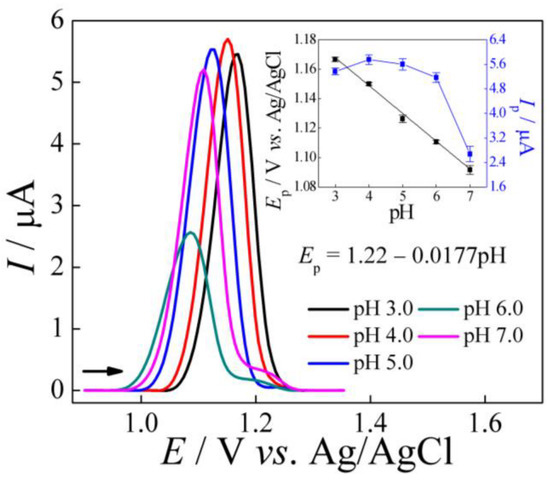
Figure 3.
SWV recorded for 5.0 μmol L−1 of TMP in 0.20 mol L−1 of phosphate buffer solution, in a pH ranging from 3.0 to 7.0, using AuNPs–Printex(6L)–CTS:EPH/GCE. Parameters: f = 20 Hz, a = 40 mV, ΔEs = 5 mV and tacc = 30 s. Inserted: graphics of Ep vs. pH and Ip vs. pH.
Furthermore, as can be seen in the inserted Figure 3 (Ep. vs. pH), with increasing pH there was a shift to less positive potentials, as expected by the direct effect of pH on the peak potential. In addition, the Ep. vs. pH showed a linear relationship, with a slope of 17.7 mV per pH unit and a value close to the theoretical value of 14.8 mV/pH at 25 °C for an oxidation process that involves four electrons [25]. Therefore, based on the literature [26,27], the possible electro-oxidation of TMP is shown in Scheme 1. However, to validate this process, other studies would need to be carried out.
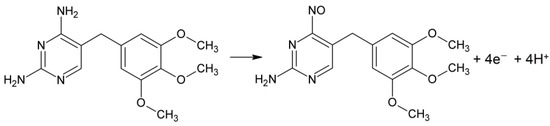
Scheme 1.
Proposed reaction for the oxidation of TMP [26].
Next, the effect of other supporting electrolytes (acetate buffer solution) with pH 4.0 was tested. As can be seen in Figure S2 (Supplementary Materials), the phosphate buffer (Ip = 5.69 μA) showed a response 2.35 times greater than the acetate buffer (Ip = 2.42 μA). Therefore, all the following measurements were carried out in phosphate buffer pH 4.0, as this was found to be an optimal condition for TMP oxidation.
3.5. Potential Scan Rate Effect
The effect of the scan rate on the TMP voltammetric response was studied using the AuNPs–Printex(6L)–CTS:EPH/GCE sensor and the cyclic voltammetry technique.
As shown in Figure 4A, with an increasing potential scan rate (10–200 mV s−1), a continuous increase in peak current magnitude was verified. From this, as can be seen in Figure 4B,C, the results showed good linearity of the plots of peak current vs. potential sweep speed (Ip vs. v) (r = 0.992) and peak current vs. square root of potential sweep speed (Ip vs. v1/2) (r = 0.997), indicating that the redox process could be governed by both adsorption and/or diffusion, respectively. Thus, in order to determine which of these processes dominated the electron transfer of the respective analyte under study, we constructed the graphic of the decimal logarithm of peak current (log Ip) vs. the decimal logarithm of the potential sweep speed (log v). Thus, as can be seen in the graph (log Ip vs. log v) in Figure 4C, a slope value equal to 0.65 was obtained. This value is between the theoretical values of 0.50, typical for diffusion-controlled mass transport, and 1.0, typically reported for redox processes governed by adsorption. Therefore, the redox process could have a mixed behavior, governed by both adsorption and diffusion.
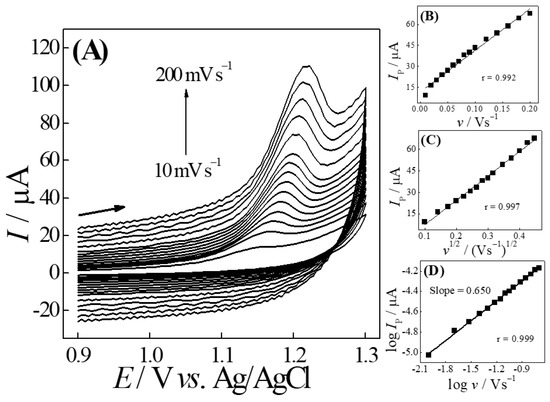
Figure 4.
(A) Cyclic voltammograms for different scan rates (10–200 mV s−1) using the AuNPs–Printex(6L)–CTS:EPH/GCE sensor, for 0.20 mmol L−1 of TMP in 0.20 mol L−1 of phosphate buffer solution (pH 4.0). Graphics of (B) Ip vs. v and (C) Ip vs. v1/2; (D) log Ip vs. log v.
3.6. Optimization of SWV Parameters
The SWV technique was selected because it presented the best peak and magnitude definitions of the analytical signal when compared to the DPV. Thus, the experimental parameters that affect the electrochemical response of SWV were optimized in 0.20 mol L−1 of phosphate buffer solution (pH 4.0). The optimal values obtained for these parameters (amplitude (a), frequency (f) and scan increment (ΔEs)) were a = 60 mV, f = 15 Hz and ΔEs = 6 mV (Table S1).
However, in view of the previous study (effect of potential scan rate), the use of the pre-concentration potential from 0 to 1.2 V (for 30 s under stirring) and the effect of the pre-concentration time were evaluated (stirring only) in the time interval of 10 to 60 s (Table S1). Thus, when applying the pre-concentration potential, no increase in peak current magnitude was observed. Therefore, no pre-concentration potential was applied. However, when the stirring time (tacc) was varied, a constant increase in the magnitude of the peak current was observed in the time interval from 10 to 40 s and remained practically constant for longer times. Therefore, the accumulation time selected for the rest of the studies was 40 s.
Furthermore, the electrode surface did not undergo any cleaning process between measurements, as no saturation process or poisoning/blocking of the electrode surface caused by the analyte between measurements was observed. This may be related to the low concentration of the analyte, which in turn should not occupy all active sites on the electrode. In addition, cleaning/regeneration of the electrode surface may also occur due to the natural desorption of the analyte on the electrode surface.
3.7. Voltammetric Determination of Trimethoprim
After optimizing the TMP detection parameters by SWV, the AuNPs–Printex(6L)–CTS:EPH/GCE sensor was used to explore the electrochemical response as a function of the drug concentration in question. In Figure 5, we can see that the profile of the calibration curve is characteristic of a linear analytical response (r = 0.999) between the measured property (peak current magnitude) and the studied variable (concentration) in the range of 0.20–6.0 μmol L−1, with the following equation of the analytical linear regression curve:
ΔIp (μA) = −(0.11 ± 0.01) + (1.88 ± 0.03)[TMP] (μmol L−1)
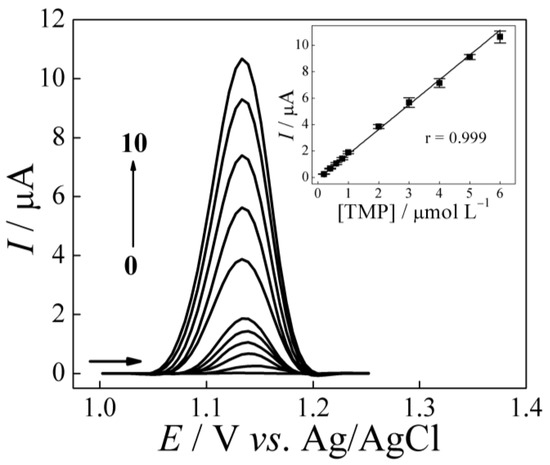
Figure 5.
SW voltammograms obtained using AuNPs–Printex(6L)–CTS:EPH/GCE for TMP at concentrations of (0–10) 0; 0.20; 0.40; 0.60; 0.80; 1.0; 2.0; 3.0; 4.0; 5.0; and 6.0 μmol L−1 in 0.20 mol L−1 of phosphate buffer (pH 4.0). SWV conditions: f = 15 Hz, a = 60 mV, ΔEs = 6 mV and tacc = 40 s.
In this perspective, the limits of detection (LOD) and quantification (LOQ) were determined from the standard deviation of the blank signal and the slope of the analytical curve, according to the criteria: LOD = “3σ”/”S” and LOQ = “10σ”/”S”, where “σ” is the standard deviation of ten measurements of the blank signal (supporting electrolyte only) and “S” is the slope of the analytical curve. Thus, the values obtained were 12.4 nmol L−1 and 41.5 nmol L−1 for LOD and LOQ, respectively.
The analytical parameters of the proposed method using the AuNPs–Printex(6L)–CTS:EPH/GCE sensor were compared with other works reported in the literature [5,6,7,21,25,28,29]. As can be seen in Table 1, the developed sensor led to a good linear concentration range and LOD for TMP, being similar to, or better than, those reported by other authors. Among the main advantages of the GCE modified with AuNPs–Printex(6L)–CTS:EPH are the high stability and selectivity, as shown below.

Table 1.
Analytical parameters comparison obtained for the AuNPs–Printex(6L)–CTS:EPH/GCE sensor and other reported sensors for the determination of TMP.
3.8. Study of Repeatability and Interference Effects
The repeatability study for the AuNPs–Printex(6L)–CTS:EPH/GCE sensor was assessed through an intra-day (same day) analysis using 3.0 μmol L−1 of TMP in 0.20 mol L−1 of phosphate buffer solution (pH 4.0). As shown in Figure S3 (Supplementary Materials), the analysis was performed through a sequence of electrochemical measurements (n = 20), where the relative standard deviation (RSD) obtained was 2.7%. From this result, it is possible to observe the good precision of the proposed method using the AuNPs–Printex(6L)–CTS:EPH/GCE sensor for the detection of TMP.
In addition, some substances commonly found in environmental and biological fluid samples (possible interferents) were introduced into the analysis system together with the target drug to identify their influence on the oxidation profile of the TMP molecule, such as humic acid, heavy metals (Pb2+ and Cd2+), caffeine, ascorbic acid, dopamine, uric acid and urea in a 1:10 ratio (analyte:possible interferent) (Table S2). Based on the results obtained by square wave voltammetry, it was observed that these molecules/compounds did not influence the determination of the investigated molecule, showing that the sensor has good selectivity for TMP detection in the presence of these concomitants.
3.9. Applications in Environmental and Biological Samples
Finally, the AuNPs–Printex(6L)–CTS:EPH/GCE sensor was applied for the quantification of TMP in three different samples: one environmental sample (river water), and two biological samples (serum and synthetic urine). The samples were enriched with two known concentrations of TMP (A = 0.40 and B = 3.0 μmol L−1) and analyzed in triplicate (n = 3) (Figure S4). As can be seen in Table 2, the application of the proposed method led to recovery percentages ranging from 93 to 105% in all cases. Thus, it can be concluded that the proposed procedure using the AuNPs–Printex(6L)–CTS:EPH/GCE sensor had no matrix effect, which suggests the great potential of the proposed method for the determination of TMP in serum, urine and river water samples.
Table 2.
Results obtained from the analysis of river water, serum and urine samples.
4. Conclusions
Due to the electrochemical properties presented by the AuNPs–Printex(6L)–CTS:EPH/GCE sensor, it is possible to consider that the GCE modified with Printex(6L) carbon and AuNPs obtained significant electrochemical behavior to oxidize the trimethoprim molecule and be applied in environmental and biological samples using the SWV method. The good results obtained with the sensor may be related to the synergistic effect obtained between the combination of different materials (Printex(6L) and AuNPs) and its high stability, as verified in the repeatability study, thus demonstrating that the proposed method for the determination of TMP in river water, serum and urine samples is highly reliable and advantageous for this type of analysis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/analytica4020013/s1.
Author Contributions
Conceptualization: M.H.A.F., A.M.S., A.W. and F.C.M.; methodology: M.H.A.F. and A.M.S.; validation: A.M.S. and A.W.; formal analysis: M.H.A.F.; investigation: M.H.A.F. and A.M.S.; resources: F.C.M.; data curation/analysis: M.H.A.F. and A.M.S.; writing—original draft preparation: M.H.A.F., A.M.S. and A.W.; writing—review and editing: F.C.M.; visualization: M.H.A.F., A.M.S. and A.W.; supervision: F.C.M.; project administration: F.C.M. and R.S.R.; funding acquisition: F.C.M. and R.S.R. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge the support by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brazil (CAPES)—Finance Code 001 and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) process number 2017/10118-0 and 2022/05454-9.
Data Availability Statement
The data are contained within the article and Supplementary Materials.
Acknowledgments
The authors gratefully acknowledge the financial support granted by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brazil (CAPES), and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP).
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Wang, Q.; Xue, Q.; Chen, T.; Li, J.; Liu, Y.; Shan, X.; Liu, F.; Jia, J. Recent advances in electrochemical sensors for antibiotics and their applications. Chin. Chem. Lett. 2021, 32, 609–619. [Google Scholar] [CrossRef]
- Yang, Y.; Song, W.; Lin, H.; Wang, W.; Du, L.; Xing, W. Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ. Int. 2018, 116, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Huerta, B.; Marti, E.; Gros, M.; López, P.; Pompêo, M.; Armengol, J.; Barceló, D.; Balcázar, J.L.; Rodríguez-Mozaz, S.; Marcé, R. Exploring the links between antibiotic occurrence, antibiotic resistance, and bacterial communities in water supply reservoirs. Sci. Total Environ. 2013, 456–457, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Lavilla Lerma, L.; Benomar, N.; Casado Muñoz, M.d.C.; Gálvez, A.; Abriouel, H. Antibiotic multiresistance analysis of mesophilic and psychrotrophic Pseudomonas spp. isolated from goat and lamb slaughterhouse surfaces throughout the meat production process. App. Environ. Microbial. 2014, 80, 6792–6806. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Li, Z.; Zhao, S. A new electrochemical sensor for simultaneous detection of sulfamethoxazole and trimethoprim antibiotics based on graphene and ZnO nanorods modified glassy carbon electrode. Microchem. J. 2020, 159, 105440. [Google Scholar] [CrossRef]
- Sgobbi, L.F.; Razzino, C.A.; Machado, S.A.S. A disposable electrochemical sensor for simultaneous detection of sulfamethoxazole and trimethoprim antibiotics in urine based on multiwalled nanotubes decorated with Prussian blue nanocubes modified screen-printed electrode. Electrochim. Acta 2016, 191, 1010–1017. [Google Scholar] [CrossRef]
- Guaraldo, T.T.; Goulart, L.A.; Moraes, F.C.; Lanza, M.R.V. Carbon black nanospheres modified with Cu (II)-phthalocyanine for electrochemical determination of Trimethoprim antibiotic. App. Surf. Sci. 2019, 470, 555–564. [Google Scholar] [CrossRef]
- Santos, A.M.; Wong, A.; Feitosa, M.H.A.; Silva, L.P.; Fatibello-Filho, O.; Moraes, F.C. using carbon paste electrode modified with graphene and nanodiamond for the determination of nimesulide in biologic and environmental samples. Electroanalysis 2022, 34, 1441–1449. [Google Scholar] [CrossRef]
- Wong, A.; Santos, A.M.; Proença, C.A.; Baldo, T.A.; Feitosa, M.H.A.; Moraes, F.C.; Sotomayor, M.D.P.T. voltammetric determination of 3-methylmorphine using glassy carbon electrode modified with rGO and bismuth film. Biosensors 2022, 12, 860. [Google Scholar] [CrossRef]
- Santos, A.M.; Wong, A.; Fatibello-Filho, O.; Moraes, F.C. Amperometric biosensor based on laccase enzyme, gold nanoparticles, and glutaraldehyde for the determination of dopamine in biological and environmental samples. C 2022, 8, 40. [Google Scholar] [CrossRef]
- Wang, Z.; Dai, Z. Carbon nanomaterial-based electrochemical biosensors: An overview. Nanoscale 2015, 7, 6420–6431. [Google Scholar] [CrossRef]
- Baptista, F.R.; Belhout, S.A.; Giordani, S.; Quinn, S.J. Recent developments in carbon nanomaterial sensors. Chem. Soc. Rev. 2015, 44, 4433–4453. [Google Scholar] [CrossRef]
- Silva, R.M.S.; Santos, A.M.; Wong, A.; Fatibello-Filho, O.; Moraes, F.C.; Farias, M.A.S. Determination of ofloxacin in the presence of dopamine, paracetamol, and caffeine using a glassy carbon electrode based on carbon nanomaterials and gold nanoparticles. Anal. Methods 2022, 14, 3859–3866. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Dhanjai; Jain, R.; Zhao, H.; Karolia, P.; Jadon, N. Voltammetric sensing based on the use of advanced carbonaceous nanomaterials: A review. Microchim. Acta 2018, 185, 89. [Google Scholar] [CrossRef]
- Yin, P.T.; Shah, S.; Chhowalla, M.; Lee, K.-B. Design, synthesis, and characterization of graphene–nanoparticle hybrid materials for bioapplications. Chem. Rev. 2015, 115, 2483–2531. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chen, X.; Ren, T.; Zhang, P.; Yang, D. Carbon nanotube based biosensors. Sens. Actuators B Chem. 2015, 207, 690–715. [Google Scholar] [CrossRef]
- Wu, B.; Kuang, Y.; Zhang, X.; Chen, J. Noble metal nanoparticles/carbon nanotubes nanohybrids: Synthesis and applications. Nano Today 2011, 6, 75–90. [Google Scholar] [CrossRef]
- Moreno-Mañas, M.; Pleixats, R. Formation of Carbon−Carbon Bonds under Catalysis by Transition-Metal Nanoparticles. Acc. Chem. Res. 2003, 36, 638–643. [Google Scholar] [CrossRef]
- Jiang, H.; Zhu, L.; Moon, K.-s.; Wong, C.P. The preparation of stable metal nanoparticles on carbon nanotubes whose surfaces were modified during production. Carbon 2007, 45, 655–661. [Google Scholar] [CrossRef]
- Lee, K.Y.; Kim, M.; Lee, Y.W.; Lee, J.-J.; Han, S.W. Fabrication of metal nanoparticles–carbon nanotubes composite materials in solution. Chem. Phys. Lett. 2007, 440, 249–252. [Google Scholar] [CrossRef]
- Martins, T.S.; Bott-Neto, J.L.; Oliveira Jr, O.N.; Machado, S.A.S. Paper-based electrochemical sensors with reduced graphene nanoribbons for simultaneous detection of sulfamethoxazole and trimethoprim in water samples. J. Electroanal. Chem. 2021, 882, 114985. [Google Scholar] [CrossRef]
- Ojea-Jiménez, I.; Romero, F.M.; Bastús, N.G.; Puntes, V. Small Gold Nanoparticles Synthesized with Sodium Citrate and Heavy Water: Insights into the Reaction Mechanism. J. Phys. Chem. C 2010, 114, 1800–1804. [Google Scholar] [CrossRef]
- Laube, N.; Mohr, B.; Hesse, A. Laser-probe-based investigation of the evolution of particle size distributions of calcium oxalate particles formed in artificial urines. J. Cryst. Growth 2001, 233, 367–374. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R.; White, H.S. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Cesarino, I.; Cesarino, V.; Lanza, M.R.V. Carbon nanotubes modified with antimony nanoparticles in a paraffin composite electrode: Simultaneous determination of sulfamethoxazole and trimethoprim. Sens. Actuators B Chem. 2013, 188, 1293–1299. [Google Scholar] [CrossRef]
- Rajith, L.; Kumar, K.G. Electroanalysis of trimethoprim on metalloporphyrin incorporated glassy carbon electrode. Drug Test. Anal. 2010, 2, 436–441. [Google Scholar] [CrossRef]
- Rajith, L.; Jissy, A.K.; Kumar, K.G.; Datta, A. Mechanistic study for the facile oxidation of trimethoprim on a manganese porphyrin incorporated glassy carbon electrode. J. Phys. Chem. C 2011, 115, 21858–21864. [Google Scholar] [CrossRef]
- da Silva, H.; Pacheco, J.G.; McS Magalhães, J.; Viswanathan, S.; Delerue-Matos, C. MIP-graphene-modified glassy carbon electrode for the determination of trimethoprim. Biosens. Bioelectron. 2014, 52, 56–61. [Google Scholar] [CrossRef]
- Carapuça, H.M.; Cabral, D.J.; Rocha, L.S. Adsorptive stripping voltammetry of trimethoprim: Mechanistic studies and application to the fast determination in pharmaceutical suspensions. J. Pharm. Biom. Anal. 2005, 38, 364–369. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).