
Liver and Vascular Involvement in Philadelphia-Negative Chronic Myeloproliferative Neoplasms—A Narrative Review


Abstract
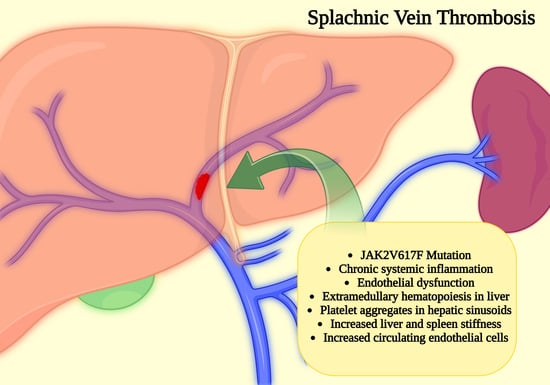
1. Introduction
2. Liver and Myelofibrosis
3. JAK/STAT Molecular Pathway
4. Liver and Spleen Stiffness
5. Extramedullary Hematopoiesis
6. Portal Hypertension
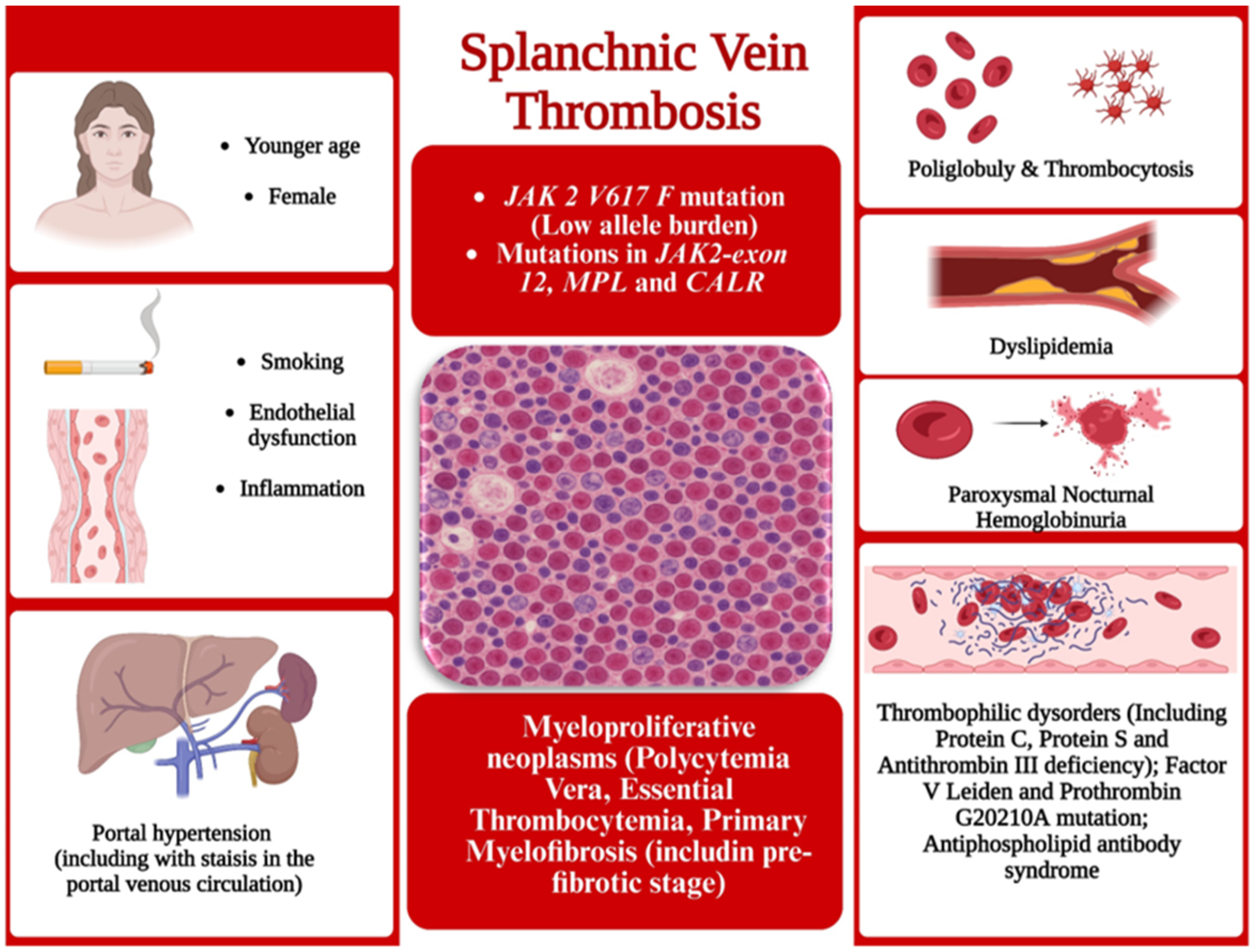
7. Splanchnic Vein Thrombosis
8. Upper Gastrointestinal Bleeding
9. Budd-Chiari Syndrome
10. Liver Diseases and Myeloproliferative Neoplasms
11. Therapeutic Interventions and Liver Consequences
11.1. Hydroxycarbamidum
11.2. Anagrelide
11.3. Interferon
11.4. Ruxolitinib
11.5. Fedratinib
11.6. Anticoagulant Treatment
12. Discussion
13. Conclusions
14. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McArthur, K.I.; Papayanis, P.N.; Nguyen, M.H.K.; Daneshbod, Y.; Akhtari, M. A Case Report on Hepatic Extramedullary Hematopoiesis as the Manifestation of Progression to Secondary Myelofibrosis in a Patient with Essential Thrombocytopenia. Hematol. Rep. 2022, 14, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Todor, S.B.; Ichim, C.; Boicean, A.; Mihaila, R.G. Cardiovascular Risk in Philadelphia-Negative Myeloproliferative Neoplasms: Mechanisms and Implications—A Narrative Review. Curr. Issues Mol. Biol. 2024, 46, 8407–8423. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ran, F.; Lin, J.; Zhang, J.; Ma, D. Genetic and Clinical Characteristics of Patients with Philadelphia-Negative Myeloproliferative Neoplasm Carrying Concurrent Mutations in JAK2V617F, CALR, and MPL. Technol. Cancer Res. Treat. 2023, 22, 15330338231154092. [Google Scholar] [CrossRef] [PubMed]
- Ammad Ud, D.M.; Liaqat, H.; Osama, M. MPL-Positive Essential Thrombocytosis Presenting as Budd-Chiari Syndrome in a Middle-Aged Woman with an Initially Normal Platelet Count. Eur. J. Case Rep. Intern. Med. 2021, 8, 003081. [Google Scholar] [CrossRef]
- Gou, P.; Zhang, W.; Giraudier, S. Insights into the Potential Mechanisms of JAK2V617F Somatic Mutation Contributing Distinct Phenotypes in Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2022, 23, 1013. [Google Scholar] [CrossRef]
- Hermouet, S. Mutations, inflammation and phenotype of myeloproliferative neoplasms. Front. Oncol. 2023, 13, 1196817. [Google Scholar] [CrossRef]
- Watanabe, H.; Fujishima, F.; Fukuhara, N.; Taniuchi, S.; Joh, K.; Sasano, H. Advanced extramedullary hematopoiesis with a marked increase in reticulin fibers and hemorrhage on various organs: The first autopsy case report. Med. Mol. Morphol. 2022, 55, 68–75. [Google Scholar] [CrossRef]
- Crawford, C.K.; Arshad, H.; Kawamoto, S.; Fishman, E.K. Periportal extramedullary hematopoiesis in a patient with primary myelofibrosis: A great mimicker of an infiltrating tumor in the liver. Radiol. Case Rep. 2025, 20, 2482–2486. [Google Scholar] [CrossRef]
- Yamazaki, K.; Suzuki, S.; Kawaguchi, T. Extramedullary haematopoiesis within hepatic sinusoids of a patient with primary myelofibrosis. Br. J. Haematol. 2022, 196, 1130. [Google Scholar] [CrossRef]
- Slot, S.; van de Donk, N.W.C.J.; Otten, R.H.J.; Boden, B.J.H.; Zijlstra, J.; Raijmakers, P.G.H.M.; Zweegman, S. The value of bone marrow, liver, and spleen imaging in diagnosis, prognostication, and follow-up monitoring of myeloproliferative neoplasms: A systematic review. Cancer Imaging 2021, 21, 36. [Google Scholar] [CrossRef]
- Pescia, C.; Lopez, G.; Cattaneo, D.; Bucelli, C.; Gianelli, U.; Iurlo, A. The molecular landscape of myeloproliferative neoplasms associated with splanchnic vein thrombosis: Current perspective. Leuk. Res. 2024, 136, 107420. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Essential thrombocythemia: 2024 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2024, 99, 697–718. [Google Scholar] [CrossRef]
- Solomon, A.; Negrea, M.O.; Cipăian, C.R.; Boicean, A.; Mihaila, R.; Rezi, C.; Cristinescu, B.A.; Berghea-Neamtu, C.S.; Popa, M.L.; Teodoru, M.; et al. Interactions between Metabolic Syndrome, MASLD, and Arterial Stiffening: A Single-Center Cross-Sectional Study. Healthcare 2023, 11, 2696. [Google Scholar] [CrossRef]
- Kjær, L. Clonal Hematopoiesis and Mutations of Myeloproliferative Neoplasms. Cancers 2020, 12, 2100. [Google Scholar] [CrossRef]
- Skov, V.; Thomassen, M.; Kjær, L.; Ellervik, C.; Larsen, M.K.; Knudsen, T.A.; Kruse, T.A.; Hasselbalch, H.C. Interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms favorably impacts deregulation of oxidative stress genes and antioxidative defense mechanisms. PLoS ONE 2022, 17, e0270669. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, Z.; Liu, Y.; Liu, Y.; Zhao, H. Function and regulation of GPX4 in the development and progression of fibrotic disease. J. Cell. Physiol. 2022, 237, 2808–2824. [Google Scholar] [CrossRef] [PubMed]
- Odell, I.D. Cross-tissue organization of myeloid cells in scleroderma and related fibrotic diseases. Curr. Opin. Rheumatol. 2024, 36, 379–386. [Google Scholar] [CrossRef]
- Mehjabin, A.; Kabir, M.; Micolucci, L.; Akhtar, M.M.; Mollah, A.K.M.M.; Islam, M.S. MicroRNA in Fibrotic Disorders: A Potential Target for Future Therapeutics. Front. Biosci. 2023, 28, 317. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F.; Luo, F. The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules 2023, 13, 119. [Google Scholar] [CrossRef]
- Takeda, K.; Tago, K.; Funakoshi-Tago, M. The indispensable role of the RNA helicase DDX5 in tumorigenesis induced by the myeloproliferative neoplasm-associated JAK2V617F mutant. Cell. Signal. 2023, 102, 110537. [Google Scholar] [CrossRef] [PubMed]
- De Moner, B.; Martinez-Sanchez, J.; Garrote, M.; Ramos, A.; Ventosa-Capell, H.; Moreno-Castaño, A.; Nomdedeu, M.; Ojeda, A.; Escolar, G.; Garcia-Pagan, J.C.; et al. Endothelial Damage in JAK2V617F Myeloproliferative Neoplasms with Splanchnic Vein Thrombosis. Thromb. Haemost. 2025. Available online: https://www.thieme-connect.com/products/ejournals/abstract/10.1055/a-2498-4849 (accessed on 19 May 2025). [CrossRef] [PubMed]
- Yakami, Y.; Yagyu, T.; Bando, T.; Hanada, M. Asymptomatic Essential Thrombocytosis Presenting with Extrahepatic Portal Vein Thrombosis: A Case Report. Am. J. Case Rep. 2023, 24, e938547. [Google Scholar] [CrossRef]
- Moisa, C.; Gaman, M.A.; Diaconu, C.C.; Gaman, A.M. Oxidative Stress Levels, JAK2V617F Mutational Status and Thrombotic Complications in Patients with Essential Thrombocythemia. Rev. Chim. 2019, 70, 2822–2825. [Google Scholar] [CrossRef]
- Kawata, E.; Siew, D.A.; Payne, J.G.; Louzada, M.; Kovacs, M.J.; Lazo-Langner, A. Splanchnic vein thrombosis: Clinical manifestations, risk factors, management, and outcomes. Thromb. Res. 2021, 202, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Sira, L.; Zsíros, N.; Bidiga, L.; Barna, S.; Kanyári, Z.; Nagy, E.B.; Guillaume, N.; Wild, D.; Rázsó, K.; Andó, S.; et al. Case report: Metastatic pancreatic neuroendocrine tumour associated with portal vein thrombosis; successful management with subsequent pregnancies. Front. Endocrinol. 2023, 14, 1095815. [Google Scholar] [CrossRef]
- Mihaila, R.G. Thrombin generation—A potentially useful biomarker of thrombotic risk in Philadelphia-negative myeloproliferative neoplasms. Biomed. Pap. 2017, 161, 50–53. [Google Scholar] [CrossRef]
- Ge, H.; Wang, C.; Tian, C.; Diao, Y.; Wang, W.; Ma, X.; Zhang, J.; Li, H.; Zhao, Z.; Zhu, L. Efficacy of WWQ-131, a highly selective JAK2 inhibitor, in mouse models of myeloproliferative neoplasms. Biomed. Pharmacother. 2022, 156, 113884. [Google Scholar] [CrossRef]
- Sansone, V.; Auteri, G.; Tovoli, F.; Mazzoni, C.; Paglia, S.; Di Pietro, C.; Vianelli, N.; Cavo, M.; Palandri, F.; Piscaglia, F. Liver and spleen shear-wave elastography in the diagnosis and severity staging of myeloproliferative diseases and myelofibrosis. J. Ultrasound. 2024, 27, 715–722. [Google Scholar] [CrossRef]
- Benedetti, E.; Tavarozzi, R.; Morganti, R.; Bruno, B.; Bramanti, E.; Baratè, C.; Balducci, S.; Iovino, L.; Ricci, F.; Ricchiuto, V.; et al. Organ Stiffness in the Work-Up of Myelofibrosis and Philadelphia-Negative Chronic Myeloproliferative Neoplasms. J. Clin. Med. 2020, 9, 2149. [Google Scholar] [CrossRef]
- Normand, A.; Le Bris, Y.; Loussouarn, D.; Gournay, J.; Mosnier, J.F. Obliteration of liver sinusoids through platelet aggregates associated to extramedullary haematopoiesis in myeloid neoplasms. Virchows Arch. 2025, 486, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.I.; Sharma, B.; John, S. Portal Hypertension. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507718/ (accessed on 19 May 2025).
- Giszas, B.; Weber, M.; Heidel, F.H.; Reuken, P.A. Recurrent Upper Gastrointestinal Bleeding from Isolated Gastric Varices as Primary Symptom of Myelofibrosis: A Case Report on Combining Interventional and Pharmacologic Treatment Options. Dig. Dis. 2022, 40, 530–534. [Google Scholar] [CrossRef]
- Chen, Y.; Kong, B.B.; Yin, H.; Liu, H.; Wu, S.; Xu, T. Acute upper gastrointestinal bleeding due to portal hypertension in a patient with primary myelofibrosis: A case report. World J. Clin. Cases 2024, 12, 2621–2626. [Google Scholar] [CrossRef]
- Gan, X.; Yu, S.; Zhu, M.; Ning, B.; He, S.; Xie, X.; Tu, L.; Yu, H. Case report: Primary myelofibrosis presenting with portal hypertension mimicking cirrhosis. Front. Med. 2024, 11, 1375571. [Google Scholar] [CrossRef]
- De Stefano, V.; Qi, X.; Betti, S.; Rossi, E. Splanchnic vein thrombosis and myeloproliferative neoplasms: Molecular-driven diagnosis and long-term treatment. Thromb. Haemost. 2016, 115, 240–249. [Google Scholar]
- Finazzi, G.; De Stefano, V.; Barbui, T. Splanchnic vein thrombosis in myeloproliferative neoplasms: Treatment algorithm 2018. Blood Cancer J. 2018, 8, 64. [Google Scholar] [CrossRef]
- Dias, E.; Liberal, R.; Costa-Moreira, P.; Príncipe, F.; Fonseca, E.; Macedo, G. Primary Myelofibrosis in the Prefibrotic Stage Presenting as Portal, Splenic, and Superior Mesenteric Vein Thrombosis: A Case Report and Review of the Literature. GE Port. J. Gastroenterol. 2021, 29, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Magaz, M.; Alvarez-Larrán, A.; Colomer, D.; López-Guerra, M.; García-Criado, M.Á.; Mezzano, G.; Belmonte, E.; Olivas, P.; Soy, G.; Cervantes, F.; et al. Next-generation sequencing in the diagnosis of non-cirrhotic splanchnic vein thrombosis. J. Hepatol. 2021, 74, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Brito, M.; Nunes, G.; Laranjo, A.; Sabino, J.; Oliveira, C.; Valle, S.; Gonçalves, D.; Fonseca, J. A unique case of bleeding from esophageal varices as the first sign of essential thrombocythemia. Clin. J. Gastroenterol. 2021, 14, 1612–1616. [Google Scholar] [CrossRef]
- Karam, D.; Iyer, V.; Agrawal, B. Occult myeloproliferative neoplasms: Not so occult any more. BMJ Case Rep. 2017, 2017, bcr2017219388. [Google Scholar] [CrossRef]
- Jeon, W.J.; Mehta, A.; Hudson, J.; Castillo, D.R.; Wang, J.; Nguyen, A.; Akhtari, M. Portal vein thrombosis as the presenting manifestation of JAK2 positive myeloproliferative neoplasm. Am. J. Med. Sci. 2023, 365, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Urano, F.; Okamura, S.; Kawashima, H. Extensive Hepatic Infarction due to Polycythemia Vera. J. Gastrointest. Liver Dis. 2024, 33, 18. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wang, Q.; Luo, B.; Chen, H.; Wang, Z.; Niu, J.; Yuan, J.; Yuan, X.; Bai, W.; He, C.; et al. Prevalence of prothrombotic factors in patients with Budd-Chiari syndrome or non-cirrhotic nonmalignant portal vein thrombosis: A hospital-based observational study. J. Gastroenterol. Hepatol. 2020, 35, 1215–1222. [Google Scholar] [CrossRef]
- Li, M.; De Stefano, V.; Song, T.; Zhou, X.; Guo, Z.; Zhu, J.; Qi, X. Prevalence of CALR mutations in splanchnic vein thrombosis: A systematic review and meta-analysis. Thromb. Res. 2018, 167, 96–103. [Google Scholar] [CrossRef]
- Poisson, J.; Plessier, A.; Kiladjian, J.J.; Turon, F.; Cassinat, B.; Andreoli, A.; De Raucourt, E.; Goria, O.; Zekrini, K.; Bureau, C.; et al. French national network for vascular liver diseases. Selective testing for calreticulin gene mutations in patients with splanchnic vein thrombosis: A prospective cohort study. J. Hepatol. 2017, 67, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Füreder, W.; Sperr, W.R.; Heibl, S.; Zebisch, A.; Pfeilstöcker, M.; Stefanzl, G.; Jäger, E.; Greiner, G.; Schwarzinger, I.; Kundi, M.; et al. Prognostic factors and follow-up parameters in patients with paroxysmal nocturnal hemoglobinuria (PNH): Experience of the Austrian PNH network. Ann. Hematol. 2020, 99, 2303–2313. [Google Scholar] [CrossRef]
- Fortea, J.I.; Carrera, I.G.; Puente, Á.; Cuadrado, A.; Huelin, P.; Tato, C.Á.; Fernández, P.Á.; Montes, M.D.R.P.; Céspedes, J.N.; López, A.B.; et al. Portal Thrombosis in Cirrhosis: Role of Thrombophilic Disorders. J. Clin. Med. 2020, 9, 2822. [Google Scholar] [CrossRef]
- Debureaux, P.E.; Cassinat, B.; Soret-Dulphy, J.; Mora, B.; Verger, E.; Maslah, N.; Plessier, A.; Rautou, P.E.; Ollivier-Hourman, I.; De Ledinghen, V.; et al. Molecular profiling and risk classification of patients with myeloproliferative neoplasms and splanchnic vein thromboses. Blood Adv. 2020, 4, 3708–3715. [Google Scholar] [CrossRef]
- Tremblay, D.; Winters, A.; Beckman, J.D.; Naymagon, L.; Patel, R.; Mascarenhas, J.; Schiano, T.D. Splanchnic vein thrombosis associated with myeloproliferative neoplasms. Thromb. Res. 2022, 218, 8–16. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Lv, W.; Hu, J.; Gou, C. Association of polycythemia vera with non-cirrhotic portal hypertension in five patients: A case series. Exp. Ther. Med. 2023, 25, 149. [Google Scholar] [CrossRef]
- Iwaki, K.; Ueda, Y.; Mishima, M.; Narukami, S.; Shiokawa, M.; Sawada, K.; Kanda, J.; Takahashi, K.; Seno, H. Portal vein thrombosis due to essential thrombocythemia with limited cutaneous systemic sclerosis. Clin. J. Gastroenterol. 2021, 14, 293–296. [Google Scholar] [CrossRef]
- Silva, L.; Moura, R.; Rocha, L.; Costa, T.; Breda, F.; Cochicho, J. A Case of Porto-Sinusoidal Vascular Disease. Eur. J. Case Rep. Intern. Med. 2023, 10, 004138. [Google Scholar] [CrossRef] [PubMed]
- Giordano, G.; Napolitano, M.; Cellurale, M.; Di Carlo, P.; Musuraca, G.; Micucci, G.; Lucchesi, A. Circulating Endothelial Cell Levels Correlate with Treatment Outcomes of Splanchnic Vein Thrombosis in Patients with Chronic Myeloproliferative Neoplasms. J. Pers. Med. 2022, 12, 364. [Google Scholar] [CrossRef]
- Zafar, M.; Heslop-Harrison, W.; Loterh, L.; Ofuafor, K. Splenic Vein Thrombosis: A Case Series of Consequential Chronic Pancreatitis and Sequential Myeloproliferative Disorder. Cureus 2022, 14, e25924. [Google Scholar] [CrossRef]
- Erdinc, B.; Ramachandran, P.; Boris, A. Polycythemia Vera Presenting with Normal Hemoglobin and Hematocrit: A Rare Variant. Cureus 2020, 12, e8404. [Google Scholar] [CrossRef] [PubMed]
- Chatzidavid, S.; Giannakopoulou, N.; Diamantopoulos, P.T.; Gavriilaki, E.; Katsiampoura, P.; Lakiotaki, E.; Sakellariou, S.; Viniou, N.A.; Dryllis, G. JAK2V617F positive polycythemia vera with paroxysmal nocturnal hemoglobinuria and visceral thromboses: A case report and review of the literature. Thromb. J. 2021, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Gao, Y.; Liu, J.W.; Dai, M.G.; Yang, S.W.; Ye, B. Gastroesophageal varices in a patient presenting with essential thrombocythemia: A case report. World J. Clin. Cases 2021, 9, 1871–1876. [Google Scholar] [CrossRef]
- Santos, S.; Dantas, E.; Veloso Gomes, F.; Luz, J.H.; Vasco Costa, N.; Bilhim, T.; Calinas, F.; Martins, A.; Coimbra, É. Retrospective Study of Transjugular Intrahepatic Portosystemic Shunt Placement for Cirrhotic Portal Hypertension. GE Port. J. Gastroenterol. 2020, 28, 5–126. [Google Scholar] [CrossRef]
- Alukal, J.J.; Zhang, T.; Thuluvath, P.J. A Nationwide Analysis of Budd-Chiari Syndrome in the United States. J. Clin. Exp. Hepatol. 2021, 11, 181–187. [Google Scholar] [CrossRef]
- Li, Z.; Li, N.; Ji, Z.; Shi, J.; Xin, G. Buddi-Chiari syndrome associated with hypereosinophilic syndrome: A case report. Medicine 2023, 102, e34291. [Google Scholar] [CrossRef]
- Camões Neves, J.; Rodrigues, F.; Apolinário, I.; Alves, M.; Sousa Caetano, O. Budd-Chiari Syndrome Caused by Polycythemia Vera: A Case Report. Cureus 2023, 15, e45527. [Google Scholar] [CrossRef] [PubMed]
- Van Wettere, M.; Bruno, O.; Rautou, P.E.; Vilgrain, V.; Ronot, M. Diagnosis of Budd-Chiari syndrome. Abdom. Radiol. 2018, 43, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Porrello, G.; Mamone, G.; Miraglia, R. Budd-Chiari Syndrome Imaging Diagnosis: State of the Art and Future Perspectives. Diagnostics 2023, 13, 2256. [Google Scholar] [CrossRef]
- Chen, H.L.; Zhang, Q.Q.; Xu, H.; Xiao, J.C.; Wei, N.; Cui, Y.F.; Liu, H.T.; Wang, W.L.; Zu, M.H. Comparison of clinical features of JAK2V617F gene mutation and non-mutation in patients with Budd-Chiari syndrome. Zhonghua Gan Zang Bing Za Zhi 2022, 30, 1365–1369. [Google Scholar]
- Kimura, K.; Osaki, A.; Hirata, Y.; Egawa, H.; Kogiso, T.; Nakamura, G.; Hashidate, H.; Wakabayashi, T.; Sato, M.; Waguri, N. A case of acute liver failure caused by Budd-Chiari syndrome salvaged by brain-dead donor liver transplantation. Clin. J. Gastroenterol. 2024, 17, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Coe, T.M.; Tanaka, M.; Bethea, E.D.; D’Alessandro, D.A.; Kimura, S.; Yeh, H.; Markmann, J.F. Liver transplantation with suprahepatic caval anastomosis including inferior vena cava stent. Transpl. Rep. 2020, 5, 100062. [Google Scholar] [CrossRef]
- Krečak, I.; Bačić, J.A.; Šimunić, N.; Bušac, V.; Pivac, L.; Čubrić, E.; Skelin, M.; Lucijanić, M. No impact of steatotic liver disease on clinical outcomes in patients with essential thrombocythemia and polycythemia vera: A pilot study. Heliyon 2024, 10, e32827. [Google Scholar] [CrossRef]
- Gonzalez-Mosquera, L.F.; Moscoso, B.; Tobar, P.; Cardenas-Maldonado, D.; Podrumar, A.I.; Mesa, R.; Cuenca, J.A. Sepsis-Related Outcomes of Patients with Philadelphia-Negative Myeloproliferative Neoplasms. Cancer Investig. 2023, 41, 423–431. [Google Scholar] [CrossRef]
- Hydroxyurea. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK547852/ (accessed on 19 May 2025).
- Enokida, K.; Tomomatsu, K.; Okada, N.; Takeuchi, T.; Takihara, T.; Amaki, J.; Hayama, N.; Oguma, T.; Aoki, T.; Asano, K. Transudative Pleural Effusion Associated with Extramedullary Hematopoiesis. Intern. Med. 2021, 60, 449–452. [Google Scholar] [CrossRef]
- Haijer, F.; Koets-Shajari, S.; Heegsma, J.; Serna-Salas, S.; Blokzijl, T.; Buist-Homan, M.; Moshage, H.; Faber, K.N. Hydroxyurea attenuates hepatic stellate cell proliferation in vitro and liver fibrogenesis in vivo. FASEB J. 2023, 37, e23124. [Google Scholar] [CrossRef]
- Lin, H.H.; Hsu, S.J.; Lu, S.N.; Chuang, W.L.; Hsu, C.W.; Chien, R.N.; Yang, S.S.; Su, W.W.; Wu, J.C.; Lee, T.H.; et al. Ropeginterferon alfa-2b in patients with genotype 1 chronic hepatitis C: Pharmacokinetics, safety, and preliminary efficacy. JGH Open 2021, 5, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.J.; Yu, M.L.; Su, C.W.; Peng, C.Y.; Chien, R.N.; Lin, H.H.; Lo, G.H.; Su, W.W.; Kuo, H.T.; Hsu, C.W.; et al. Ropeginterferon Alfa-2b administered every two weeks for patients with genotype 2 chronic hepatitis C. J. Formos. Med. Assoc. 2021, 120, 956–964. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chuang, W.L.; Qin, A.; Zhang, W.H.; Zhu, L.Y.; Zhang, G.Q.; Chen, J.J.; Lo, C.C.; Zhou, X.; Mao, X.; et al. A Phase 3 clinical trial validating the potency and safety of an innovative, extra-long-acting interferon in chronic hepatitis C. JGH Open 2022, 6, 782–791. [Google Scholar] [CrossRef]
- Shao, T.; Leung, P.S.C.; Zhang, W.; Tsuneyama, K.; Ridgway, W.M.; Young, H.A.; Shuai, Z.; Ansari, A.A.; Gershwin, M.E. Treatment with a JAK1/2 inhibitor ameliorates murine autoimmune cholangitis induced by IFN overexpression. Cell. Mol. Immunol. 2022, 19, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Ruxolitinib. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/homepage (accessed on 12 May 2025).
- Rampal, R.K.; Pinzon-Ortiz, M.; Somasundara, A.V.H.; Durham, B.; Koche, R.; Spitzer, B.; Mowla, S.; Krishnan, A.; Li, B.; An, W.; et al. Therapeutic Efficacy of Combined JAK1/2, Pan-PIM, and CDK4/6 Inhibition in Myeloproliferative Neoplasms. Clin. Cancer Res. 2021, 27, 3456–3468. [Google Scholar] [CrossRef]
- Shaker, M.E.; Hendawy, O.M.; El-Mesery, M.; Hazem, S.H. The JAK inhibitor ruxolitinib abrogates immune hepatitis instigated by concanavalin A in mice. Int. Immunopharmacol. 2022, 103, 108463. [Google Scholar] [CrossRef]
- Le Vée, M.; Bruyère, A.; Jouan, E.; Fardel, O. Janus kinase-dependent regulation of drug detoxifying protein expression by interleukin-22 in human hepatic cells. Int. Immunopharmacol. 2020, 83, 106439. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, X.; Zhang, W.; Luo, Y.; Xiao, H.; Liu, Y.; Dai, G.; Hong, J.; Li, A. Ruxolitinib suppresses liver fibrosis progression and accelerates fibrosis reversal via selectively targeting Janus kinase 1/2. J. Transl. Med. 2022, 20, 157. [Google Scholar] [CrossRef]
- Kirito, K.; Sakamoto, M.; Enomoto, N. Elevation of the Hepatitis B Virus DNA during the Treatment of Polycythemia Vera with the JAK Kinase Inhibitor Ruxolitinib. Intern. Med. 2016, 55, 1341–1344. [Google Scholar] [CrossRef]
- Kurumazaki, M.; Ogawa, N.; Kobayashi, M.; Ikejiri, F.; Kanasaki, K. A Case of Severe Hypocalcemia During JAK1/2 Inhibitor Therapy for Myelofibrosis in a Patient with Liver Cirrhosis. Intern. Med. 2025. Available online: https://www.jstage.jst.go.jp/article/internalmedicine/advpub/0/advpub_4723-24/_article (accessed on 19 May 2025). [CrossRef]
- Siddiqui, M.U.; Siddiqui, M.D.; Hameed, S.; Naeem, Z.; Siddiqui, M. Platelet Anti-Aggregator Drowns the Heart. S D Med. 2021, 74, 502–505. [Google Scholar] [PubMed]
- Interferon. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/homepage (accessed on 12 May 2025).
- Ropeginterferon Alpha2b. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/homepage (accessed on 12 May 2025).
- Appeldoorn, T.Y.J.; Munnink, T.H.O.; Morsink, L.M.; Hooge, M.N.L.; Touw, D.J. Pharmacokinetics and Pharmacodynamics of Ruxolitinib: A Review. Clin. Pharmacokinet. 2023, 62, 559–571. [Google Scholar] [CrossRef]
- Tremblay, D.; Putra, J.; Vogel, A.; Winters, A.; Hoffman, R.; Schiano, T.D.; Fiel, M.I.; Mascarenhas, J.O. The Implications of Liver Biopsy Results in Patients with Myeloproliferative Neoplasms Being Treated with Ruxolitinib. Case Rep. Hematol. 2019, 6, 3294046. [Google Scholar] [CrossRef]
- Sandhu, S.; Chaudhry, H.; Zhang, S.; Prajapati, D. Marked Hyperbilirubinemia Associated with Primary Myelofibrosis Responsive to Ruxolitinib. Case Rep. Hepatol. 2022, 2022, 9630996. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro da Cunha, M.; Marques, T. A Case of Hepatitis E Persistence in a Patient with Myelofibrosis Under Ruxolitinib. ACG Case Rep. J. 2021, 8, e00674. [Google Scholar] [CrossRef]
- Dold, L.; Lutz, P.; Heine, A.; Weismüller, T.J.; Strassburg, C.P.; Spengler, U. Ruxolitinib for treatment of polycythemia vera and myelofibrosis in patients after liver transplantation. Clin. Case Rep. 2021, 9, e04782. [Google Scholar] [CrossRef]
- Schiffer, M.; Kowalski, A.; Zhao, J.; Bewersdorf, J.P.; Lewis, R.S., Jr.; Zeidan, A.M. Fedratinib hydrochloride to treat intermediate-2 or high-risk primary or secondary myelofibrosis. Drugs Today 2020, 56, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Tohamy, H.G.; El-Neweshy, M.S.; Soliman, M.M.; Sayed, S.; Shukry, M.; Ghamry, H.I.; Abd-Ellatieff, H. Protective potential of royal jelly against hydroxyurea -induced hepatic injury in rats via antioxidant, anti-inflammatory, and anti-apoptosis properties. PLoS ONE 2022, 17, e0265261. [Google Scholar] [CrossRef]
- Huang, Y.W.; Hsu, C.W.; Lu, S.N.; Yu, M.L.; Su, C.W.; Su, W.W.; Chien, R.N.; Hsu, C.S.; Hsu, S.J.; Lai, H.C.; et al. Ropeginterferon alfa-2b every 2 weeks as a novel pegylated interferon for patients with chronic hepatitis B. Hepatol. Int. 2020, 14, 997–1008. [Google Scholar] [CrossRef]
- Peginterferon Alfa-2a. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/homepage (accessed on 12 May 2025).
- Mesa, R.A. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. IDrugs 2010, 13, 394–403. [Google Scholar]
- Pardanani, A.; Tefferi, A. Targeting myeloproliferative neoplasms with JAK inhibitors. Curr. Opin. Hematol. 2011, 18, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Ruxolitinib. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK547852/ (accessed on 12 May 2025).
- Zhang, M.Y.; Zhao, P.; Zhang, Y.; Wang, J.S. Efficacy and safety of ruxolitinib for steroid-refractory graft-versus-host disease: Systematic review and meta-analysis of randomised and non-randomised studies. PLoS ONE 2022, 17, e0271979. [Google Scholar] [CrossRef]
- Pan, C.; Cao, M.; Yan, C.; Ou, X.; Zhang, X.; Xu, W.; Xu, Y.; Cui, X. Hepatitis B virus reactivation associated with Janus kinase (JAK) inhibitors: A retrospective study of pharmacovigilance databases and review of the literature. Expert. Opin. Drug Saf. 2023, 22, 469–476. [Google Scholar] [CrossRef]
- Ali, H.; Tsai, N.C.; Synold, T.; Mokhtari, S.; Tsia, W.; Palmer, J.; Stiller, T.; Al Malki, M.; Aldoss, I.; Salhotra, A.; et al. Peritransplantation ruxolitinib administration is safe and effective in patients with myelofibrosis: A pilot open-label study. Blood Adv. 2022, 6, 1444–1453. [Google Scholar] [CrossRef]
- Kim, E.; Adeel, A.; Bozorgzadeh, A.; Amano, S.; Barry, C.T.; Daly, J.S.; Devuni, D.; Elaba, Z.; Houk, L.; Martins, P.N.; et al. Treatment of Acute Graft-versus-Host Disease in Liver Transplant Recipients. Case Rep. Transplant. 2021, 2021, 8981429. [Google Scholar] [CrossRef] [PubMed]
- Fedratinib. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK547852/ (accessed on 12 May 2025).
- Ogasawara, K.; Smith, W.B.; Xu, C.; Yin, J.; Palmisano, M.; Krishna, G. Pharmacokinetics and tolerability of fedratinib, an oral, selective Janus kinase 2 inhibitor, in subjects with renal or hepatic impairment. Cancer Chemother. Pharmacol. 2020, 85, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Naymagon, L.; Tremblay, D.; Zubizarreta, N.; Moshier, E.; Schiano, T.; Mascarenhas, J. Portal vein thrombosis patients harboring JAK2V617F have poor long-term outcomes despite anticoagulation. J. Thromb. Thrombolysis 2020, 50, 652–660. [Google Scholar] [CrossRef]
- Premkumar, M.; Bhujade, H.; Sharma, P.; Nain, J.; Ahluwalia, J.; Sandhu, A.; Kumar, Y.; Rathi, S.; Taneja, S.; Duseja, A.K.; et al. Experience With Dabigatran on Rate of Portal Vein Thrombosis Recanalization, Disease Progression and Survival. Aliment. Pharmacol. Ther. 2025, 6, 971–987. [Google Scholar] [CrossRef]
- Guy, A.; Gourdou-Latyszenok, V.; Le Lay, N.; Peghaire, C.; Kilani, B.; Dias, J.V.; Duplaa, C.; Renault, M.-A.; Denis, C.; Villeval, J.L.; et al. Vascular endothelial cell expression of JAK2 V617F is sufficient to promote a pro-thrombotic state due to increased P-selectin expression. Haematologica 2019, 104, 70–81. [Google Scholar] [CrossRef]
- Guadall, A.; Lesteven, E.; Letort, G.; Awan Toor, S.; Delord, M.; Pognant, D.; Brusson, M.; Verger, E.; Maslah, N.; Giraudier, S.; et al. Endothelial Cells Harbouring the JAK2V617F Mutation Display Pro-Adherent and Pro-Thrombotic Features. Thromb. Haemost. 2018, 118, 1586–1599. [Google Scholar] [CrossRef]
- Najem, M.Y.; Couturaud, F.; Lemarié, C.A. Cytokine and chemokine regulation of venous thromboembolism. J. Thromb. Haemost. 2020, 18, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Bjørn, M.E.; Hasselbalch, H.C. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediat. Inflamm. 2015, 2015, 648090. [Google Scholar] [CrossRef] [PubMed]
- Ferdinande, K.; Raevens, S.; Decaestecker, J.; De Vloo, C.; Seynhaeve, L.; Hoof, L.; Verhelst, X.; Geerts, A.; Devreese, K.M.J.; Degroote, H.; et al. Unravelling the coagulation paradox in liver cirrhosis: Challenges and insights. Acta Clin. Belg. 2024, 79, 451–461. [Google Scholar] [CrossRef]
- Mihaila, R.; Boicean, A.; Birlutiu, V. The Connection between Hypercoagulability and Fibrogenesis in Chronic Liver Diseases. Rom. Biotechnol. Lett. 2020, 25, 1851–1860. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Parameter | Clinical Role | Comments | Reference(s) |
|---|---|---|---|
| JAK2V617F mutation | Most frequent mutation in PV, ET, PMF; driver mutation of JAK/STAT pathway overactivation | Present in ~90% PV, ~50% ET, ~50% PMF; associated with higher cardiovascular risk | [1,2,3,4,14] |
| CALR mutation | Rare in SVT patients; testing recommended selectively in SVT with certain criteria | Important mutation in ET and PMF; useful for diagnosis when JAK2 negative | [2,3,11,44,45,46] |
| MPL mutation | Usually exclusive with other mutations | Less frequent driver mutation in MPNs | [1,2,3] |
| JAK2 exon 12 mutation | Less common | Rare; found in some SVT and JAK2-negative MPN cases | [36] |
| Concurrent mutations | Generally, not used diagnostically due to rarity | May be present; associated with older age, higher platelet count, palpable splenomegaly, leukocytosis | [3] |
| Bone marrow biopsy | Gold standard for diagnosis; detects marrow fibrosis and cellularity | Risk of sampling error; invasive | [1,7,10] |
| Splenic transient elastography | Limited by body habitus, ascites | Non-invasive prediction of bone marrow fibrosis grade | [10,12,29,30] |
| Dynamic contrast-enhanced MRI | Expensive, less available | Differentiates MF from ET and healthy controls; estimates BM fat content (cellularity/fibrosis) | [10] |
| 18-FDG and 18-FLT PET/CT | Radiation exposure, limited specificity | Estimates bone marrow fibrosis; assesses residual disease | [10] |
| LS and SS via elastography | Needs further validation in large cohorts | LS and SS correlate with bone marrow fibrosis grade; SS higher in MF and PV vs ET | [12,29,30] |
| EMH in liver | May cause microthromboses and portal hypertension independent of mutations | Seen in PMF; causes hepatosplenomegaly and portal hypertension | [7,8,9,31,35] |
| Portal hypertension | Gradient > 6 mmHg suggests portal hypertension; >10 mmHg clinically significant | Occurs due to portal vein thrombosis, extramedullary hematopoiesis, or cirrhosis | [32,33,34,35] |
| Inflammatory cytokines and chronic inflammation | Related to aberrant leukocytes and platelets | Contribute to thrombo-hemorrhagic complications and cardiovascular risk | [2,6,11,12,13] |
| Drug | Liver Beneficial Effects | References |
|---|---|---|
| Hydroxycarbamidum | Inhibits HSC proliferation and suppresses accumulation of desmin-positive HSCs and liver collagen deposition after CCl4 treatment; attenuates early development of hepatic fibrosis in vivo while preserving hepatocyte regeneration | [72] |
| PEG-IFN-α2a | Useful for the treatment of genotype 1 and 2 of chronic HCV | [73,74] |
| Ropeginterferon α-2b | Useful for the treatment of genotype 2 of chronic HCV and chronic HBV | [74,75] |
| Ruxolitinib | Ameliorates murine autoimmune cholangitis induced by IFN overexpression by inhibiting the secretion of IL-6, TNF, and monocyte chemoattractant protein-1, as well as the expression of STAT1; reduces marked hyperbilirubinemia; reduces liver size in murine models of MPN in combination with a CDK4/6 inhibitor (LEE011) and a PIM kinase inhibitor (PIM447); produces significant liver protection against Concanavalin A toxicity, via curbing the inflammatory cytokine storm; fully prevents the IL-22-mediated CYP3A4, CYP2B6, and NTCP repression in HepaRG cells (96); could offer liver protection of MPN patients during various infections, but clinical studies are needed to confirm this hypothesis; significantly attenuates fibrosis progression, improves cell damage, and accelerates fibrosis reversal in the liver of mice treated with CCl4 or Thioacetamide; improves outcomes in steroid refractory cases of acute GvHD after allogenic hematopoietic stem cell transplantation | [76,77,78,79,80,81,82,83] |
| Drug | Liver Side Effects | References |
|---|---|---|
| Hydroxycarbamidum | Transient serum enzymes and bilirubin elevations, rare cases of clinically apparent acute hepatic injury with jaundice | [72] |
| Anagrelide | Increased serum transaminase levels | [84] |
| Interferon | Liver function abnormalities | [85] |
| Ropeginterferon α-2b | Very frequently: increased plasma levels of gamma-glutamyltransferase; frequently: increased serum ALT, serum AST, blood alkaline phosphatase; infrequently: hepatotoxicity, toxic hepatitis, hepatomegaly; rarely: hepatic failure | [86] |
| Ruxolitinib | Frequently: elevated plasma concentrations of alanine aminotransferase and aspartate aminotransferase; rare instances of self-limited acute liver injury; it may favor opportunistic infections, including reactivation of the HBV and persistent HEV replication in immunosuppressed patients | [87,88,89,90,91] |
| Fedratinib | Frequently: serum liver enzyme elevations; rarely: clinically apparent acute liver injury | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihăilă, R.G.; Todor, S.B.; Mihăilă, M.D. Liver and Vascular Involvement in Philadelphia-Negative Chronic Myeloproliferative Neoplasms—A Narrative Review. Livers 2025, 5, 29. https://doi.org/10.3390/livers5030029
Mihăilă RG, Todor SB, Mihăilă MD. Liver and Vascular Involvement in Philadelphia-Negative Chronic Myeloproliferative Neoplasms—A Narrative Review. Livers. 2025; 5(3):29. https://doi.org/10.3390/livers5030029
Chicago/Turabian StyleMihăilă, Romeo G., Samuel B. Todor, and Marius D. Mihăilă. 2025. "Liver and Vascular Involvement in Philadelphia-Negative Chronic Myeloproliferative Neoplasms—A Narrative Review" Livers 5, no. 3: 29. https://doi.org/10.3390/livers5030029
APA StyleMihăilă, R. G., Todor, S. B., & Mihăilă, M. D. (2025). Liver and Vascular Involvement in Philadelphia-Negative Chronic Myeloproliferative Neoplasms—A Narrative Review. Livers, 5(3), 29. https://doi.org/10.3390/livers5030029