
The Evolution of Circulating Biomarkers for Use in Acetaminophen/Paracetamol-Induced Liver Injury in Humans: A Scoping Review



Abstract
:1. Introduction
2. Uses of Biomarkers in APAP Overdose
3. ALT, AST, and Other Enzymes
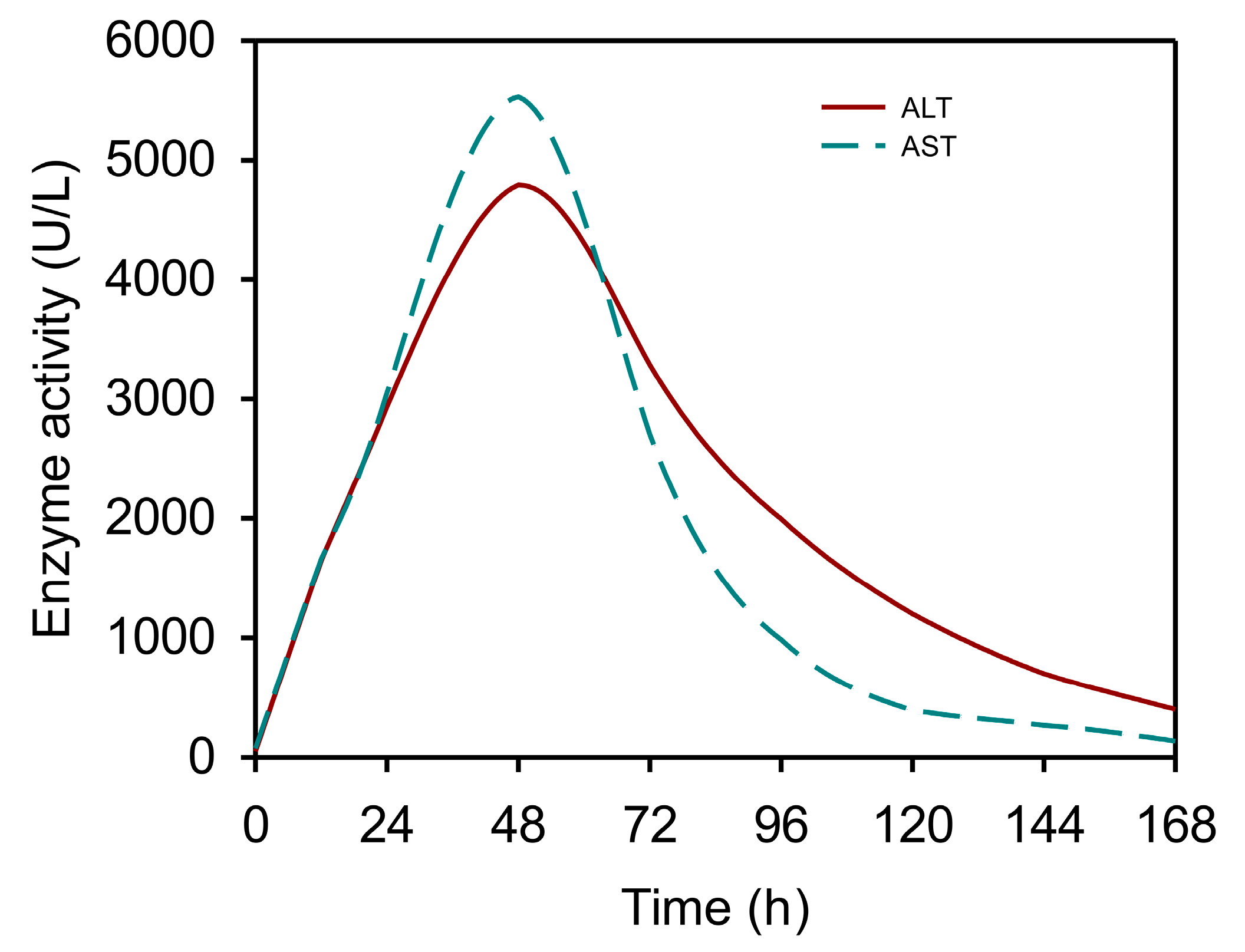
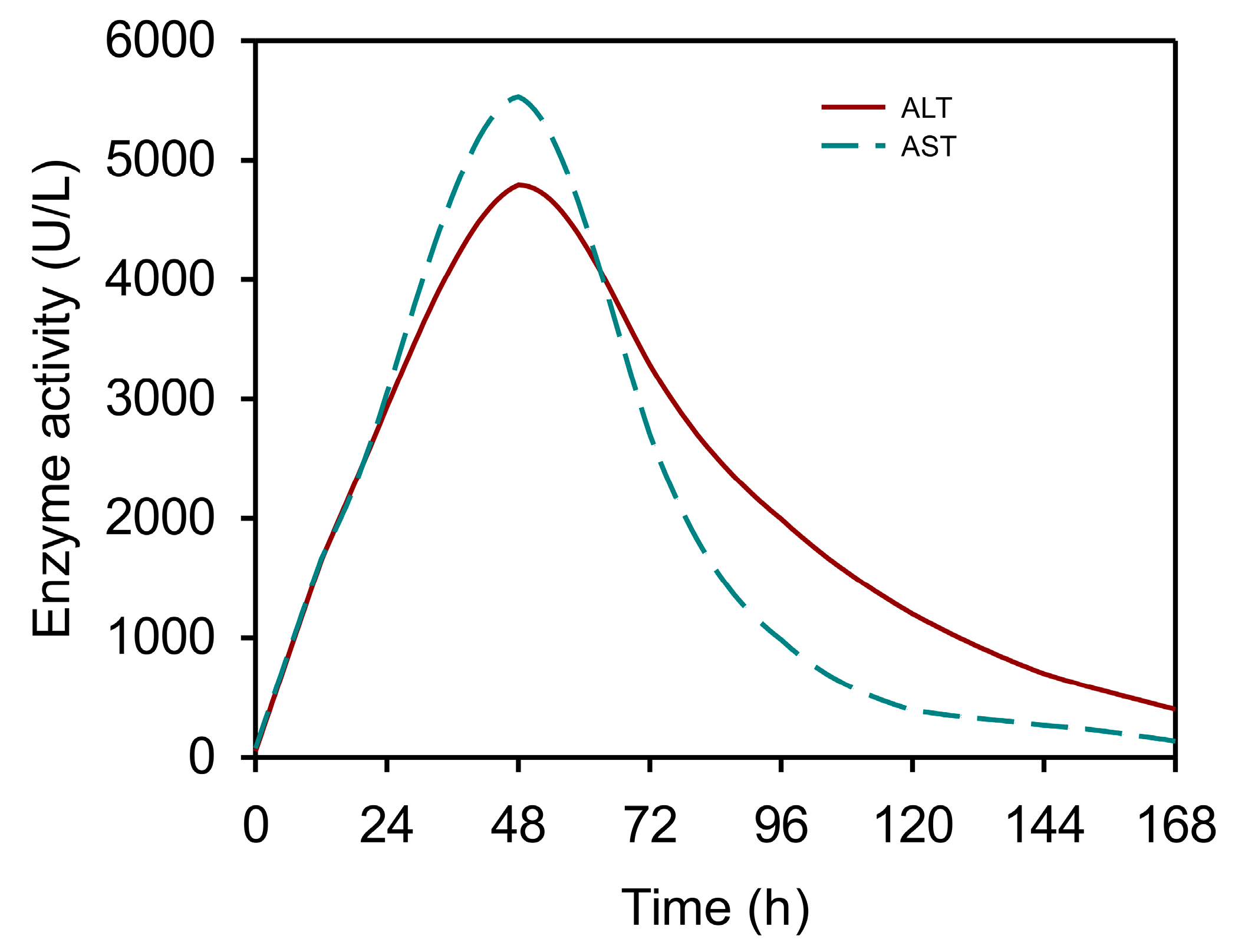
3.1. The Kinetics of Serum Aminotransferases after APAP Overdose
3.2. Uses and Limitations of Serum Aminotransferases in APAP Toxicity
3.3. Other Early Enzymes
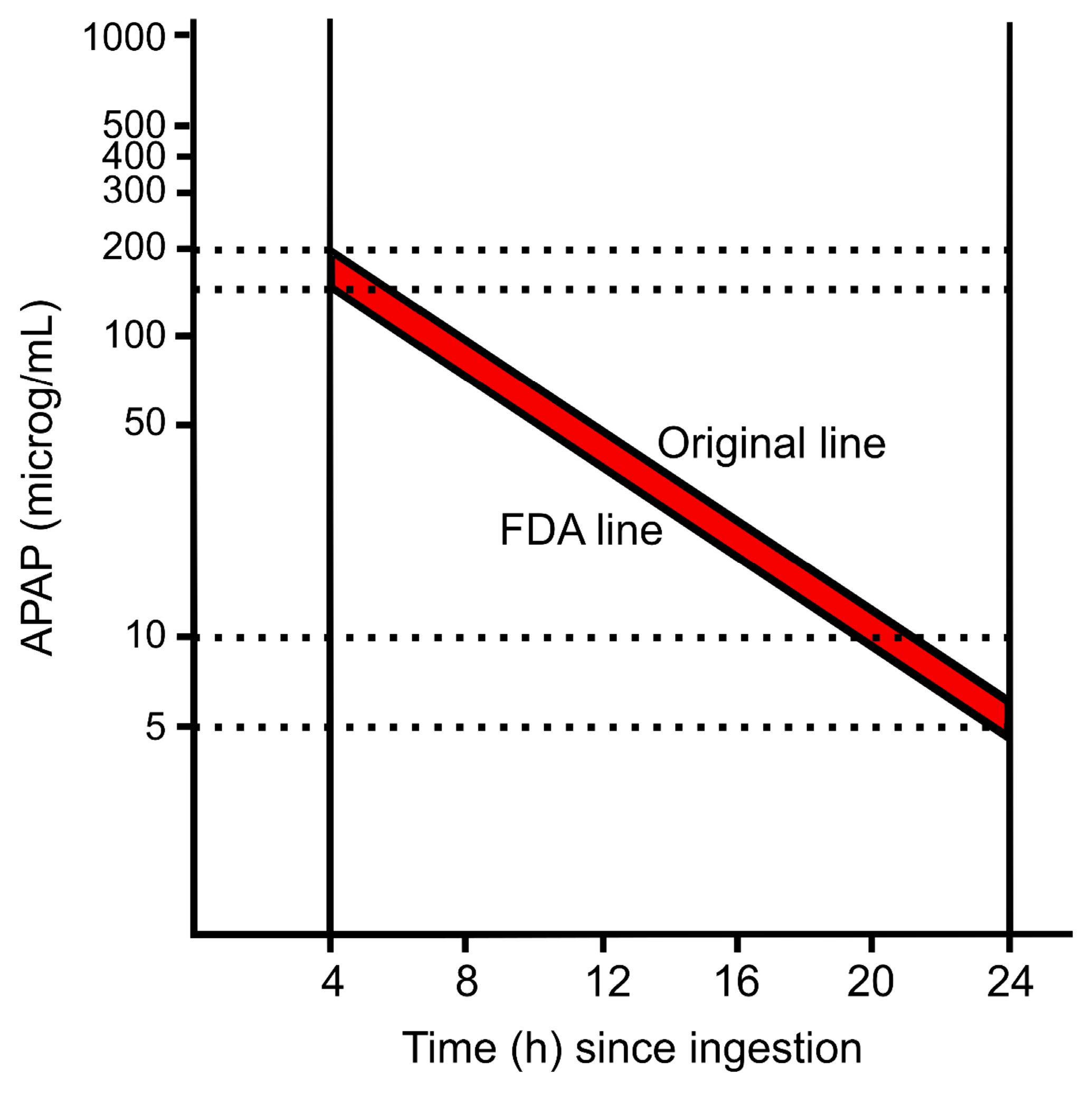
4. Serum APAP and the Rumack–Matthew Nomogram
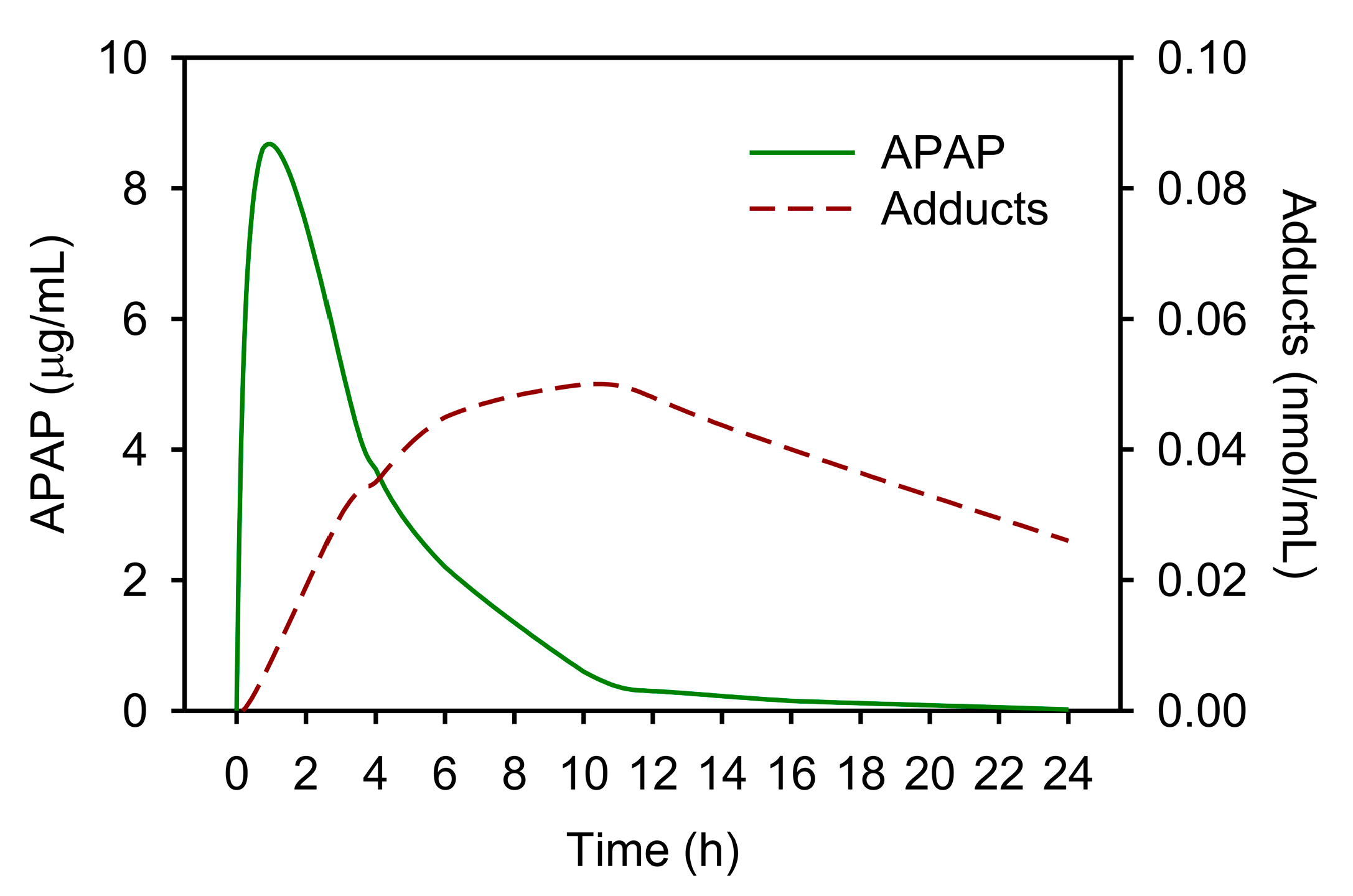
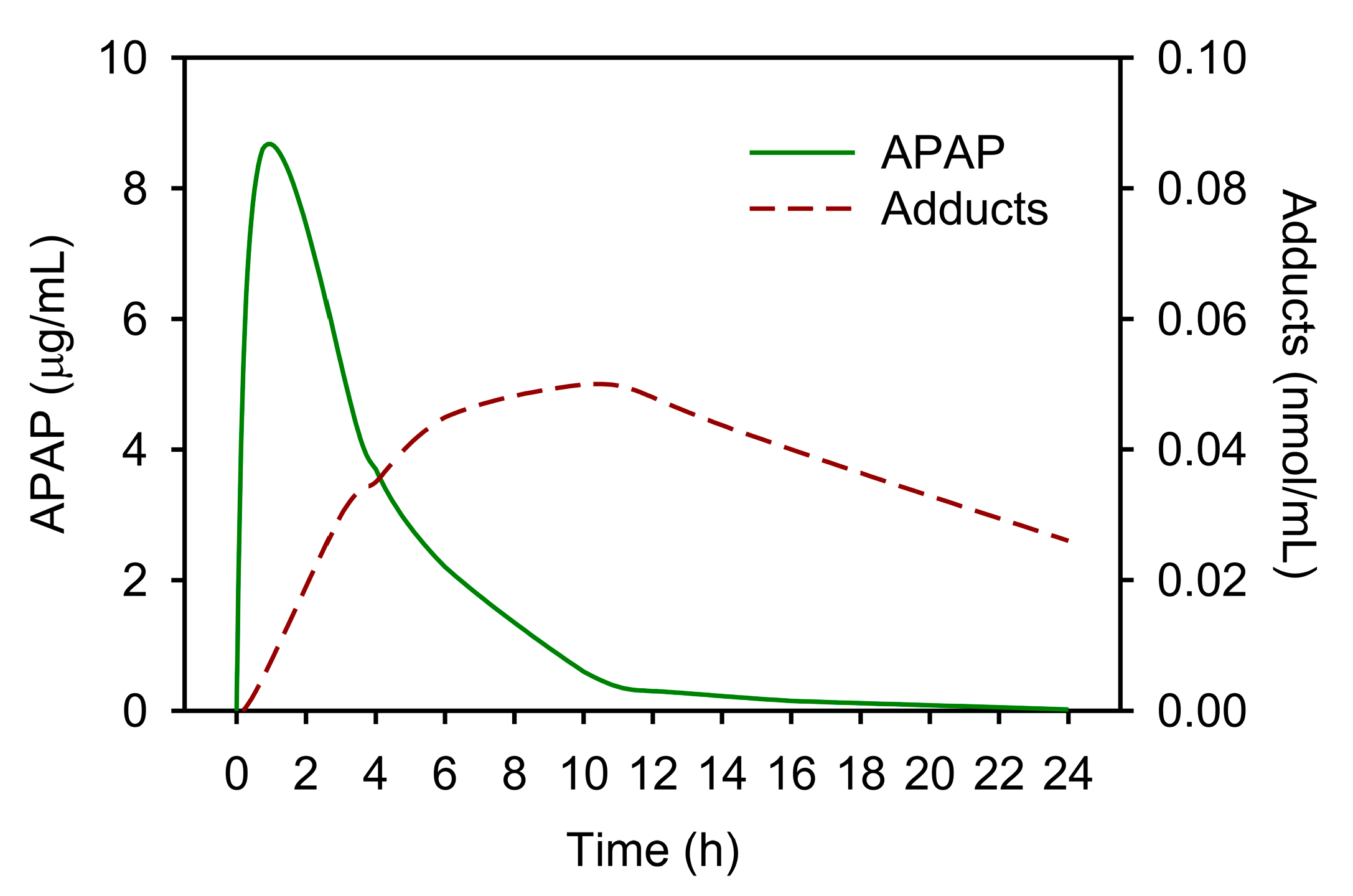
5. APAP-Protein Adducts
5.1. Diagnostic Utility of APAP-Protein Adducts as a Biomarker
5.2. Evolution of APAP-Protein Adduct Methods
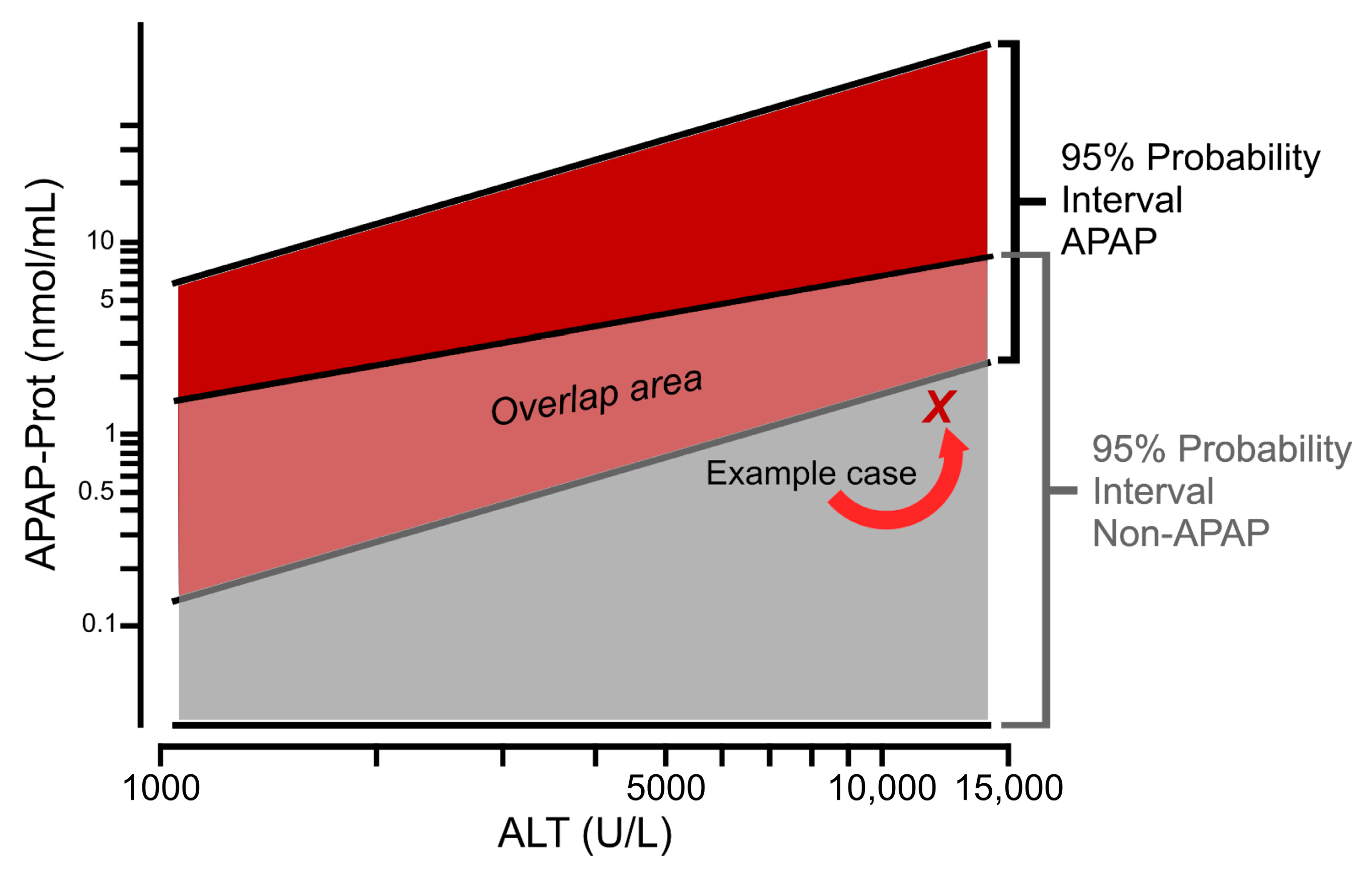
5.3. Limitations of APAP-Protein Adducts for Diagnosis and Proposed Solutions
5.4. Factors Affecting Serum Adduct Levels at Therapeutic Doses of Acetaminophen
6. The Rise of Mechanism-Based Biomarkers
6.1. Mitotoxicity Biomarkers
6.2. Cell Death Mode Markers
6.3. Other Mechanistic Biomarkers
7. Inflammation Biomarkers
8. miRNA-Based Biomarkers
9. Liver Regeneration and Repair Biomarkers
10. Miscellaneous Other Biomarkers
11. Conclusions and Future Directions in Biomarker Development
11.1. New Models for Biomarker Discovery
11.2. Understanding What Makes a Good Biomarker
11.3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Proudfoot, A.T.; Good, A.M.; Bateman, D.N. Clinical toxicology in Edinburgh, two centuries of progress. Clin. Toxicol. 2013, 51, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.G.; Eastham, W.N. Acute liver necrosis following overdose of paracetamol. Br. Med. J. 1966, 2, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Eder, H. Chronic Toxicity Studies on Phenacetin, N-Acetyl-p-Aminophenol (NAPA) and Acetylsalicylic Acid on Cats. Acta Pharmacol. Toxicol. 1964, 21, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.M.; Bereczky, G.M. Liver Necrosis from Paracetamol. Br. J. Pharmacol. Chemother. 1966, 26, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Bone, J.M. Aspirin and Gastric Bleeding. Br. Med. J. 1967, 3, 810–811. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.S.; Prescott, L.F. Liver damage and impaired glucose tolerance after paracetamol overdosage. Br. Med. J. 1966, 2, 506–507. [Google Scholar] [CrossRef] [PubMed]
- Toghill, P.; Williams, R.; Stephens, J.; Carroll, J. Acute Hepatic Necrosis Following an Overdose of Paracetamol. Gastroenterology 1969, 56, 773–776. [Google Scholar] [CrossRef]
- Boyer, T.D.; Rouff, S.L. Acetaminophen-Induced Hepatic Necrosis and Renal Failure. JAMA J. Am. Med. Assoc. 1971, 218, 440–441. [Google Scholar] [CrossRef]
- James, O.; Roberts, S.; Douglas, A.; Lesna, M.; Pulman, L.; Smith, P.; Watson, A. Liver Damage after Paracetamol Overdose. Comparison of Liver-Function Tests, Fasting Serum Bile Acids, and Liver Histology. Lancet 1975, 306, 579–581. [Google Scholar] [CrossRef]
- McJunkin, B.; Barwick, K.W.; Little, W.C.; Winfield, J.B. Fatal Massive Hepatic Necrosis Following Acetaminophen Overdose. JAMA 1976, 236, 1874–1875. [Google Scholar] [CrossRef]
- Barker, J.D.; de Carle, D.J.; Anuras, S. Chronic Excessive Acetaminophen Use and Liver Damage. Ann. Intern. Med. 1977, 87, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Krenzelok, E.P.; Best, L.; Manoguerra, A.S. Acetaminophen Toxity. Am. J. Hosp. Pharm. 1977, 34, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.R.; Snyder, S.K.; Cameron, A.J. Hepatotoxicity in Acetaminophen Poisoning. Mayo Clin. Proc. 1977, 52, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Groarke, J.F.; Averett, J.M.; Hirschowitz, B.I. Acetaminophen and Hepatic Necrosis. N. Engl. J. Med. 1977, 296, 233. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Acetaminophen Toxicity: A History of Serendipity and Unintended Consequences. Clin. Liver Dis. 2020, 16, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Manthripragada, A.D.; Zhou, E.H.; Budnitz, D.S.; Lovegrove, M.C.; Willy, M.E. Characterization of acetaminophen overdose-related emergency department visits and hospitalizations in the United States. Pharmacoepidemiol. Drug Saf. 2011, 20, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Martin, B.C. Trends in emergency department visits attributable to acetaminophen overdoses in the United States: 1993–2007. Pharmacoepidemiol. Drug Saf. 2011, 20, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Budnitz, D.S.; Lovegrove, M.C.; Crosby, A.E. Emergency Department Visits for Overdoses of Acetaminophen-Containing Products. Am. J. Prev. Med. 2011, 40, 585–592. [Google Scholar] [CrossRef]
- Stravitz, R.T.; Fontana, R.J.; Karvellas, C.; Durkalski, V.; McGuire, B.; Rule, J.A.; Tujios, S.; Lee, W.M.; Acute Liver Failure Study Group. Future directions in acute liver failure. Hepatology 2023, 78, 1266–1289. [Google Scholar] [CrossRef]
- Karmen, A.; Wróblewski, F.; LaDue, J.S. Transaminase Activity in Human Blood. J. Clin. Investig. 1955, 34, 126–133. [Google Scholar] [CrossRef]
- Wróblewski, F.; Ladue, J.S. Serum Glutamic Pyruvic Transaminase SGP-T in Hepatic Disease: A Preliminary Report. Ann. Intern. Med. 1956, 45, 801–811. [Google Scholar] [CrossRef]
- De Ritis, F.; Coltori, M.; Giusti, G. Transaminase Activity of Human Serum in Viral Hepatitis; Preventive Note. Minerva Medica 1955, 46, 1207–1209. [Google Scholar] [PubMed]
- Alfred, D. Reagents and Method for Assaying Biological Samples. U.S. Patent 3,413,198, 26 November 1968. [Google Scholar]
- Kessler, G.; Rush, R.; Leon, L.; Delea, A.; Cupiola, R. Automated 340 Nm Measurement of SGOT, SGPT and LDH. Clin. Chem. 1970, 16, 530–531. [Google Scholar]
- Smith, A.F.; Taylor, R.H. Comparison of two-point (AutoAnalyzer II) with kinetic methods for transaminase assay. J. Clin. Pathol. 1973, 26, 42–47. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Jaeschke, H. Metabolism and Disposition of Acetaminophen: Recent Advances in Relation to Hepatotoxicity and Diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Hinson, J.A. The development and hepatotoxicity of acetaminophen: Reviewing over a century of progress. Drug Metab. Rev. 2020, 52, 472–500. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ramachandran, A. Central mechanisms of acetaminophen hepatotoxicity: Mitochondrial dysfunction by protein adducts and oxidant stress. Drug Metab. Dispos. 2023, 51. [Google Scholar] [CrossRef]
- Rej, R.; Keese, C.R.; Giaever, I. Direct immunological determination of aspartate aminotransferase isoenzymes. Clin. Chem. 1981, 27, 1597–1601. [Google Scholar] [CrossRef]
- Panteghini, M.; Pagani, F.; Cuccia, C. Activity of serum aspartate aminotransferase isoenzymes in patients with acute myocardial infarction. Clin. Chem. 1987, 33, 67–71. [Google Scholar] [CrossRef]
- Zoppini, G.; Cacciatori, V.; Negri, C.; Stoico, V.; Lippi, G.; Targher, G.; Bonora, E. The aspartate aminotransferase-to-alanine aminotransferase ratio predicts all-cause and cardiovascular mortality in patients with type 2 diabetes. Medicine 2016, 95, e4821. [Google Scholar] [CrossRef]
- Mcgovern, A.J.; Vitkovitsky, I.V.; Jones, D.L.; Mullins, M.E. Can AST/ALT Ratio Indicate Recovery after Acute Paracetamol Poisoning? Clin. Toxicol. 2015, 53, 164–167. [Google Scholar] [CrossRef]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Maclean, D.; Peters, T.J.; Brown, R.A.; McCathie, M.; Baines, G.F.; Robertson, P.G. Treatment of Acute Paracetamol Poisoning. Lancet 1968, 292, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Carracio, T.R.; Mofenson, H.C. The Temporal Profile of Increased Transaminase Levels in Patients with Acetaminophen-Induced Liver Dysfunction. Ann. Emerg. Med. 1995, 26, 49–53. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.M.; Sivilotti, M.L.A. A descriptive analysis of aspartate and alanine aminotransferase rise and fall following acetaminophen overdose. Clin. Toxicol. 2015, 53, 849–855. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Staggs, V.S.; Sharpe, M.R.; Lee, W.M.; Jaeschke, H. Serum Mitochondrial Biomarkers and Damage-Associated Molecular Patterns Are Higher in Acetaminophen Overdose Patients with Poor Outcome. Hepatology 2014, 60, 1336–1345. [Google Scholar] [CrossRef]
- Price, J.R.; Hagrass, H.; Filip, A.B.; McGill, M.R. LDH and the MELD-LDH in Severe Acute Liver Injury and Acute Liver Failure: Preliminary Confirmation of a Novel Prognostic Score for Risk Stratification. J. Appl. Lab. Med. 2023, 8, 504–513. [Google Scholar] [CrossRef]
- Green, T.J.; Sivilotti, M.L.; Langmann, C.; Yarema, M.; Juurlink, D.; Burns, M.J.; Johnson, D.W. When Do the Aminotransferases Rise after Acute Acetaminophen Overdose. Clin. Toxicol. 2010, 48, 787–792. [Google Scholar] [CrossRef]
- Al-Hourani, K.; Mansi, R.; Pettie, J.; Dow, M.; Bateman, D.N.; Dear, J.W. The predictive value of hospital admission serum alanine transaminase activity in patients treated for paracetamol overdose. QJM 2013, 106, 541–546. [Google Scholar] [CrossRef]
- Curry, S.C.; Padilla-Jones, A.; Kang, A.; Snow, J.; Heise, C. Serum ALT Activity at Commencement of N-Acetylecysteine Therapy as a Predictor of Hepatotoxocity, Hepatic Encephalopathy, and Mortality in Patients with Acetaminophen Toxicity. J. Med. Toxicol. 2019, 15, 71. [Google Scholar] [CrossRef]
- Harrison, P.; Keays, R.; Bray, G.; Alexander, G.; Williams, R. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet 1990, 335, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Keays, R.; Harrison, P.M.; Wendon, J.A.; Forbes, A.; Gove, C.; Alexander, G.J.; Williams, R. Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: A prospective controlled trial. BMJ 1991, 303, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Mullins, M.E.; Schwarz, E. Re: Remien Etal.: Mathematical Modeling of Liver Injury and Dysfunction after Acetaminophen Overdose. Hepatology 2012, 56, 2427–2428. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; McCullough, S.S.; Lamps, L.W.; Hinson, J.A. Effect of N-Acetylcysteine on Acetaminophen Toxicity in Mice: Relationship to Reactive Nitrogen and Cytokine Formation. Toxicol. Sci. 2003, 75, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; McGill, M.R.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Jaeschke, H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol. Appl. Pharmacol. 2014, 279, 266–274. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Lebofsky, M.; Norris, H.-R.; Slawson, M.H.; Bajt, M.L.; Xie, Y.; Williams, C.D.; Wilkins, D.G.; Rollins, D.E.; Jaeschke, H. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: Dose–response, mechanisms, and clinical implications. Toxicol. Appl. Pharmacol. 2013, 269, 240–249. [Google Scholar] [CrossRef]
- Vazquez, J.H.; Kennon-McGill, S.; Byrum, S.D.; Mackintosh, S.G.; Jaeschke, H.; Williams, D.K.; Lee, W.M.; Dranoff, J.A.; McGill, M.R. Proteomics Indicates Lactate Dehydrogenase Is Prognostic in Acetaminophen-Induced Acute Liver Failure Patients and Reveals Altered Signaling Pathways. Toxicol. Sci. 2022, 187, 25–34. [Google Scholar] [CrossRef]
- Sivilotti, M.L.A.; Green, T.J.; Langmann, C.; Yarema, M.; Juurlink, D.; Johnson, D. Multiplying the Serum Aminotransferase by the Acetaminophen Concentration to Predict Toxicity Following Overdose. Clin. Toxicol. 2010, 48, 793–799. [Google Scholar] [CrossRef]
- Wróblewski, F.; Ladue, J.S. Lactic Dehydrogenase Activity in Blood. Proc. Soc. Exp. Biol. Med. 1955, 90, 210–213. [Google Scholar] [CrossRef]
- Hsieh, K.M.; Blumenthal, H.T. Serum Lactic Dehydrogenase Levels in Various Disease States. Proc. Soc. Exp. Biol. Med. 1956, 91, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Hammond, K.D.; Balinsky, D. Isozyme Studies of Several Enzymes of Carbohydrate Metabolism in Human Adult and Fetal Tissues, Tumor Tissues, and Cell Cultures. Cancer Res. 1978, 38, 1323–1328. [Google Scholar] [PubMed]
- Milne, E.; Doxey, D. Lactate dehydrogenase and its isoenzymes in the tissues and sera of clinically normal dogs. Res. Vet. Sci. 1987, 43, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Farhana, A.; Lappin, S.L. Biochemistry, Lactate Dehydrogenase (LDH); StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Knezevic, C.E.; Ness, M.A.; Tsang, P.H.T.; Tenney, B.J.; Marzinke, M.A. Establishing hemolysis and lipemia acceptance thresholds for clinical chemistry tests. Clin. Chim. Acta 2020, 510, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Bing, R.J.; Castellanos, A.; Siegel, A. Diagnostic Value of Activity of Malic Dehydrogenase and Phosphohexose Isomerase; Preliminary Report of Findings in Patients with Myocardial Infarction and Liver Disease. J. Am. Med. Assoc. 1957, 164, 647–650. [Google Scholar] [CrossRef]
- Schomaker, S.; Warner, R.; Bock, J.; Johnson, K.; Potter, D.; Van Winkle, J.; Aubrecht, J. Assessment of Emerging Biomarkers of Liver Injury in Human Subjects. Toxicol. Sci. 2013, 132, 276–283. [Google Scholar] [CrossRef]
- Lester, D.; Greenberg, L.A. The Metabolic Fate of Acetanilid and Other Aniline Derivatives; Major Metabolites of Acetanilid Appearing in the Blood. J. Pharmacol. Exp. Ther. 1947, 90, 68–75. [Google Scholar]
- Brodie, B.B.; Axelrod, J. The Estimation of Acetanilide and Its Metabolic Products, Aniline, n-Acetyl p-Aminophenol and p-Aminophenol (Free and Total Conjugated) in Biological Fluids and Tissues. J. Pharmacol. Exp. Ther. 1948, 94, 22–28. [Google Scholar]
- Prescott, L.; Roscoe, P.; Wright, N.; Brown, S. Plasma-Paracetamol Half-Life and Hepatic Necrosis in Patients with Paracetamol Overdosage. Lancet 1971, 297, 519–522. [Google Scholar] [CrossRef]
- Done, A.K. Salicylate Intoxication. Significance of Measurements of Salicylate in Blood in Cases of Acute Ingestion. Pediatrics 1960, 26, 800–807. [Google Scholar] [CrossRef]
- Rumack, B.H.; Matthew, H. Acetaminophen Poisoning and Toxicity. Pediatrics 1975, 55, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Chiew, A.L.; Isbister, G.K.; Kirby, K.A.; Page, C.B.; Chan, B.S.H.; Buckley, N.A. Massive paracetamol overdose: An observational study of the effect of activated charcoal and increased acetylcysteine dose (ATOM-2). Clin. Toxicol. 2017, 55, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Chiew, A.L.; James, L.P.; Isbister, G.K.; Pickering, J.W.; McArdle, K.; Chan, B.S.; Buckley, N.A. Early acetaminophen-protein adducts predict hepatotoxicity following overdose (ATOM-5). J. Hepatol. 2020, 72, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.C.; Lim, M.; Lai, L.; Mendoza, E.; Albertson, T.E.; Chenoweth, J.A. Evaluation of N-acetylcysteine dose for the treatment of massive acetaminophen ingestion. Clin. Toxicol. 2022, 60, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.C.; Padilla-Jones, A.; Ruha, A.M.; O’Connor, A.D.; Kang, A.M.; Wilkins, D.G.; Jaeschke, H.; Wilhelms, K.; Gerkin, R.D.; Graeme, K.A.; et al. The Relationship Between Circulating Acetaminophen-Protein Adduct Concentrations and Alanine Aminotransferase Activities in Patients with and Without Acetaminophen Overdose and Toxicity. J. Med. Toxicol. 2019, 15, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kellmeyer, K.; Yates, C.; Parker, S.; Hilligoss, D. Bilirubin interference with kit determination of acetaminophen. Clin. Chem. 1982, 28, 554–555. [Google Scholar] [CrossRef] [PubMed]
- Bertholf, R.L.; Johannsen, L.M.; Bazooband, A.; Mansouri, V. False-Positive Acetaminophen Results in a Hyperbilirubinemic Patient. Clin. Chem. 2003, 49, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Beuhler, M.; Curry, S. False Positive Acetaminophen Levels Associated with Hyperbilirubinemia. Clin. Toxicol. 2005, 43, 167–170. [Google Scholar] [CrossRef]
- Polson, J.; Wians, F.H.; Orsulak, P.; Fuller, D.; Murray, N.G.; Koff, J.M.; Khan, A.I.; Balko, J.A.; Hynan, L.S.; Lee, W.M. False positive acetaminophen concentrations in patients with liver injury. Clin. Chim. Acta 2008, 391, 24–30. [Google Scholar] [CrossRef]
- Chan, B.; Tsang, H.; Ng, C.W.; Ling, W.H.; Leung, D.C.; Lee, H.H.; Mak, C.M. Performance evaluation of five commercial assays for detection of acetaminophen. J. Clin. Lab. Anal. 2019, 33, e22683. [Google Scholar] [CrossRef]
- Tyhach, R.J.; Mayer, M.; Salpeter, L. More on Interference of N-Acetylcysteine in Measurement of Acetaminophen [3] (Multiple Letters). Clin. Chem. 1999, 45, 584. [Google Scholar] [CrossRef] [PubMed]
- Rumack, B.H.; Peterson, R.C.; Koch, G.G.; Amara, I.A. Acetaminophen Overdose: 662 Cases with Evaluation of Oral Acetylcysteine Treatment. Arch. Intern. Med. 1981, 141, 380. [Google Scholar] [CrossRef] [PubMed]
- Smilkstein, M.J.; Knapp, G.L.; Kulig, K.W.; Rumack, B.H. Efficacy of Oral N-Acetylcysteine in the Treatment of Acetaminophen Overdose. N. Engl. J. Med. 1988, 319, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Makin, A.J.; Wendon, J.; Williams, R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987–1993). Gastroenterology 1995, 109, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Sivilotti, M.L.A.; Good, A.M.; Yarema, M.C.; Juurlink, D.N.; Johnson, D.W. A New Predictor of Toxicity Following Acetaminophen Overdose Based on Pretreatment Exposure. Clin. Toxicol. 2005, 43, 229–234. [Google Scholar] [CrossRef]
- Sivilotti, M.L.; Yarema, M.C.; Juurlink, D.N.; Good, A.M.; Johnson, D.W. A Risk Quantification Instrument for Acute Acetaminophen Overdose Patients Treated with N-Acetylcysteine. Ann. Emerg. Med. 2005, 46, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Chomchai, S.; Chomchai, C.; Anusornsuwan, T. Acetaminophen psi parameter: A useful tool to quantify hepatotoxicity risk in acute acetaminophen overdose. Clin. Toxicol. 2011, 49, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Chomchai, S.; Chomchai, C. Predicting acute acetaminophen hepatotoxicity with acetaminophen-aminotransferase multiplication product and the Psi parameter. Clin. Toxicol. 2014, 52, 506–511. [Google Scholar] [CrossRef]
- James, L.P.; Chiew, A.; Abdel-Rahman, S.M.; Letzig, L.; Graudins, A.; Day, P.; Roberts, D. Acetaminophen protein adduct formation following low-dose acetaminophen exposure: Comparison of immediate-release vs extended-release formulations. Eur. J. Clin. Pharmacol. 2013, 69, 851–857. [Google Scholar] [CrossRef]
- McGill, M.R.; James, L.P.; McCullough, S.S.; Moran, J.H.; Mathews, S.E.; Peterson, E.C.; Fleming, D.P.; Tripod, M.E.; Vazquez, J.H.; Kennon-McGill, S.; et al. Short-Term Safety of Repeated Acetaminophen Use in Patients with Compensated Cirrhosis. Hepatol. Commun. 2022, 6, 361–373. [Google Scholar] [CrossRef]
- Davern, T.J.; James, L.P.; Hinson, J.A.; Polson, J.; Larson, A.M.; Fontana, R.J.; Lalani, E.; Munoz, S.; Shakil, A.O.; Lee, W.M. Measurement of Serum Acetaminophen–Protein Adducts in Patients with Acute Liver Failure. Gastroenterology 2006, 130, 687–694. [Google Scholar] [CrossRef] [PubMed]
- James, L.; Capparelli, E.; Simpson, P.; Letzig, L.; Roberts, D.; Hinson, J.; Kearns, G.; Blumer, J.; Sullivan, J. Acetaminophen-Associated Hepatic Injury: Evaluation of Acetaminophen Protein Adducts in Children and Adolescents with Acetaminophen Overdose. Clin. Pharmacol. Ther. 2008, 84, 684–690. [Google Scholar] [CrossRef]
- James, L.P.; Letzig, L.; Simpson, P.M.; Capparelli, E.; Roberts, D.W.; Hinson, J.A.; Davern, T.J.; Lee, W.M. Pharmacokinetics of Acetaminophen-Protein Adducts in Adults with Acetaminophen Overdose and Acute Liver Failure. Drug Metab. Dispos. 2009, 37, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.W.; Lee, W.M.; Hinson, J.A.; Bai, S.; Swearingen, C.J.; Stravitz, R.T.; Reuben, A.; Letzig, L.; Simpson, P.M.; Rule, J.; et al. An Immunoassay to Rapidly Measure Acetaminophen Protein Adducts Accurately Identifies Patients with Acute Liver Injury or Failure. Clin. Gastroenterol. Hepatol. 2017, 15, 555–562.e3. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen Induced Hepatic Necrosis. I. Role of Drug Metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185–194. [Google Scholar] [PubMed]
- Jollow, D.J.; Mitchell, J.R.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen Induced Hepatic Necrosis. II. Role of Covalent Binding in Vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202. [Google Scholar]
- Potter, W.Z.; Davis, D.C.; Mitchell, J.R.; Jollow, D.J.; Gillette, J.R.; Brodie, B.B. Acetaminophen Induced Hepatic Necrosis. III. Cytochrome P 450 Mediated Covalent Binding in Vitro. J. Pharmacol. Exp. Ther. 1973, 187, 203–210. [Google Scholar]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen Induced Hepatic Necrosis. IV. Protective Role of Glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Potter, W.; Thorgeirsson, S.; Jollow, D.; Mitchell, J. Acetaminophen-Induced Hepatic Necrosis V. Correlation of Hepatic Necrosis, Covalent Binding and Glutathione Depletion in Hamsters. Pharmacology 1974, 12, 129–143. [Google Scholar] [CrossRef]
- Roberts, D.W.; Pumford, N.R.; Potter, D.W.; Benson, R.W.; Hinson, J.A. A Sensitive Immunochemical Assay for Acetaminophen-Protein Adducts. J. Pharmacol. Exp. Ther. 1987, 241, 527–533. [Google Scholar]
- Muldrew, K.L.; James, L.P.; Coop, L.; McCullough, S.S.; Hendrickson, H.P.; Hinson, J.A.; Mayeux, P.R. Determination of Acetaminophen-Protein Adducts in Mouse Liver and Serum and Human Serum after Hepatotoxic Doses of Acetaminophen Using High-Performance Liquid Chromatography with Electrochemical Detection. Drug Metab. Dispos. 2002, 30, 446–451. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Yan, H.-M.; Ramachandran, A.; Murray, G.J.; Rollins, D.E.; Jaeschke, H. HepaRG cells: A human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 2011, 53, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.F.; King, A.D.; Chang, Y.; Murray, G.J.; Norris, H.-R.K.; Dart, R.C.; Green, J.L.; Curry, S.C.; Rollins, D.E.; Wilkins, D.G. Quantification of a biomarker of acetaminophen protein adducts in human serum by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry: Clinical and animal model applications. J. Chromatogr. B 2015, 985, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Heard, K.J.; Green, J.L.; James, L.P.; Judge, B.S.; Zolot, L.; Rhyee, S.; Dart, R.C. Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterol. 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Kennon_McGill, S.; McGill, M. Extrahepatic toxicity of acetaminophen: Critical evaluation of the evidence and proposed mechanisms. J. Clin. Transl. Res. 2017, 3, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.C.; Padilla-Jones, A.; O’Connor, A.D.; Ruha, A.M.; Bikin, D.S.; Wilkins, D.G.; Rollins, D.E.; Slawson, M.H.; Gerkin, R.D. Prolonged Acetaminophen-Protein Adduct Elimination During Renal Failure, Lack of Adduct Removal by Hemodiafiltration, and Urinary Adduct Concentrations After Acetaminophen Overdose. J. Med. Toxicol. 2015, 11, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Heard, K.; Green, J.L.; Anderson, V.; Bucher-Bartelson, B.; Dart, R.C. Paracetamol (acetaminophen) protein adduct concentrations during therapeutic dosing. Br. J. Clin. Pharmacol. 2016, 81, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Monte, A.A.; Sonn, B.; Saben, J.; Rumack, B.H.; Reynolds, K.M.; Dart, R.C.; Heard, K.J. The Genomics of Elevated ALT and Adducts in Therapeutic Acetaminophen Treatment: A Pilot Study. J. Med. Toxicol. 2020, 17, 160–167. [Google Scholar] [CrossRef]
- Thomas, K.C.; Wilkins, D.G.; Curry, S.C.; Grey, T.C.; Andrenyak, D.M.; McGill, L.D.; Rollins, D.E. Detection of Acetaminophen–Protein Adducts in Decedents with Suspected Opioid–Acetaminophen Combination Product Overdose. J. Forensic Sci. 2016, 61, 1301–1306. [Google Scholar] [CrossRef]
- Petersen, P.; Vilstrup, H. Relation between Liver Function and Hepatocyte Ultrastructure in a Case of Paracetamol Intoxication. Digestion 1979, 19, 415–419. [Google Scholar] [CrossRef]
- Meyers, L.L.; Beierschmitt, W.P.; Khairallah, E.A.; Cohen, S.D. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol. Appl. Pharmacol. 1988, 93, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Glutathione Disulfide Formation and Oxidant Stress during Acetaminophen-Induced Hepatotoxicity in Mice in Vivo: The Protective Effect of Allopurinol. J. Pharmacol. Exp. Ther. 1990, 255, 935–941. [Google Scholar] [PubMed]
- Kon, K.; Kim, J.-S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.B.; Kurten, R.C.; McCullough, S.S.; Brock, R.W.; Hinson, J.A. Mechanisms of Acetaminophen-Induced Hepatotoxicity: Role of Oxidative Stress and Mitochondrial Permeability Transition in Freshly Isolated Mouse Hepatocytes. J. Pharmacol. Exp. Ther. 2005, 312, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Cover, C.; Mansouri, A.; Knight, T.R.; Bajt, M.L.; Lemasters, J.J.; Pessayre, D.; Jaeschke, H. Peroxynitrite-Induced Mitochondrial and Endonuclease-Mediated Nuclear DNA Damage in Acetaminophen Hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 315, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Bajt, M.L.; Cover, C.; Lemasters, J.J.; Jaeschke, H. Nuclear Translocation of Endonuclease G and Apoptosis-Inducing Factor during Acetaminophen-Induced Liver Cell Injury. Toxicol. Sci. 2006, 94, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Lebofsky, M.; Baines, C.P.; Lemasters, J.J.; Jaeschke, H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free. Radic. Res. 2011, 45, 156–164. [Google Scholar] [CrossRef]
- McGill, M.R.; Williams, C.D.; Xie, Y.; Ramachandran, A.; Jaeschke, H. Acetaminophen-induced liver injury in rats and mice: Comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol. Appl. Pharmacol. 2012, 264, 387–394. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Du, K.; Akakpo, J.Y.; Umbaugh, D.S.; Jaeschke, H.; Ramachandran, A. Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol. Lett. 2021, 338, 21–31. [Google Scholar] [CrossRef]
- Gunawan, B.K.; Liu, Z.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-Terminal Kinase Plays a Major Role in Murine Acetaminophen Hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef]
- Henderson, N.C.; Pollock, K.J.; Frew, J.; Mackinnon, A.C.; Flavell, R.A.; Davis, R.J.; Sethi, T.; Simpson, K.J. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut 2007, 56, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Min, R.W.M.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Min, R.W.; Chen, C.Q.; Zhang, J.; Chen, Y.; Li, M.; Suzuki, A.; Abdelmalek, M.F.; Wang, Y.; Aghajan, M.; et al. Expression of mitochondrial membrane–linked SAB determines severity of sex-dependent acute liver injury. J. Clin. Investig. 2019, 129, 5278–5293. [Google Scholar] [CrossRef] [PubMed]
- Placke, M.E.; Ginsberg, G.L.; Wyand, D.S.; Cohen, S.D. Ultrastructural Changes during Acute Acetaminophen-Induced Hepatotoxicity in the Mouse: A Time and Dose Study. Toxicol. Pathol. 1987, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.G.; Card, J.W.; Racz, W.J. The role of mitochondrial injury in bromobenzene and furosemide induced hepatotoxicity. Toxicol. Lett. 2000, 116, 171–181. [Google Scholar] [CrossRef]
- Church, R.J.; Schomaker, S.J.; Eaddy, J.S.; Boucher, G.G.; Kreeger, J.M.; Aubrecht, J.; Watkins, P.B. Glutamate dehydrogenase as a biomarker for mitotoxicity; insights from furosemide hepatotoxicity in the mouse. PLoS ONE 2020, 15, e0240562. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Jaeschke, H. Biomarkers of Mitotoxicity after Acute Liver Injury: Further Insights into the Interpretation of Glutamate Dehydrogenase. J. Clin. Transl. Res. 2021, 7, 5. [Google Scholar] [CrossRef]
- McGill, M.R.; Li, F.; Sharpe, M.R.; Williams, C.D.; Curry, S.C.; Ma, X.; Jaeschke, H. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch. Toxicol. 2014, 88, 391–401. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Yan, K.; Pence, L.; Simpson, P.M.; Gill, P.; Letzig, L.G.; Beger, R.D.; Sullivan, J.E.; Kearns, G.L.; Reed, M.D.; et al. Targeted liquid chromatography–mass spectrometry analysis of serum acylcarnitines in acetaminophen toxicity in children. Biomark. Med. 2014, 8, 147–159. [Google Scholar] [CrossRef]
- Xie, Y.; McGill, M.R.; Du, K.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Ding, W.-X.; Jaeschke, H. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol. Appl. Pharmacol. 2015, 289, 213–222. [Google Scholar] [CrossRef]
- Craig, D.G.N.; Lee, P.; Pryde, E.A.; Masterton, G.S.; Hayes, P.C.; Simpson, K.J. Circulating apoptotic and necrotic cell death markers in patients with acute liver injury. Liver Int. 2011, 31, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, S.V.W.; Jang, Y.-J.; Fontana, R.J.; Omary, M.B. Carbamoyl phosphate synthetase-1 is a rapid turnover biomarker in mouse and human acute liver injury. Am. J. Physiol. Liver Physiol. 2014, 307, G355–G364. [Google Scholar] [CrossRef] [PubMed]
- Kwan, R.; Chen, L.; Park, M.-J.; Su, Z.; Weerasinghe, S.V.; Lee, W.M.; Durkalski-Mauldin, V.L.; Fontana, R.J.; Omary, M.B. The Role of Carbamoyl Phosphate Synthetase 1 as a Prognostic Biomarker in Patients with Acetaminophen-induced Acute Liver Failure. Clin. Gastroenterol. Hepatol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Julian, M.W.; Huff, J.E.; Struck, J.; Cook, C.H. Carbamoyl phosphate synthase-1: A marker of mitochondrial damage and depletion in the liver during sepsis. Crit. Care Med. 2006, 34, 2439–2446. [Google Scholar] [CrossRef] [PubMed]
- Leers, M.P.G.; Bergman, T.; Tribbick, G.; Persson, B.; Ramaekers, F.C.S.; Nap, M.; Schutte, B.; Kölgen, W.; Björklund, V.; Björklund, P.; et al. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. J. Pathol. 1999, 187, 567–572. [Google Scholar] [CrossRef]
- Ueno, T.; Toi, M.; Bivén, K.; Bando, H.; Ogawa, T.; Linder, S. Measurement of an apoptotic product in the sera of breast cancer patients. Eur. J. Cancer 2003, 39, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Havelka, A.M.; Ueno, T.; Shoshan, M.C. Determining tumor apoptosis and necrosis in patient serum using cytokeratin 18 as a biomarker. Cancer Lett. 2004, 214, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, A.E.; Hynan, L.S.; Borges, C.B.; Forcione, D.G.; Blackard, J.T.; Lin, W.; Gorman, A.R.; Shaikh, O.S.; Reuben, A.; Harrison, E.; et al. Serum Apoptosis Markers in Acute Liver Failure: A Pilot Study. Clin. Gastroenterol. Hepatol. 2007, 5, 1477–1483. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Marquitan, G.; Jochum, C.; Saner, F.; Gerken, G.; Canbay, A. Apoptosis versus necrosis rate as a predictor in acute liver failure following acetaminophen intoxication compared with acute-on-chronic liver failure. Liver Int. 2008, 28, 713–716. [Google Scholar] [CrossRef]
- Volkmann, X.; Anstaett, M.; Hadem, J.; Stiefel, P.; Bahr, M.J.; Lehner, F.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Caspase activation is associated with spontaneous recovery from acute liver failure. Hepatology 2008, 47, 1624–1633. [Google Scholar] [CrossRef]
- Gujral, J.S.; Knight, T.R.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Mode of Cell Death after Acetaminophen Overdose in Mice: Apoptosis or Oncotic Necrosis? Toxicol. Sci. 2002, 67, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Koerner, M.R.; Lampe, J.N.; Farhood, A.; Jaeschke, H. Mouse strain-dependent caspase activation during acetaminophen hepatotoxicity does not result in apoptosis or modulation of inflammation. Toxicol. Appl. Pharmacol. 2011, 257, 449–458. [Google Scholar] [CrossRef] [PubMed]
- McGregor, A.H.; More, L.J.; Simpson, K.J.; Harrison, D.J. Liver death and regeneration in paracetamol toxicity. Hum. Exp. Toxicol. 2003, 22, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Church, R.J.; Kullak-Ublick, G.A.; Aubrecht, J.; Bonkovsky, H.L.; Chalasani, N.; Fontana, R.J.; Goepfert, J.C.; Hackman, F.; King, N.M.P.; Kirby, S.; et al. Candidate biomarkers for the diagnosis and prognosis of drug-induced liver injury: An international collaborative effort. Hepatology 2019, 69, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Dear, J.W.; Clarke, J.I.; Francis, B.; Allen, L.; Wraight, J.; Shen, J.; Dargan, P.I.; Wood, D.; Cooper, J.; Thomas, S.H.L.; et al. Risk stratification after paracetamol overdose using mechanistic biomarkers: Results from two prospective cohort studies. Lancet Gastroenterol. Hepatol. 2018, 3, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Del Turco, S.; Navarra, T.; Lee, W.M. Circulating Levels of Soluble Receptor for Advanced Glycation End Products and Ligands of the Receptor for Advanced Glycation End Products in Patients with Acute Liver Failure. Liver Transplant. 2015, 21, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Ghallab, A.; Hassan, R.; Hofmann, U.; Friebel, A.; Hobloss, Z.; Brackhagen, L.; Begher-Tibbe, B.; Myllys, M.; Reinders, J.; Overbeck, N.; et al. Interruption of bile acid uptake by hepatocytes after acetaminophen overdose ameliorates hepatotoxicity. J. Hepatol. 2022, 77, 71–83. [Google Scholar] [CrossRef]
- Bhushan, B.; Borude, P.; Edwards, G.; Walesky, C.; Cleveland, J.; Li, F.; Ma, X.; Apte, U. Role of Bile Acids in Liver Injury and Regeneration following Acetaminophen Overdose. Am. J. Pathol. 2013, 183, 1518–1526. [Google Scholar] [CrossRef]
- Woolbright, B.L.; McGill, M.R.; Staggs, V.S.; Winefield, R.D.; Gholami, P.; Olyaee, M.; Sharpe, M.R.; Curry, S.C.; Lee, W.M.; Jaeschke, H.; et al. Glycodeoxycholic Acid Levels as Prognostic Biomarker in Acetaminophen-Induced Acute Liver Failure Patients. Toxicol. Sci. 2014, 142, 436–444. [Google Scholar] [CrossRef]
- Luo, L.; Aubrecht, J.; Li, D.; Warner, R.L.; Johnson, K.J.; Kenny, J.; Colangelo, J.L. Assessment of serum bile acid profiles as biomarkers of liver injury and liver disease in humans. PLoS ONE 2018, 13, e0193824. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, H.P.; Vaidya, V.S.; Wang, Z.; Peng, Q.; Hyde, C.; Potter, D.; Wang, J.; Zong, Q.; Arat, S.; Martin, M.; et al. Evaluating the Sensitivity and Specificity of Promising Circulating Biomarkers to Diagnose Liver Injury in Humans. Toxicol. Sci. 2021, 181, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J.H.; McGill, M.R. Redrawing the Map to Novel DILI Biomarkers in Circulation: Where Are We, Where Should We Go, and How Can We Get There? Livers 2021, 1, 286–293. [Google Scholar] [CrossRef]
- Jaeschke, H.; Williams, C.D.; Ramachandran, A.; Bajt, M.L. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 2012, 32, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A. Mechanisms and pathophysiological significance of sterile inflammation during acetaminophen hepatotoxicity. Food Chem. Toxicol. 2020, 138, 111240. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Pilaro, A.M.; Ji, S. Potential role of activated macrophages in acetaminophen hepatotoxicity: II. Mechanism of macrophage accumulation and activation. Toxicol. Appl. Pharmacol. 1986, 86, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Pilaro, A.M. Potential role of activated macrophages in acetaminophen hepatotoxicity: I. Isolation and characterization of activated macrophages from rat liver. Toxicol. Appl. Pharmacol. 1986, 86, 204–215. [Google Scholar] [CrossRef]
- Laskin, D.L.; Gardner, C.R.; Price, V.F.; Jollow, D.J. Modulation of macrophage functioning abrogates the acute hepatotoxicity of acetaminophen. Hepatology 1995, 21, 1045–1050. [Google Scholar] [CrossRef]
- Ju, C.; Reilly, T.P.; Bourdi, M.; Radonovich, M.F.; Brady, J.N.; George, J.W.; Pohl, L.R. Protective Role of Kupffer Cells in Acetaminophen-Induced Hepatic Injury in Mice. Chem. Res. Toxicol. 2002, 15, 1504–1513. [Google Scholar] [CrossRef]
- Clemens, M.M.; Vazquez, J.H.; Kennon-McGill, S.; McCullough, S.S.; James, L.P.; McGill, M.R. Pre-treatment twice with liposomal clodronate protects against acetaminophen hepatotoxicity through a pre-conditioning effect. Liver Res. 2020, 4, 145–152. [Google Scholar] [CrossRef]
- James, L.P.; McCullough, S.S.; Knight, T.R.; Jaeschke, H.; Hinson, J.A. Acetaminophen Toxicity in Mice Lacking NADPH Oxidase Activity: Role of Peroxynitrite Formation and Mitochondrial Oxidant Stress. Free. Radic. Res. 2003, 37, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.P.; Cheng, L.; Ju, C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 2008, 84, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Investig. 2009, 119, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Antoine, D.J.; Shaw, P.J.; Benson, C.; Farhood, A.; Williams, D.P.; Kanneganti, T.-D.; Park, B.K.; Jaeschke, H. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol. Appl. Pharmacol. 2011, 252, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Bajt, M.L.; Sharpe, M.R.; McGill, M.R.; Farhood, A.; Jaeschke, H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol. Appl. Pharmacol. 2014, 275, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Umbaugh, D.S.; Smith, S.; Adelusi, O.B.; Sanchez-Guerrero, G.; Ramachandran, A.; Jaeschke, H. Dose-dependent pleiotropic role of neutrophils during acetaminophen-induced liver injury in male and female mice. Arch. Toxicol. 2023, 97, 1397–1412. [Google Scholar] [CrossRef]
- James, L.P.; Farrar, H.C.; Darville, T.L.; Sullivan, J.E.; Givens, T.G.; Kearns, G.L.; Do, G.S.W.; Simpson, P.M.; Hinson, J.A. Elevation of serum interleukin 8 levels in acetaminophen overdose in children and adolescents. Clin. Pharmacol. Ther. 2001, 70, 280–286. [Google Scholar] [CrossRef]
- Williams, A.M.; Langley, P.G.; Osei-Hwediah, J.; Wendon, J.A.; Hughes, R.D. Hyaluronic acid and endothelial damage due to paracetamol-induced hepatotoxicity. Liver Int. 2003, 23, 110–115. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Nguyen, N.T.; McGill, M.R.; Sharpe, M.R.; Curry, S.C.; Jaeschke, H. Generation of pro-and anti-inflammatory mediators after acetaminophen overdose in surviving and non-surviving patients. Toxicol. Lett. 2022, 367, 59–66. [Google Scholar] [CrossRef]
- James, L.P.; Simpson, P.M.; Farrar, H.C.; Kearns, G.L.; Wasserman, G.S.; Blumer, J.L.; Reed, M.D.; Sullivan, J.E.; Hinson, J.A. Cytokines and Toxicity in Acetaminophen Overdose. J. Clin. Pharmacol. 2005, 45, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Steuerwald, N.M.; Foureau, D.M.; Norton, H.J.; Zhou, J.; Parsons, J.C.; Chalasani, N.; Fontana, R.J.; Watkins, P.B.; Lee, W.M.; Reddy, K.R.; et al. Profiles of Serum Cytokines in Acute Drug-Induced Liver Injury and Their Prognostic Significance. PLoS ONE 2013, 8, e81974. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.G.; Lee, P.; Pryde, E.A.; Walker, S.W.; Beckett, G.J.; Hayes, P.C.; Simpson, K.J. Elevated levels of the long pentraxin 3 in paracetamol-induced human acute liver injury. Eur. J. Gastroenterol. Hepatol. 2013, 25, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Slowik, V.; the Acute Liver Failure Study Group; Borude, P.; Jaeschke, H.; Woolbright, B.L.; Lee, W.M.; Apte, U. Leukocyte cell derived chemotaxin-2 (Lect2) as a predictor of survival in adult acute liver failure. Transl. Gastroenterol. Hepatol. 2019, 4, 17. [Google Scholar] [CrossRef]
- Craig, D.G.; Lee, P.; Pryde, E.A.; Hayes, P.C.; Simpson, K.J. Serum Neopterin and Soluble CD163 as Markers of Macrophage Activation in Paracetamol (Acetaminophen)-Induced Human Acute Liver Injury. Aliment. Pharmacol. Ther. 2013, 38, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Marzolf, B.; Troisch, P.; Brightman, A.; Hu, Z.; Hood, L.E.; Galas, D.J. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc. Natl. Acad. Sci. USA 2009, 106, 4402–4407. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.J.S.; Dear, J.; Platt, V.; Simpson, K.J.; Craig, D.G.; Antoine, D.J.; French, N.S.; Dhaun, N.; Webb, D.J.; Costello, E.M.; et al. Circulating microRNAs as potential markers of human drug-induced liver injury. Hepatology 2011, 54, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Kanchagar, C.; Veksler-Lublinsky, I.; Lee, R.C.; McGill, M.R.; Jaeschke, H.; Curry, S.C.; Ambros, V.R. Circulating microRNA profiles in human patients with acetaminophen hepatotoxicity or ischemic hepatitis. Proc. Natl. Acad. Sci. USA 2014, 111, 12169–12174. [Google Scholar] [CrossRef]
- Krauskopf, J.; Caiment, F.; Claessen, S.M.; Johnson, K.J.; Warner, R.L.; Schomaker, S.J.; Burt, D.A.; Aubrecht, J.; Kleinjans, J.C. Application of High-Throughput Sequencing to Circulating microRNAs Reveals Novel Biomarkers for Drug-Induced Liver Injury. Toxicol. Sci. 2015, 143, 268–276. [Google Scholar] [CrossRef]
- Yang, X.; Salminen, W.F.; Shi, Q.; Greenhaw, J.; Gill, P.S.; Bhattacharyya, S.; Beger, R.D.; Mendrick, D.L.; Mattes, W.B.; James, L.P. Potential of extracellular microRNAs as biomarkers of acetaminophen toxicity in children. Toxicol. Appl. Pharmacol. 2015, 284, 180–187. [Google Scholar] [CrossRef]
- Krauskopf, J.; De Kok, T.M.; Schomaker, S.J.; Gosink, M.; Burt, D.A.; Chandler, P.; Warner, R.L.; Johnson, K.J.; Caiment, F.; Kleinjans, J.C.; et al. Serum microRNA signatures as "liquid biopsies" for interrogating hepatotoxic mechanisms and liver pathogenesis in human. PLoS ONE 2017, 12, e0177928. [Google Scholar] [CrossRef] [PubMed]
- Carreiro, S.; Marvel-Coen, J.; Lee, R.; Chapman, B.; Ambros, V. Circulating microRNA Profiles in Acetaminophen Toxicity. J. Med. Toxicol. 2020, 16, 177–187. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Jaeschke, H. MicroRNAs as Signaling Mediators and Biomarkers of Drug- and Chemical-Induced Liver Injury. J. Clin. Med. 2015, 4, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W. Biomarkers for risk assessment in paracetamol hepatotoxicity. Lancet Gastroenterol. Hepatol. 2018, 3, 76–77. [Google Scholar] [CrossRef] [PubMed]
- McCrae, J.C.; Sharkey, N.; Webb, D.J.; Vliegenthart, A.D.B.; Dear, J.W. Ethanol consumption produces a small increase in circulating miR-122 in healthy individuals. Clin. Toxicol. 2016, 54, 53–55. [Google Scholar] [CrossRef] [PubMed]
- López-Longarela, B.; Morrison, E.E.; Tranter, J.D.; Chahman-Vos, L.; Léonard, J.-F.; Gautier, J.-C.; Laurent, S.; Lartigau, A.; Boitier, E.; Sautier, L.; et al. Direct Detection of miR-122 in Hepatotoxicity Using Dynamic Chemical Labeling Overcomes Stability and isomiR Challenges. Anal. Chem. 2020, 92, 3388–3395. [Google Scholar] [CrossRef] [PubMed]
- Kersaudy-Kerhoas, M.; Liga, A.; Roychoudhury, A.; Stamouli, M.; Grant, R.; Carrera, D.S.; Schulze, H.; Mielczarek, W.; Oosthuyzen, W.; Quintana, J.F.; et al. Microfluidic system for near-patient extraction and detection of miR-122 microRNA biomarker for drug-induced liver injury diagnostics. Biomicrofluidics 2022, 16, 024108. [Google Scholar] [CrossRef]
- Roychoudhury, A.; Dear, J.W.; Kersaudy-Kerhoas, M.; Bachmann, T.T. Amplification-free electrochemical biosensor detection of circulating microRNA to identify drug-induced liver injury. Biosens. Bioelectron. 2023, 231, 115298. [Google Scholar] [CrossRef]
- Tavabie, O.D.; Karvellas, C.J.; Salehi, S.; Speiser, J.L.; Rose, C.F.; Menon, K.; Prachalias, A.; Heneghan, M.A.; Agarwal, K.; Lee, W.M.; et al. A novel microRNA-based prognostic model outperforms standard prognostic models in patients with acetaminophen-induced acute liver failure. J. Hepatol. 2021, 75, 424–434. [Google Scholar] [CrossRef]
- John, K.; Hadem, J.; Krech, T.; Wahl, K.; Manns, M.P.; Dooley, S.; Batkai, S.; Thum, T.; Schulze-Osthoff, K.; Bantel, H. MicroRNAs play a role in spontaneous recovery from acute liver failure. Hepatology 2014, 60, 1346–1355. [Google Scholar] [CrossRef]
- Salehi, S.; Brereton, H.C.; Arno, M.J.; Darling, D.; Quaglia, A.; O’grady, J.; Heaton, N.; Aluvihare, V.R. Human Liver Regeneration Is Characterized by the Coordinated Expression of Distinct MicroRNA Governing Cell Cycle Fate. Am. J. Transplant. 2013, 13, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Salehi, S.; Tavabie, O.D.; Verma, S.; McPhail, M.J.W.; Farzaneh, F.; Bernal, W.; Menon, K.; Agarwal, K.; Aluvihare, V.R. Serum MicroRNA Signatures in Recovery from Acute and Chronic Liver Injury and Selection for Liver Transplantation. Liver Transplant. 2020, 26, 811–822. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, J.G.; Alexander, G.J.; Hayllar, K.M.; Williams, R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989, 97, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Malinchoc, M.; Kamath, P.S.; Gordon, F.D.; Peine, C.J.; Rank, J.; ter Borg, P.C. A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology 2000, 31, 864–871. [Google Scholar] [CrossRef]
- Koch, D.G.; Tillman, H.; Durkalski, V.; Lee, W.M.; Reuben, A. Development of a Model to Predict Transplant-free Survival of Patients with Acute Liver Failure. Clin. Gastroenterol. Hepatol. 2016, 14, 1199–1206.e2. [Google Scholar] [CrossRef] [PubMed]
- Walesky, C.M.; Kolb, K.E.; Winston, C.L.; Henderson, J.; Kruft, B.; Fleming, I.; Ko, S.; Monga, S.P.; Mueller, F.; Apte, U.; et al. Functional compensation precedes recovery of tissue mass following acute liver injury. Nat. Commun. 2020, 11, 5785. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.E.; Dalhoff, K. Alpha-fetoprotein is a predictor of outcome in acetaminophen-induced liver injury. Hepatology 2005, 41, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.E.; Dalhoff, K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002, 36, 659–665. [Google Scholar] [CrossRef]
- Rudnick, D.A.; Dietzen, D.J.; Turmelle, Y.P.; Shepherd, R.; Zhang, S.; Belle, S.H.; Squires, R.; Karpen, S.; Dell Olio, D.; Squires, R.; et al. Serum α-NH2-Butyric Acid May Predict Spontaneous Survival in Pediatric Acute Liver Failure. Pediatr. Transplant. 2009, 13, 223–230. [Google Scholar] [CrossRef]
- Schmelzle, M.; Splith, K.; Andersen, L.W.; Kornek, M.; Schuppan, D.; Jones-Bamman, C.; Nowak, M.; Toxavidis, V.; Salhanick, S.D.; Han, L.; et al. Increased Plasma Levels of Microparticles Expressing CD39 and CD133 in Acute Liver Injury. Transplantation 2013, 95, 63–69. [Google Scholar] [CrossRef]
- Umbaugh, D.; Nguyen, N.; Guerrero, G.; Villaneuva, C.; Hagenbuch, B.; Ramachandran, A.; Jaeschke, H. Hepatocyte Senescence Is Initiated through a Klf6-P21 Mechanism That Mediates the Production of Cxcl14, a Novel Prognostic Biomarker of Acute Liver Failure. Toxicologist 2023, S3, 587. [Google Scholar]
- Lutkewitte, A.J.; Schweitzer, G.G.; Kennon-McGill, S.; Clemens, M.M.; James, L.P.; Jaeschke, H.; Finck, B.N.; McGill, M.R. Lipin deactivation after acetaminophen overdose causes phosphatidic acid accumulation in liver and plasma in mice and humans and enhances liver regeneration. Food Chem. Toxicol. 2018, 115, 273–283. [Google Scholar] [CrossRef]
- Clemens, M.M.; Kennon-McGill, S.; Apte, U.; James, L.P.; Finck, B.N.; McGill, M.R. The inhibitor of glycerol 3-phosphate acyltransferase FSG67 blunts liver regeneration after acetaminophen overdose by altering GSK3β and Wnt/β-catenin signaling. Food Chem. Toxicol. 2019, 125, 279–288. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Cao, M.; Svetlov, A.; Sharpe, M.R.; Williams, C.D.; Curry, S.C.; Farhood, A.; Jaeschke, H.; Svetlov, S.I. Argininosuccinate synthetase as a plasma biomarker of liver injury after acetaminophen overdose in rodents and humans. Biomarkers 2014, 19, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J.H.; Clemens, M.M.; Allard, F.D.; Yee, E.U.; Kennon-McGill, S.; Mackintosh, S.G.; Jaeschke, H.; Hambuchen, M.D.; McGill, M.R. Identification of Serum Biomarkers to Distinguish Hazardous and Benign Aminotransferase Elevations. Toxicol. Sci. 2019, 173, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Rule, J.A.; Hynan, L.S.; Attar, N.; Sanders, C.; Korzun, W.J.; Lee, W.M.; Larson, A.M.; Liou, I.; Davern, T.; Fix, O.; et al. Procalcitonin Identifies Cell Injury, Not Bacterial Infection, in Acute Liver Failure. PLoS ONE 2015, 10, e0138566. [Google Scholar] [CrossRef] [PubMed]
- Mallet, M.; Haq, M.; Tripon, S.; Bernard, M.; Benosman, H.; Thabut, D.; Rudler, M. Elevated procalcitonin is associated with bacterial infection during acute liver failure only when unrelated to acetaminophen intoxication. Eur. J. Gastroenterol. Hepatol. 2017, 29, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, A.; Salem, S.; Malissin, I.; Diallo, A.; Deye, N.; Goury, A.; Gourlain, H.; Péron, N.; Vicaut, E.; Voicu, S.; et al. Plasma procalcitonin may be an early predictor of liver injury in acetaminophen poisoning: A prospective cohort study. United Eur. Gastroenterol. J. 2021, 9, 571–580. [Google Scholar] [CrossRef]
- Mikus, M.; Drobin, K.; Gry, M.; Bachmann, J.; Lindberg, J.; Yimer, G.; Aklillu, E.; Makonnen, E.; Aderaye, G.; Roach, J.; et al. Elevated levels of circulating CDH5 and FABP1 in association with human drug-induced liver injury. Liver Int. 2017, 37, 132–140. [Google Scholar] [CrossRef]
- Ghandforoush-Sattari, M.; Mashayekhi, S. Evaluation of taurine as a biomarker of liver damage in paracetamol poisoning. Eur. J. Pharmacol. 2008, 581, 171–176. [Google Scholar] [CrossRef]
- Gai, Z.; Samodelov, S.L.; Alecu, I.; Hornemann, T.; Grove, J.I.; Aithal, G.P.; Visentin, M.; Kullak-Ublick, G.A. Plasma Sphingoid Base Profiles of Patients Diagnosed with Intrinsic or Idiosyncratic Drug-induced Liver Injury. Int. J. Mol. Sci. 2023, 24, 3013. [Google Scholar] [CrossRef] [PubMed]
- Westerink, W.M.; Schoonen, W.G. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. Vitr. 2007, 21, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.N.; Li, Y.; Nakamoto, K.; Subileau, E.-A.; Steen, D.; Zhong, X.-B. A Comparison of Whole Genome Gene Expression Profiles of HepaRG Cells and HepG2 Cells to Primary Human Hepatocytes and Human Liver Tissues. Drug Metab. Dispos. 2010, 38, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Ewart, L.; Apostolou, A.; Briggs, S.A.; Carman, C.V.; Chaff, J.T.; Heng, A.R.; Jadalannagari, S.; Janardhanan, J.; Jang, K.-J.; Joshipura, S.R.; et al. Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology. Commun. Med. 2022, 2, 154. [Google Scholar] [CrossRef] [PubMed]
- Maschmeyer, I.; Hasenberg, T.; Jaenicke, A.; Lindner, M.; Lorenz, A.K.; Zech, J.; Garbe, L.-A.; Sonntag, F.; Hayden, P.; Ayehunie, S.; et al. Chip-based human liver–intestine and liver–skin co-cultures—A first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 2015, 95, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Zhou, X.; Geng, X.; Drewell, C.; Hübner, J.; Li, Z.; Zhang, Y.; Xue, M.; Marx, U.; Li, B. Repeated dose multi-drug testing using a microfluidic chip-based coculture of human liver and kidney proximal tubules equivalents. Sci. Rep. 2020, 10, 8879. [Google Scholar] [CrossRef] [PubMed]
- Novak, R.; Ingram, M.; Marquez, S.; Das, D.; Delahanty, A.; Herland, A.; Maoz, B.M.; Jeanty, S.S.F.; Somayaji, M.R.; Burt, M.; et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng. 2020, 4, 407–420. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Teles, D.; Yeager, K.; Tavakol, D.N.; Zhao, Y.; Chramiec, A.; Tagore, S.; Summers, M.; Stylianos, S.; Tamargo, M.; et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 2022, 6, 351–371. [Google Scholar] [CrossRef]
- Schomaker, S.; Potter, D.; Warner, R.; Larkindale, J.; King, N.; Porter, A.C.; Owens, J.; Tomlinson, L.; Sauer, J.-M.; Johnson, K.; et al. Serum glutamate dehydrogenase activity enables early detection of liver injury in subjects with underlying muscle impairments. PLoS ONE 2020, 15, e0229753. [Google Scholar] [CrossRef]
- Miyazaki, M.; Rosenblum, J.S.; Kasahara, Y.; Nakagawa, I.; Patricelli, M.P. Determination of enzymatic source of alanine aminotransferase activity in serum from dogs with liver injury. J. Pharmacol. Toxicol. Methods 2009, 60, 307–315. [Google Scholar] [CrossRef]
- Yang, R.-Z.; Park, S.; Reagan, W.J.; Goldstein, R.; Zhong, S.; Lawton, M.; Rajamohan, F.; Qian, K.; Liu, L.; Gong, D.-W. Alanine aminotransferase isoenzymes: Molecular cloning and quantitative analysis of tissue expression in rats and serum elevation in liver toxicity. Hepatology 2009, 49, 598–607. [Google Scholar] [CrossRef]
- Rafter, I.; Gråberg, T.; Kotronen, A.; Strömmer, L.; Mattson, C.M.; Kim, R.W.; Ehrenborg, E.; Andersson, H.B.; Yki-Järvinen, H.; Schuppe-Koistinen, I.; et al. Isoform-specific alanine aminotransferase measurement can distinguish hepatic from extrahepatic injury in humans. Int. J. Mol. Med. 2012, 30, 1241–1249. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker (s)/Tool (s) | Purpose | Reference (s) |
|---|---|---|
| * ALT, AST | Detection of ALI | [20,22,32,33] |
| # ALT >50 U/L at admission | Prediction of ALI (high NPV) | [41,42] |
| # Declining transaminases or AST/ALT <0.4 | Discontinuation of NAC | [32,37] |
| # APAP × AT | Prediction of ALI | [50] |
| # LDH | Prediction of death | [39,49] |
| MDH1 | Detection of ALI; prediction of death | [49,57,58] |
| * Rumack–Matthew nomogram | Prediction of ALI | [63] |
| # Psi parameter | Prediction of ALI | [77,78,79,80] |
| * APAP-protein adducts | Mechanistic: Protein alkylation; Diagnosis of APAP hepatotoxicity | [81,82,83,84,85,86] |
| # 95% probability intervals for APAP-protein adducts | Diagnosis of APAP hepatotoxicity | [67] |
| GLDH | Detection of ALI; Mechanistic: Mitochondrial damage; Prediction of ALI | [36,38,119,137] |
| mtDNA | Mechanistic: Mitochondrial damage | [36,38] |
| Nuclear DNA fragments | Mechanistic: Mitochondrial damage, DNA fragmentation | [36,38,123] |
| Acylcarnitines | Mechanistic: Mitochondrial damage | [120,121] |
| CPS1 | Mechanistic: Mitochondrial damage; Prediction of death | [124,125] |
| Total K18 | Mechanistic: Cell death mode; Prediction of ALI | [123,131,132,137] |
| ccK18 | Mechanistic: Cell death mode | [123,131,132] |
| Caspase 3 activity | Mechanistic: Cell death mode | [36] |
| HMGB1 | Mechanistic: Cell death mode; Prediction of ALI | [123,137,138] |
| Bile acids (e.g., glycodeoxycholic acid) | Mechanistic: Bile acid toxicity?; Prediction of death | [140,142,143] |
| Cytokines/chemokines | Inflammation markers | [160,161,162,163,165,166,167] |
| miR-122 | Prediction of ALI | [137] |
| miRNA regeneration profiles | Regeneration markers | [181,182,183,184] |
| # AFP | Regeneration marker | [189] |
| # Phosphate | Regeneration marker | [190] |
| α-NH-butyric acid | Regeneration marker | [191] |
| Lect2 | Regeneration marker | [166] |
| CXCL14 | Regeneration marker | [193] |
| Phosphatidic acid species | Possible regeneration markers | [194,195] |
| ASS1 | Detection of ALI | [49,196,197] |
| ADH1, ALDH1A1, FBP1 | Detection of ALI | [197] |
| FABP1 | Detection of ALI; Regeneration marker | [201] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGill, M.R.; Curry, S.C. The Evolution of Circulating Biomarkers for Use in Acetaminophen/Paracetamol-Induced Liver Injury in Humans: A Scoping Review. Livers 2023, 3, 569-596. https://doi.org/10.3390/livers3040039
McGill MR, Curry SC. The Evolution of Circulating Biomarkers for Use in Acetaminophen/Paracetamol-Induced Liver Injury in Humans: A Scoping Review. Livers. 2023; 3(4):569-596. https://doi.org/10.3390/livers3040039
Chicago/Turabian StyleMcGill, Mitchell R., and Steven C. Curry. 2023. "The Evolution of Circulating Biomarkers for Use in Acetaminophen/Paracetamol-Induced Liver Injury in Humans: A Scoping Review" Livers 3, no. 4: 569-596. https://doi.org/10.3390/livers3040039
APA StyleMcGill, M. R., & Curry, S. C. (2023). The Evolution of Circulating Biomarkers for Use in Acetaminophen/Paracetamol-Induced Liver Injury in Humans: A Scoping Review. Livers, 3(4), 569-596. https://doi.org/10.3390/livers3040039