
Oxidative Stress in Non-Alcoholic Fatty Liver Disease

 , ,
, ,  ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Evidences on the Presence of Oxidative Stress in NAFLD
2.1. Evidence for a Role of Oxidative Stress in NAFLD
2.2. Redox Regulation of Lipid Metabolism in NAFLD
2.3. Oxidative Stress and Antioxidant Markers in NAFLD and Other Metabolic Diseases
3. Evidences on the Role of Oxidative Stress on NAFLD Progression
3.1. Markers of Immunological Responses to Inflammation and Oxidative Stress during NAFLD Evolution
3.2. Mitochondrial Oxidative Injury as a Key Pathway That Links Saturated Fat Intake to the Development and Progression of NAFLD
3.2.1. Mitochondrial Dysfunction and Oxidative Stress in NAFLD/NASH
3.2.2. Diagnostic Tools for Mitochondrial Dysfunction in NAFLD
3.3. Gut Microbiota and Oxidative Stress in NAFLD, with Possible Therapeutic Implications
3.3.1. Role of Microbiota in Hepatic Steatosis and Oxidative Stress
3.3.2. Modulation of Gut-Liver Axis: Effects on Oxidative Stress
3.4. Relationship of Oxidative Stress and the Development and/or Progression of NAFLD-HCC
3.5. Biomarkers of Oxidative Stress in NAFLD/NASH
4. Genetic Polymorphisms and Oxidative Stress in NAFLD/NASH
4.1. General Premises
4.2. Single Nucleotide Polymorphisms
4.3. Other Genetic Variants
4.4. Conclusive Remarks
5. Therapeutic Potential of Targeting Oxidative Stress in NAFLD Patients
5.1. Classical Antioxidants: Vitamin C and Vitamin E
5.2. Coffee Components: Caffeine and Coffee Polyphenols
5.3. Metformin
5.4. Hesperetin
5.5. Silymarin
5.6. Other Compounds
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Khan, F.Z.; Perumpail, R.B.; Wong, R.J.; Ahmed, A. Advances in hepatocellular carcinoma: Nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2155–2161. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Kojima, T.; Itoh, Y.; Harano, Y.; Fujii, K.; Nakajima, T.; Kato, T.; Takeda, N.; Okuda, J.; Ida, K.; et al. The severity of ultrasonographic findings in nonalcoholic fatty liver disease reflects the metabolic syndrome and visceral fat accumulation. Am. J. Gastroenterol. 2007, 102, 2708–2715. [Google Scholar] [CrossRef]
- Shenoy-Bhangle, A.; Baliyan, V.; Kordbacheh, H.; Guimaraes, A.R.; Kambadakone, A. Diffusion weighted magnetic resonance imaging of liver: Principles, clinical applications and recent updates. World J. Hepatol. 2017, 9, 1081–1091. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Mato, J.M.; Lu, S.C. Nonalcoholic fatty liver disease: Update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp. Biol. Med. 2015, 240, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, B.; Zhao, J.; Feng, Q.; Peng, J.; Hu, Y.; Zhao, Y. Biological mechanisms and related natural modulators of liver X receptor in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2019, 113, 108778. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Targher, G.; Marchesini, G.; Byrne, C.D. Risk of type 2 diabetes in patients with non-alcoholic fatty liver disease: Causal association or epiphenomenon? Diabetes Metab. 2016, 42, 142–156. [Google Scholar] [CrossRef]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef]
- Dai, W.; Ye, L.; Liu, A.; Wen, S.W.; Deng, J.; Wu, X.; Lai, Z. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: A meta-analysis. Medicine 2017, 96, e8179. [Google Scholar] [CrossRef]
- Tesfay, M.; Goldkamp, W.J.; Neuschwander-Tetri, B.A. NASH: The Emerging Most Common Form of Chronic Liver Disease. Mo. Med. 2018, 115, 225. [Google Scholar]
- Polimeni, L.; del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336. [Google Scholar] [CrossRef]
- Vergara, D.; Casadei-Gardini, A.; Giudetti, A.M. Oxidative Molecular Mechanisms Underlying Liver Diseases: From Systems Biology to the Personalized Medicine. Oxid. Med. Cell. Longev. 2019, 2019, 7864316. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Hruszkewycz, A.M. Evidence for mitochondrial DNA damage by lipid peroxidation. Biochem. Biophys. Res. Commun. 1988, 153, 191–197. [Google Scholar] [CrossRef]
- Chen, J.; Schenker, S.; Frosto, T.A.; Henderson, G.I. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme subunits. Biochim. Biophys. Acta 1998, 1380, 336–344. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Gambino, R.; Musso, G.; Cassader, M. Redox balance in the pathogenesis of nonalcoholic fatty liver disease: Mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1325–1365. [Google Scholar] [CrossRef]
- Sastre, J.; Pallardó, F.V.; Llopis, J.; Furukawa, T.; Vinã, J.R.; Viña, J. Glutathione depletion by hyperphagia-induced obesity. Life Sci. 1989, 45, 183–187. [Google Scholar] [CrossRef]
- Strauss, R.S.; Barlow, S.E.; Dietz, W.H. Prevelance of abnormal serum aminotransferase values in overweight and obese adolescents. J. Pediatr. 2000, 136, 727–733. [Google Scholar] [CrossRef]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef]
- George, D.K.; Goldwurm, S.; Macdonald, G.A.; Cowley, L.L.; Walker, N.I.; Ward, P.J.; Jazwinska, E.C.; Powell, L.W. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998, 114, 311–318. [Google Scholar] [CrossRef]
- Chitturi, S.; Weltman, M.; Farrell, G.C.; McDonald, D.; Liddle, C.; Samarasinghe, D.; Lin, R.; Abeygunasekera, S.; George, J. HFE mutations, hepatic iron, and fibrosis: Ethnic-specific association of NASH with C282Y but not with fibrotic severity. Hepatology 2002, 36, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Crawford, D.H.; Stuart, K.; House, M.J.; St. Pierre, T.G.; Webb, M.; Ching, H.L.I.; Kava, J.; Bynevelt, M.; Macquillan, G.C.; et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective, randomized, controlled trial. Hepatology 2015, 61, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Caputi, A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J. Gastroenterol. 2011, 17, 3785–3794. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lee, W.Y.; Lee, Y.S.; Rhee, E.J.; Kim, S.W. The relative effects of obesity and insulin resistance on cardiovascular risk factors in nondiabetic and normotensive men. Korean J. Intern. Med. 2004, 19, 75–80. [Google Scholar] [CrossRef]
- Loomba, R.; Schork, N.; Chen, C.H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Genetic Factors in the Pathogenesis of Nonalcoholic Fatty Liver and Steatohepatitis. Biomed Res. Int. 2015, 2015, 460190. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.S.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef]
- Wigg, A.J.; Roberts-Thomson, I.C.; Grose, R.H.; Cummins, A.G.; Dymock, R.B.; McCarthy, P.J. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 38–42. [Google Scholar] [CrossRef] [PubMed]
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology 2009, 136, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Kurtjones, E.A.; Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011, 140, 697–708.e4. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Csak, T.; Ganz, M.; Szabo, G. Differences in innate immune signaling between alcoholic and non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2013, 28, 93. [Google Scholar] [CrossRef]
- Konerman, M.A.; Jones, J.C.; Harrison, S.A. Pharmacotherapy for NASH: Current and emerging. J. Hepatol. 2018, 68, 362–375. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, Á.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Corrigendum to “Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease”. Oxid. Med. Cell. Longev. 2021, 2021, 9757921. [Google Scholar] [CrossRef]
- Klisic, A.; Kavaric, N.; Ninic, A.; Kotur-Stevuljevic, J. Oxidative stress and cardiometabolic biomarkers in patients with non-alcoholic fatty liver disease. Sci. Rep. 2021, 11, S41598–S416021. [Google Scholar] [CrossRef] [PubMed]
- Warolin, J.; Coenen, K.R.; Kantor, J.L.; Whitaker, L.E.; Wang, L.; Acra, S.A.; Roberts, L.J.; Buchowski, M.S. The relationship of oxidative stress, adiposity and metabolic risk factors in healthy Black and White American youth. Pediatr. Obes. 2014, 9, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Codoñer-Franch, P.; Tavárez-Alonso, S.; Murria-Estal, R.; Tortajada-Girbés, M.; Simó-Jordá, R.; Alonso-Iglesias, E. Elevated advanced oxidation protein products (AOPPs) indicate metabolic risk in severely obese children. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Hermsdorff, H.H.M.; Barbosa, K.B.; Volp, A.C.P.; Puchau, B.; Bressan, J.; Zulet, M.Á.; Martínez, J.A. Gender-specific relationships between plasma oxidized low-density lipoprotein cholesterol, total antioxidant capacity, and central adiposity indicators. Eur. J. Prev. Cardiol. 2014, 21, 884–891. [Google Scholar] [CrossRef]
- Karaouzene, N.; Merzouk, H.; Aribi, M.; Merzouk, S.A.; Yahia Berrouiguet, A.; Tessier, C.; Narce, M. Effects of the association of aging and obesity on lipids, lipoproteins and oxidative stress biomarkers: A comparison of older with young men. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 792–799. [Google Scholar] [CrossRef]
- Gabbia, D.; Cannella, L.; Martin, S. De The Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef]
- Sies, H.; Stahl, W.; Sevanian, A. Nutritional, dietary and postprandial oxidative stress. J. Nutr. 2005, 135, 96–972. [Google Scholar] [CrossRef]
- Dandona, P.; Ghanim, H.; Chaudhuri, A.; Dhindsa, S.; Kim, S.S. Macronutrient intake induces oxidative and inflammatory stress: Potential relevance to atherosclerosis and insulin resistance. Exp. Mol. Med. 2010, 42, 245–253. [Google Scholar] [CrossRef]
- Serra, D.; Mera, P.; Malandrino, M.I.; Mir, J.F.; Herrero, L. Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 2013, 19, 269–284. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Kobayashi, S.; Seo, H.G. Mutual interaction between oxidative stress and endoplasmic reticulum stress in the pathogenesis of diseases specifically focusing on non-alcoholic fatty liver disease. World J. Biol. Chem. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Mlinar, B.; Marc, J. New insights into adipose tissue dysfunction in insulin resistance. Clin. Chem. Lab. Med. 2011, 49, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Ghanim, H.; Ravishankar, S.; Chang, L.S.; Viswanathan, P.; Mohanty, P.; Dandona, P. Prolonged reactive oxygen species generation and nuclear factor-kappaB activation after a high-fat, high-carbohydrate meal in the obese. J. Clin. Endocrinol. Metab. 2007, 92, 4476–4479. [Google Scholar] [CrossRef]
- Bełtowski, J. Leptin and the regulation of endothelial function in physiological and pathological conditions. Clin. Exp. Pharmacol. Physiol. 2012, 39, 168–178. [Google Scholar] [CrossRef]
- Bondia-Pons, I.; Ryan, L.; Martinez, J.A. Oxidative stress and inflammation interactions in human obesity. J. Physiol. Biochem. 2012, 68, 701–711. [Google Scholar] [CrossRef]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic syndrome: A comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef]
- Crujeiras, A.B.; Díaz-Lagares, A.; Carreira, M.C.; Amil, M.; Casanueva, F.F. Oxidative stress associated to dysfunctional adipose tissue: A potential link between obesity, type 2 diabetes mellitus and breast cancer. Free Radic. Res. 2013, 47, 243–256. [Google Scholar] [CrossRef]
- Li, S.; Hong, M.; Tan, H.Y.; Wang, N.; Feng, Y. Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 4234061. [Google Scholar] [CrossRef] [PubMed]
- Karkucinska-Wieckowska, A.; Simoes, I.C.M.; Kalinowski, P.; Lebiedzinska-Arciszewska, M.; Zieniewicz, K.; Milkiewicz, P.; Górska-Ponikowska, M.; Pinton, P.; Malik, A.N.; Krawczyk, M.; et al. Mitochondria, oxidative stress and nonalcoholic fatty liver disease: A complex relationship. Eur. J. Clin. Investig. 2021, e13622. [Google Scholar] [CrossRef] [PubMed]
- Kanikowska, D.; Kanikowska, A.; Swora-Cwynar, E.; Grzymisławski, M.; Sato, M.; Bręborowicz, A.; Witowski, J.; Korybalska, K. Moderate Caloric Restriction Partially Improved Oxidative Stress Markers in Obese Humans. Antioxidants 2021, 10, 1018. [Google Scholar] [CrossRef] [PubMed]
- Bigornia, S.J.; Mott, M.M.; Hess, D.T.; Apovian, C.M.; McDonnell, M.E.; Duess, M.A.; Kluge, M.A.; Fiscale, A.J.; Vita, J.A.; Gokce, N. Long-term successful weight loss improves vascular endothelial function in severely obese individuals. Obesity 2010, 18, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Świderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic. Res. 2019, 53, 841–850. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, Y.; Fang, S.; Kim, W.; Kim, H.J.; Kim, J. woo GPx7 ameliorates non-alcoholic steatohepatitis by regulating oxidative stress. BMB Rep. 2020, 53, 317–322. [Google Scholar] [CrossRef]
- Simon, J.; Nuñez-García, M.; Fernández-Tussy, P.; Barbier-Torres, L.; Fernández-Ramos, D.; Gómez-Santos, B.; Buqué, X.; Lopitz-Otsoa, F.; Goikoetxea-Usandizaga, N.; Serrano-Macia, M.; et al. Targeting hepatic Glutaminase 1 ameliorates Non-Alcoholic Steatohepatitis by restoring Very-Low Density Lipoproteins triglyceride assembly. Cell Metab. 2020, 31, 605. [Google Scholar] [CrossRef]
- Milaciu, M.V.; Vesa, Ș.C.; Bocșan, I.C.; Ciumărnean, L.; Sâmpelean, D.; Negrean, V.; Pop, R.M.; Matei, D.M.; Pașca, S.; Răchișan, A.L.; et al. Paraoxonase-1 Serum Concentration and PON1 Gene Polymorphisms: Relationship with Non-Alcoholic Fatty Liver Disease. J. Clin. Med. 2019, 8, 2200. [Google Scholar] [CrossRef]
- Shin, S.K.; Cho, H.W.; Song, S.E.; Song, D.K. Catalase and nonalcoholic fatty liver disease. Pflugers Arch. 2018, 470, 1721–1737. [Google Scholar] [CrossRef]
- Hwang, I.; Uddin, M.J.; Pak, E.S.; Kang, H.; Jin, E.J.; Jo, S.; Kang, D.; Lee, H.; Ha, H. The impaired redox balance in peroxisomes of catalase knockout mice accelerates nonalcoholic fatty liver disease through endoplasmic reticulum stress. Free Radic. Biol. Med. 2020, 148, 22–32. [Google Scholar] [CrossRef]
- Nerstedt, A.; Kurhe, Y.; Cansby, E.; Caputo, M.; Gao, L.; Vorontsov, E.; Ståhlman, M.; Nuñez-Durán, E.; Borén, J.; Marschall, H.U.; et al. Lipid droplet-associated kinase STK25 regulates peroxisomal activity and metabolic stress response in steatotic liver. J. Lipid Res. 2020, 61, 178–191. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Gardner, H.W. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic. Biol. Med. 1989, 7, 65–86. [Google Scholar] [CrossRef]
- Demeilliers, C.; Maisonneuve, C.; Grodet, A.; Mansouri, A.; Nguyen, R.; Tinel, M.; Lettéron, P.; Degott, C.; Feldmann, G.; Pessayre, D.; et al. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology 2002, 123, 1278–1290. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Lukosz, M.; Jakob, S.; Büchner, N.; Zschauer, T.C.; Altschmied, J.; Haendeler, J. Nuclear redox signaling. Antioxid. Redox Signal. 2010, 12, 713–742. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Debeer, L.J.; Mannaerts, G.P.; De Schepper, P.J. Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates from control and clofibrate-treated rats [proceedings]. Arch. Int. Physiol. Biochim. 1979, 87, 209–210. [Google Scholar] [PubMed]
- Robertson, G.; Leclercq, I.; Farrell, G.C. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1135–G1139. [Google Scholar] [CrossRef]
- De Craemer, D.; Pauwels, M.; Van den Branden, C. Alterations of peroxisomes in steatosis of the human liver: A quantitative study. Hepatology 1995, 22, 744–752. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Eapen, C.E.; Pullimood, A.B.; Balasubramanian, K.A. Oxidative stress in experimental liver microvesicular steatosis: Role of mitochondria and peroxisomes. J. Gastroenterol. Hepatol. 2006, 21, 1240–1249. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef]
- Machado, M.V.; Cortez-Pinto, H. Diet, Microbiota, Obesity, and NAFLD: A Dangerous Quartet. Int. J. Mol. Sci. 2016, 17, 481. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Clement, K.; Pépin, J.L. Nonalcoholic fatty liver disease and obstructive sleep apnea. Metabolism 2016, 65, 1124–1135. [Google Scholar] [CrossRef]
- Del Ben, M.; Polimeni, L.; Carnevale, R.; Bartimoccia, S.; Nocella, C.; Baratta, F.; Loffredo, L.; Pignatelli, P.; Violi, F.; Angelico, F. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2014, 14, 81. [Google Scholar] [CrossRef]
- De Minicis, S.; Bataller, R.; Brenner, D.A. NADPH oxidase in the liver: Defensive, offensive, or fibrogenic? Gastroenterology 2006, 131, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; MacK, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Dang, P.M.C.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef]
- De Minicis, S.; Seki, E.; Oesterreicher, C.; Schnabl, B.; Schwabe, R.F.; Brenner, D.A. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology 2008, 48, 2016–2026. [Google Scholar] [CrossRef]
- Pastori, D.; Baratta, F.; Carnevale, R.; Cangemi, R.; Del Ben, M.; Bucci, T.; Polimeni, L.; Labbadia, G.; Nocella, C.; Scardella, L.; et al. Similar Reduction of Cholesterol-Adjusted Vitamin E Serum Levels in Simple Steatosis and Non-Alcoholic Steatohepatitis. Clin. Transl. Gastroenterol. 2015, 6, e113. [Google Scholar] [CrossRef]
- Pastori, D.; Loffredo, L.; Perri, L.; Baratta, F.; Scardella, L.; Polimeni, L.; Pani, A.; Brancorsini, M.; Albanese, F.; Catasca, E.; et al. Relation of nonalcoholic fatty liver disease and Framingham Risk Score to flow-mediated dilation in patients with cardiometabolic risk factors. Am. J. Cardiol. 2015, 115, 1402–1406. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, X.; Guo, J.; Roberts, C.K.; McKenzie, S.; Wu, W.C.; Liu, S.; Song, Y. Effects of Exercise Training on Cardiorespiratory Fitness and Biomarkers of Cardiometabolic Health: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2015, 4, e002014. [Google Scholar] [CrossRef]
- Violi, F.; Loffredo, L.; Carnevale, R.; Pignatelli, P.; Pastori, D. Atherothrombosis and Oxidative Stress: Mechanisms and Management in Elderly. Antioxid. Redox Signal. 2017, 27, 1083–1124. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Valle, V.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2019, 31, 189–206.e8. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.R. TLR9 in MAFLD and NASH: At the Intersection of Inflammation and Metabolism. Front. Endocrinol. 2021, 11, 613639. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Ouyang, X.; Han, S.N.; Zhang, J.Y.; Nemeth, B.T.; Pacher, P.; Feng, D.; Bataller, R.; Cabezas, J.; Stärkel, P.; Caballeria, J.; et al. Digoxin Suppresses Pyruvate Kinase M2-Promoted HIF-1α Transactivation in Steatohepatitis. Cell Metab. 2018, 27, 339–350.e3. [Google Scholar] [CrossRef]
- Peters, K.M.; Wilson, R.B.; Borradaile, N.M. Non-parenchymal hepatic cell lipotoxicity and the coordinated progression of non-alcoholic fatty liver disease and atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 417–422. [Google Scholar] [CrossRef]
- Bataller, R.; Schwabe, R.F.; Choi, Y.H.; Yang, L.; Paik, Y.H.; Lindquist, J.; Qian, T.; Schoonhoven, R.; Hagedorn, C.H.; Lemasters, J.J.; et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Investig. 2003, 112, 1383–1394. [Google Scholar] [CrossRef]
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Österreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Thuy, L.T.T.; Hai, H.; Kawada, N. Role of cytoglobin, a novel radical scavenger, in stellate cell activation and hepatic fibrosis. Clin. Mol. Hepatol. 2020, 26, 280. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. Innov. Hepatol. 2021, 3, 100301. [Google Scholar] [CrossRef]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-kappaB activation: From inducer to modulator. Antioxid. Redox Signal. 2009, 11, 2223–2243. [Google Scholar] [CrossRef]
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858. [Google Scholar] [CrossRef]
- Komeili-Movahhed, T.; Bassirian, M.; Changizi, Z.; Moslehi, A. SIRT1/NFκB pathway mediates anti-inflammatory and anti-apoptotic effects of rosmarinic acid on in a mouse model of nonalcoholic steatohepatitis (NASH). J. Recept. Signal Transduct. Res. 2021, 1–10. [Google Scholar] [CrossRef]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova-Kostova, A.T.; Georgiev, M.I. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760. [Google Scholar] [CrossRef]
- Solano-Urrusquieta, A.; Morales-González, J.A.; Castro-Narro, G.E.; Cerda-Reyes, E.; Flores-Rangel, P.D.; Fierros-Oceguera, R. NRF-2 and nonalcoholic fatty liver disease. Ann. Hepatol. 2020, 19, 458–465. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 367–398. [Google Scholar] [CrossRef]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L.J. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [PubMed]
- Mohs, A.; Otto, T.; Schneider, K.M.; Peltzer, M.; Boekschoten, M.; Holland, C.H.; Hudert, C.A.; Kalveram, L.; Wiegand, S.; Saez-Rodriguez, J.; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021, 74, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jiang, X.; Dong, F.; Zhang, F.; Ji, X.; Xue, M.; Yang, F.; Chen, J.; Hu, X.; Bao, Z. Lipid accumulation-induced hepatocyte senescence regulates the activation of hepatic stellate cells through the Nrf2-antioxidant response element pathway. Exp. Cell Res. 2021, 405, 112689. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Paul, A.; Lahiri, A.; Adak, M.; Maity, S.K.; Sarkar, A.; Paul, S.; Chakrabarti, P. Proteasome dysfunction under compromised redox metabolism dictates liver injury in NASH through ASK1/PPARγ binodal complementary modules. Redox Biol. 2021, 45, 102043. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Luo, X.Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.K. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G891–G899. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Xiong, X.Z. ICOS + Tregs: A Functional Subset of Tregs in Immune Diseases. Front. Immunol. 2020, 11, 2104. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Qin, Q.; Zhang, P.; Wang, G.; Liu, M.; Ding, Q.; Qin, Y.; Shen, Q. Reverse signaling using an inducible costimulator to enhance immunogenic function of dendritic cells. Cell. Mol. Life Sci. 2009, 66, 3067–3080. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, S.; Dianzani, C.; Chiocchetti, A.; Boggio, E.; Clemente, N.; Gigliotti, C.L.; Soluri, M.F.; Minelli, R.; Fantozzi, R.; Yagi, J.; et al. Triggering of B7h by the ICOS modulates maturation and migration of monocyte-derived dendritic cells. J. Immunol. 2013, 190, 1125–1134. [Google Scholar] [CrossRef]
- Gigliotti, C.L.; Boggio, E.; Clemente, N.; Shivakumar, Y.; Toth, E.; Sblattero, D.; D’Amelio, P.; Isaia, G.C.; Dianzani, C.; Yagi, J.; et al. ICOS-Ligand Triggering Impairs Osteoclast Differentiation and Function In Vitro and In Vivo. J. Immunol. 2016, 197, 3905–3916. [Google Scholar] [CrossRef]
- Koh, K.H.; Cao, Y.; Mangos, S.; Tardi, N.J.; Dande, R.R.; Lee, H.W.; Samelko, B.; Altintas, M.M.; Schmitz, V.P.; Lee, H.; et al. Nonimmune cell-derived ICOS ligand functions as a renoprotective αvβ3 integrin-selective antagonist. J. Clin. Investig. 2019, 129, 1713–1726. [Google Scholar] [CrossRef]
- Ramavath, N.N.; Gadipudiaik, L.I.; Provera, A.; Gigliotti, L.C.; Boggio, E.; Clemente, N.; Dianzani, U.; Albano, E.; Sutti, S. Co-stimulatory signals mediated by ICOS/ICOS-L dyad influence the differentiation of liver NASH-associated macrophages (NAMs). In Abstracts of The International Liver Congress™ 2021, Proceedings of the International Liver Congress (ILC), Digital, 23–26 June 2021. J. Hepatol. 2021, 75, S598. [Google Scholar]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef] [PubMed]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic. Biol. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef] [PubMed]
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in non-alcoholic fatty liver disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Blaak, E.E. Mitochondrial Dysfunction is a Key Pathway that Links Saturated Fat Intake to the Development and Progression of NAFLD. Mol. Nutr. Food Res. 2021, 65, e1900942. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Satapati, S.; Kucejova, B.; Duarte, J.A.G.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Arya Nair, L.; Livingston, K.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef]
- Grossini, E.; Garhwal, D.P.; Calamita, G.; Romito, R.; Rigamonti, C.; Minisini, R.; Smirne, C.; Surico, D.; Bellan, M.; Pirisi, M. Exposure to Plasma from Non-alcoholic Fatty Liver Disease Patients Affects Hepatocyte Viability, Generates Mitochondrial Dysfunction, and Modulates Pathways Involved in Fat Accumulation and Inflammation. Front. Med. 2021, 8, 693997. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated with Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.M.; Pennington, E.R.; Green, W.D.; Beck, M.A.; Brown, D.A.; Shaikh, S.R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. 2018, 9, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 2002, 286, 135–141. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Willem Greve, J.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef]
- Sullivan, E.M.; Fix, A.; Crouch, M.J.; Sparagna, G.C.; Zeczycki, T.N.; Brown, D.A.; Shaikh, S.R. Murine diet-induced obesity remodels cardiac and liver mitochondrial phospholipid acyl chains with differential effects on respiratory enzyme activity. J. Nutr. Biochem. 2017, 45, 94–103. [Google Scholar] [CrossRef]
- Han, X.; Yang, J.; Yang, K.; Zhongdan, Z.; Abendschein, D.R.; Gross, R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: A shotgun lipidomics study. Biochemistry 2007, 46, 6417–6428. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Calamita, G.; Shanmugam, H.; Khalil, M.; Bonfrate, L.; Wang, D.Q.H.; Baffy, G.; Portincasa, P. Mitochondria Matter: Systemic Aspects of Nonalcoholic Fatty Liver Disease (NAFLD) and Diagnostic Assessment of Liver Function by Stable Isotope Dynamic Breath Tests. Int. J. Mol. Sci. 2021, 22, 7702. [Google Scholar] [CrossRef]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, P.; Wright, M.; Wootton, S.A.; Jackson, A.A. Clinical utility of 13C-liver-function breath tests for assessment of hepatic function. Dig. Dis. Sci. 2013, 58, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Gorowska-Kowolik, K.; Chobot, A.; Kwiecien, J. 13 C Methacetin Breath Test for Assessment of Microsomal Liver Function: Methodology and Clinical Application. Gastroenterol. Res. Pract. 2017, 2017, 7397840. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, P.R.; Scorletti, E.; Smith, D.E.; Almehmadi, A.A.; Calder, P.C.; Byrne, C.D. The characterisation of hepatic mitochondrial function in patients with non-alcoholic fatty liver disease (NAFLD) using the 13 C-ketoisocaproate breath test. J. Breath Res. 2018, 12, 046002. [Google Scholar] [CrossRef]
- Campo, L.; Eiseler, S.; Apfel, T.; Pyrsopoulos, N. Fatty Liver Disease and Gut Microbiota: A Comprehensive Update. J. Clin. Transl. Hepatol. 2019, 7, 56–60. [Google Scholar] [CrossRef]
- Lau, E.; Carvalho, D.; Freitas, P. Gut Microbiota: Association with NAFLD and Metabolic Disturbances. Biomed Res. Int. 2015, 2015, 979515. [Google Scholar] [CrossRef]
- Zhou, J.; Tripathi, M.; Sinha, R.A.; Singh, B.K.; Yen, P.M. Gut microbiota and their metabolites in the progression of non-alcoholic fatty liver disease. Hepatoma Res. 2021, 7, 11. [Google Scholar] [CrossRef]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Burz, S.D.; Monnoye, M.; Philippe, C.; Farin, W.; Ratziu, V.; Strozzi, F.; Paillarse, J.M.; Chêne, L.; Blottière, H.M.; Gérard, P. Fecal Microbiota Transplant from Human to Mice Gives Insights into the Role of the Gut Microbiota in Non-Alcoholic Fatty Liver Disease (NAFLD). Microorganisms 2021, 9, 199. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.J.; Smits, L.P.; Pekmez, C.T.; Prodan, A.; Meijnikman, A.S.; Troelstra, M.A.; Bouter, K.E.C.; Herrema, H.; Levin, E.; Holleboom, A.G.; et al. Donor Fecal Microbiota Transplantation Alters Gut Microbiota and Metabolites in Obese Individuals With Steatohepatitis. Hepatol. Commun. 2020, 4, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Hrncir, T.; Hrncirova, L.; Kverka, M.; Hromadka, R.; Machova, V.; Trckova, E.; Kostovcikova, K.; Kralickova, P.; Krejsek, J.; Tlaskalova-Hogenova, H. Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions. Microorganisms 2021, 9, 957. [Google Scholar] [CrossRef] [PubMed]
- Jennison, E.; Byrne, C.D. The role of the gut microbiome and diet in the pathogenesis of non-alcoholic fatty liver disease. Clin. Mol. Hepatol. 2021, 27, 22–43. [Google Scholar] [CrossRef]
- Bakhshimoghaddam, F.; Alizadeh, M. Contribution of gut microbiota to nonalcoholic fatty liver disease: Pathways of mechanisms. Clin. Nutr. ESPEN 2021, 44, 61–68. [Google Scholar] [CrossRef]
- Compare, D.; Coccoli, P.; Rocco, A.; Nardone, O.M.; De Maria, S.; Cartenì, M.; Nardone, G. Gut—Liver axis: The impact of gut microbiota on non alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 471–476. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Auguet, T.; Bertran, L.; Binetti, J. Intestinal Dysbiosis and Non-Alcoholic Fatty Liver Disease. In Human Microbiome; Beloborodova, N.V., Grechko, A.V., Eds.; Intech Open Limited: London, UK, 2020; pp. 1–24. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Jasirwan, O.M.C.; Muradi, A.; Hasan, I.; Simadibrata, M.; Rinaldi, I. Correlation of gut Firmicutes/Bacteroidetes ratio with fibrosis and steatosis stratified by body mass index in patients with non-alcoholic fatty liver disease. Biosci. Microbiota Food Health 2021, 40, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, R.D.; Baker, S.S. Gut microbiome and nonalcoholic fatty liver diseases. Pediatr. Res. 2015, 77, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, S.; Babbar, A.; Drescher, H.K.; Hitch, T.C.A.; Clavel, T.; Muschaweck, M.; Ritz, T.; Kroy, D.C.; Trautwein, C.; Wagner, N.; et al. Upregulation of Anti-Oxidative Stress Response Improves Metabolic Changes in L-Selectin-Deficient Mice but Does Not Prevent NAFLD Progression or Fecal Microbiota Shifts. Int. J. Mol. Sci. 2021, 22, 7314. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Q.; Chai, W.; Sun, C.; Zhang, T.; Zhao, C.; Yuan, Y.; Wang, X.; Liu, H.; Ye, H. Lactobacillus paracasei Jlus66 extenuate oxidative stress and inflammation via regulation of intestinal flora in rats with non alcoholic fatty liver disease. Food Sci. Nutr. 2019, 7, 2636–2646. [Google Scholar] [CrossRef]
- Park, E.; Jeong, J.J.; Won, S.M.; Sharma, S.P.; Gebru, Y.A.; Ganesan, R.; Gupta, H.; Suk, K.T.; Kim, D.J. Gut Microbiota-Related Cellular and Molecular Mechanisms in the Progression of Nonalcoholic Fatty Liver Disease. Cells 2021, 10, 2634. [Google Scholar] [CrossRef]
- Shen, J.; Obin, M.S.; Zhao, L. The gut microbiota, obesity and insulin resistance. Mol. Asp. Med. 2013, 34, 39–58. [Google Scholar] [CrossRef]
- Diamant, M.; Blaak, E.E.; de Vos, W.M. Do nutrient-gut-microbiota interactions play a role in human obesity, insulin resistance and type 2 diabetes? Obes. Rev. 2011, 12, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Cassard, A.-M.; Gérard, P.; Perlemuter, G. Microbiota, Liver Diseases, and Alcohol. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Kullisaar, T.; Songisepp, E.; Mikelsaar, M.; Zilmer, K.; Vihalemm, T.; Zilmer, M. Antioxidative probiotic fermented goats’ milk decreases oxidative stress-mediated atherogenicity in human subjects. Br. J. Nutr. 2003, 90, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Arora, T.; Singh, S.; Sharma, R.K. Probiotics: Interaction with gut microbiome and antiobesity potential. Nutrition 2013, 29, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Park, D.Y.; Ahn, Y.T.; Huh, C.S.; Mcgregor, R.A.; Choi, M.S. Dual probiotic strains suppress high fructose-induced metabolic syndrome. World J. Gastroenterol. 2013, 19, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Castillo, V.; Figueroa, F.; González-Pizarro, K.; Jopia, P.; Ibacache-Quiroga, C. Probiotics and Prebiotics as a Strategy for Non-Alcoholic Fatty Liver Disease, a Narrative Review. Foods 2021, 10, 1719. [Google Scholar] [CrossRef] [PubMed]
- Moszak, M.; Szulińska, M.; Walczak-Gałęzewska, M.; Bogdański, P. Nutritional Approach Targeting Gut Microbiota in NAFLD-To Date. Int. J. Environ. Res. Public Health 2021, 18, 1616. [Google Scholar] [CrossRef]
- Nagashimada, M.; Honda, M. Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics. Int. J. Mol. Sci. 2021, 22, 8008. [Google Scholar] [CrossRef]
- Khan, A.; Ding, Z.; Ishaq, M.; Bacha, A.S.; Khan, I.; Hanif, A.; Li, W.; Guo, X. Understanding the Effects of Gut Microbiota Dysbiosis on Nonalcoholic Fatty Liver Disease and the Possible Probiotics Role: Recent Updates. Int. J. Biol. Sci. 2021, 17, 818–833. [Google Scholar] [CrossRef]
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702. [Google Scholar] [CrossRef]
- Porras, D.; Nistal, E.; Martínez-Flórez, S.; González-Gallego, J.; García-Mediavilla, M.V.; Sánchez-Campos, S. Intestinal Microbiota Modulation in Obesity-Related Non-alcoholic Fatty Liver Disease. Front. Physiol. 2018, 9, 1813. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Roeb, E.; Weiskirchen, R. Fructose and Non-Alcoholic Steatohepatitis. Front. Pharmacol. 2021, 12, 634344. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef]
- Xie, C.; Halegoua-Demarzio, D. Role of Probiotics in Non-alcoholic Fatty Liver Disease: Does Gut Microbiota Matter? Nutrients 2019, 11, 2837. [Google Scholar] [CrossRef]
- Nabavi, S.; Rafraf, M.; Somi, M.H.; Homayouni-Rad, A.; Asghari-Jafarabadi, M. Effects of probiotic yogurt consumption on metabolic factors in individuals with nonalcoholic fatty liver disease. J. Dairy Sci. 2014, 97, 7386–7393. [Google Scholar] [CrossRef]
- Famouri, F.; Shariat, Z.; Hashemipour, M.; Keikha, M.; Kelishadi, R. Effects of Probiotics on Nonalcoholic Fatty Liver Disease in Obese Children and Adolescents. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 413–417. [Google Scholar] [CrossRef]
- Ahn, S.B.; Jun, D.W.; Kang, B.K.; Lim, J.H.; Lim, S.; Chung, M.J. Randomized, Double-blind, Placebo-controlled Study of a Multispecies Probiotic Mixture in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 5688. [Google Scholar] [CrossRef]
- Javadi, L.; Khoshbaten, M.; Safaiyan, A.; Ghavami, M.; Abbasi, M.M.; Gargari, B.P. Pro- and prebiotic effects on oxidative stress and inflammatory markers in non-alcoholic fatty liver disease. Asia Pac. J. Clin. Nutr. 2018, 27, 1031–1039. [Google Scholar] [CrossRef]
- Lei, K.; Li, Y.L.; Wang, Y.; Wen, J.; Wu, H.Z.; Yu, D.Y.; Li, W.F. Effect of dietary supplementation of Bacillus subtilis B10 on biochemical and molecular parameters in the serum and liver of high-fat diet-induced obese mice. J. Zhejiang Univ. Sci. B 2015, 16, 487–495. [Google Scholar] [CrossRef]
- Kozmus, C.E.P.; Moura, E.; Serrão, M.P.; Real, H.; Guimarães, J.T.; Guedes-de-Pinho, P.; Duarte, B.P.; Marques, F.; Martins, M.J.; Vieira-Coelho, M.A. Influence of dietary supplementation with dextrin or oligofructose on the hepatic redox balance in rats. Mol. Nutr. Food Res. 2011, 55, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Ekhlasi, G.; Zarrati, M.; Agah, S.; Hosseini, A.F.; Hosseini, S.; Shidfar, S.; Aarbshahi, S.S.S.; Razmpoosh, E.; Shidfar, F. Effects of symbiotic and vitamin E supplementation on blood pressure, nitric oxide and inflammatory factors in non-alcoholic fatty liver disease. EXCLI J. 2017, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tsao, R. Dietary polyphenols, oxidative stress and antioxidant and anti-inflammatory effects. Curr. Opin. Food Sci. 2016, 8, 33–42. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, M.; Wang, Z.; Qin, F.; Chen, J.; He, Z. Dietary Polyphenols to Combat Nonalcoholic Fatty Liver Disease via the Gut-Brain-Liver Axis: A Review of Possible Mechanisms. J. Agric. Food Chem. 2021, 69, 3585–3600. [Google Scholar] [CrossRef]
- Abenavoli, L.; Larussa, T.; Corea, A.; Procopio, A.C.; Boccuto, L.; Dallio, M.; Federico, A.; Luzza, F. Dietary Polyphenols and Non-Alcoholic Fatty Liver Disease. Nutrients 2021, 13, 494. [Google Scholar] [CrossRef]
- Houghton, D.; Stewart, C.J.; Day, C.P.; Trenell, M. Gut Microbiota and Lifestyle Interventions in NAFLD. Int. J. Mol. Sci. 2016, 17, 447. [Google Scholar] [CrossRef]
- Evans, C.C.; LePard, K.J.; Kwak, J.W.; Stancukas, M.C.; Laskowski, S.; Dougherty, J.; Moulton, L.; Glawe, A.; Wang, Y.; Leone, V.; et al. Exercise prevents weight gain and alters the gut microbiota in a mouse model of high fat diet-induced obesity. PLoS ONE 2014, 9, e92193. [Google Scholar] [CrossRef]
- Campbell, S.C.; Wisniewski, P.J.; Noji, M.; McGuinness, L.R.; Häggblom, M.M.; Lightfoot, S.A.; Joseph, L.B.; Kerkhof, L.J. The Effect of Diet and Exercise on Intestinal Integrity and Microbial Diversity in Mice. PLoS ONE 2016, 11, e0150502. [Google Scholar] [CrossRef]
- Petriz, B.A.; Castro, A.P.; Almeida, J.A.; Gomes, C.P.C.; Fernandes, G.R.; Kruger, R.H.; Pereira, R.W.; Franco, O.L. Exercise induction of gut microbiota modifications in obese, non-obese and hypertensive rats. BMC Genomics 2014, 15, 511. [Google Scholar] [CrossRef]
- Lambert, J.E.; Myslicki, J.P.; Bomhof, M.R.; Belke, D.D.; Shearer, J.; Reimer, R.A. Exercise training modifies gut microbiota in normal and diabetic mice. Appl. Physiol. Nutr. Metab. 2015, 40, 749–752. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Porras, D.; Garcia-Mediavilla, M.V.; Martinez-Florez, S.; Juarez-Fernandez, M.; Cuevas, M.J.; Mauriz, J.L.; Gonzalez-Gallego, J.; Nistal, E.; Sanchez-Campos, S. Beneficial effects of exercise on gut microbiota functionality and barrier integrity, and gut-liver crosstalk in an in vivo model of early obesity and non-alcoholic fatty liver disease. Dis. Model. Mech. 2019, 12, dmm039206. [Google Scholar] [CrossRef] [PubMed]
- Winn, N.C.; Liu, Y.; Rector, R.S.; Parks, E.J.; Ibdah, J.A.; Kanaley, J.A. Energy-matched moderate and high intensity exercise training improves nonalcoholic fatty liver disease risk independent of changes in body mass or abdominal adiposity—A randomized trial. Metabolism 2018, 78, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Atkin, S.L.; Simental-Mendía, L.E.; Sahebkar, A. Molecular mechanisms by which aerobic exercise induces insulin sensitivity. J. Cell. Physiol. 2019, 234, 12385–12392. [Google Scholar] [CrossRef] [PubMed]
- Houttu, V.; Boulund, U.; Grefhorst, A.; Soeters, M.R.; Pinto-Sietsma, S.J.; Nieuwdorp, M.; Holleboom, A.G. The role of the gut microbiome and exercise in non-alcoholic fatty liver disease. Therap. Adv. Gastroenterol. 2020, 13, 1756284820941745. [Google Scholar] [CrossRef] [PubMed]
- Uchida, D.; Takaki, A.; Oyama, A.; Adachi, T.; Wada, N.; Onishi, H.; Okada, H. Oxidative Stress Management in Chronic Liver Diseases and Hepatocellular Carcinoma. Nutrients 2020, 12, 1576. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- McLoughlin, M.R.; Orlicky, D.J.; Prigge, J.R.; Krishna, P.; Talago, E.A.; Cavigli, I.R.; Eriksson, S.; Miller, C.G.; Kundert, J.A.; Sayin, V.I.; et al. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. Proc. Natl. Acad. Sci. USA. 2019, 116, 11408–11417. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther. 2018, 26, 29–38. [Google Scholar] [CrossRef]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef]
- Tao, J.; Krutsenko, Y.; Moghe, A.; Singh, S.; Poddar, M.; Bell, A.; Oertel, M.; Singhi, A.D.; Geller, D.; Chen, X.; et al. Nuclear factor erythroid 2-related factor 2 and β-Catenin Coactivation in Hepatocellular Cancer: Biological and Therapeutic Implications. Hepatology 2021, 74, 741–759. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. P53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2012, 57, 837–843. [Google Scholar] [CrossRef]
- Wilson, G.K.; Tennant, D.A.; McKeating, J.A. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: Current understanding and future directions. J. Hepatol. 2014, 61, 1397–1406. [Google Scholar] [CrossRef]
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, Y.; Ge, L.; Lin, Y.; Kwok, H.F. The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma. Cancers 2018, 10, 82. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; De Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Zhu, C.; Ho, Y.J.; Salomao, M.A.; Dapito, D.H.; Bartolome, A.; Schwabe, R.F.; Lee, J.S.; Lowe, S.W.; Pajvani, U.B. Notch activity characterizes a common hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. J. Hepatol. 2021, 74, 613–626. [Google Scholar] [CrossRef]
- Pervez, M.A.; Khan, D.A.; Slehria, A.U.R.; Ijaz, A. Delta-tocotrienol supplementation improves biochemical markers of hepatocellular injury and steatosis in patients with nonalcoholic fatty liver disease: A randomized, placebo-controlled trial. Complement. Ther. Med. 2020, 52, 102494. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Y.; Liang, Z.; Duan, W.; Yang, J.; Yan, J.; Wang, N.; Feng, W.; Ding, M.; Nie, Y.; et al. Silybin-mediated inhibition of Notch signaling exerts antitumor activity in human hepatocellular carcinoma cells. PLoS ONE 2013, 8, e83699. [Google Scholar] [CrossRef]
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef]
- Tzikas, S.; Vassilikos, V.; Keller, T. GDF-15 as a risk stratification biomarker for cardiovascular disease. Int. J. Cardiol. 2019, 292, 246–247. [Google Scholar] [CrossRef]
- Ago, T.; Sadoshima, J. GDF15, a cardioprotective TGF-beta superfamily protein. Circ. Res. 2006, 98, 294–297. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef]
- Chung, H.K.; Kim, J.T.; Kim, H.W.; Kwon, M.; Kim, S.Y.; Shong, M.; Kim, K.S.; Yi, H.S. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci. Rep. 2017, 7, 17238. [Google Scholar] [CrossRef]
- Lee, H.Y.; Nga, H.T.; Tian, J.; Yi, H.S. Mitochondrial Metabolic Signatures in Hepatocellular Carcinoma. Cells 2021, 10, 1901. [Google Scholar] [CrossRef]
- Si, Y.; Liu, X.; Cheng, M.; Wang, M.; Gong, Q.; Yang, Y.; Wang, T.; Yang, W. Growth differentiation factor 15 is induced by hepatitis C virus infection and regulates hepatocellular carcinoma-related genes. PLoS ONE 2011, 6, e19967. [Google Scholar] [CrossRef]
- Liu, X.; Chi, X.; Gong, Q.; Gao, L.; Niu, Y.; Chi, X.; Cheng, M.; Si, Y.; Wang, M.; Zhong, J.; et al. Association of serum level of growth differentiation factor 15 with liver cirrhosis and hepatocellular carcinoma. PLoS ONE 2015, 10, e0127518. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kim, S.H.; Kim, H.J.; Kim, K.H.; Lee, B.S.; Ku, B.J. Growth Differentiation Factor 15 Predicts Chronic Liver Disease Severity. Gut Liver 2017, 11, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Um, S.H.; Seo, D.S.; Joo, S.K.; Bae, J.M.; Park, J.H.; Chang, M.S.; Kim, J.H.; Lee, J.; Jeong, W., II; et al. Growth differentiation factor 15 predicts advanced fibrosis in biopsy-proven non-alcoholic fatty liver disease. Liver Int. 2018, 38, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef]
- Smirne, C.; Rigamonti, C.; De Benedittis, C.; Sainaghi, P.P.; Bellan, M.; Burlone, M.E.; Castello, L.M.; Avanzi, G.C. Gas6/TAM Signaling Components as Novel Biomarkers of Liver Fibrosis. Dis. Markers 2019, 2019, 2304931. [Google Scholar] [CrossRef]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef]
- Roskams, T.; Yang, S.Q.; Koteish, A.; Durnez, A.; DeVos, R.; Huang, X.; Achten, R.; Verslype, C.; Diehl, A.M. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am. J. Pathol. 2003, 163, 1301–1311. [Google Scholar] [CrossRef]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef]
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239. [Google Scholar] [CrossRef]
- Couchie, D.; Lafdil, F.; Martin-Garcia, N.; Laperche, Y.; Zafrani, E.S.; Mavier, P. Expression and role of Gas6 protein and of its receptor Axl in hepatic regeneration from oval cells in the rat. Gastroenterology 2005, 129, 1633–1642. [Google Scholar] [CrossRef]
- Lafdil, F.; Chobert, M.N.; Deveaux, V.; Zafrani, E.S.; Mavier, P.; Nakano, T.; Laperche, Y.; Brouillet, A. Growth arrest-specific protein 6 deficiency impairs liver tissue repair after acute toxic hepatitis in mice. J. Hepatol. 2009, 51, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Fourcot, A.; Couchie, D.; Chobert, M.N.; Zafrani, E.S.; Mavier, P.; Laperche, Y.; Brouillet, A. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1043–G1053. [Google Scholar] [CrossRef] [PubMed]
- Gogoi-Tiwari, J.; Köhn-Gaone, J.; Giles, C.; Schmidt-Arras, D.; Gratte, F.D.; Elsegood, C.L.; McCaughan, G.W.; Ramm, G.A.; Olynyk, J.K.; Tirnitz-Parker, J.E.E. The Murine Choline-Deficient, Ethionine-Supplemented (CDE) Diet Model of Chronic Liver Injury. J. Vis. Exp. 2017, 128, 56138. [Google Scholar] [CrossRef] [PubMed]
- Soret, P.A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2020, 10, 36. [Google Scholar] [CrossRef]
- Eng, J.M.; Estall, J.L. Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells 2021, 10, 1805. [Google Scholar] [CrossRef]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.M.; Van Rooijen, N.; Staels, B.; Kersten, S.; Müller, M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef]
- Maquoi, E.; Vörös, G.; Carmeliet, P.; Collen, D.; Lijnen, H.R. Role of Gas-6 in adipogenesis and nutritionally induced adipose tissue development in mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1002–1007. [Google Scholar] [CrossRef]
- Castrillo, A.; Tontonoz, P. Nuclear receptors in macrophage biology: At the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell Dev. Biol. 2004, 20, 455–480. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Díaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Cohen, P.L. The PPAR-γ antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: A key role for PPAR-γ in human macrophage polarization. J. Inflamm. 2015, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.A.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Szanto, A.; Roszer, T. Nuclear receptors in macrophages: A link between metabolism and inflammation. FEBS Lett. 2008, 582, 106–116. [Google Scholar] [CrossRef]
- Repa, J.J.; Mangelsdorf, D.J. The liver X receptor gene team: Potential new players in atherosclerosis. Nat. Med. 2002, 8, 1243–1248. [Google Scholar] [CrossRef]
- Pastore, M.; Grimaudo, S.; Pipitone, R.M.; Lori, G.; Raggi, C.; Petta, S.; Marra, F. Role of Myeloid-Epithelial-Reproductive Tyrosine Kinase and Macrophage Polarization in the Progression of Atherosclerotic Lesions Associated With Nonalcoholic Fatty Liver Disease. Front. Pharmacol. 2019, 10, 604. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Pouresmail, V.; Simon, T.; Blanc-Brude, O.; Kinugawa, K.; Merval, R.; Offenstadt, G.; Lesèche, G.; Cohen, P.L.; Tedgui, A.; et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1429–1431. [Google Scholar] [CrossRef]
- Garbin, U.; Baggio, E.; Stranieri, C.; Pasini, A.; Manfro, S.; Mozzini, C.; Vallerio, P.; Lipari, G.; Merigo, F.; Guidi, G.; et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: A role for oxidized polyunsaturated fatty acids? Cardiovasc. Res. 2013, 97, 125–133. [Google Scholar] [CrossRef]
- Kamada, Y.; Ono, M.; Hyogo, H.; Fujii, H.; Sumida, Y.; Yamada, M.; Mori, K.; Tanaka, S.; Maekawa, T.; Ebisutani, Y.; et al. Use of Mac-2 binding protein as a biomarker for nonalcoholic fatty liver disease diagnosis. Hepatol. Commun. 2017, 1, 780–791. [Google Scholar] [CrossRef]
- Hayashi, S.; Nagaoka, K.; Tanaka, Y. Blood-Based Biomarkers in Hepatitis B Virus-Related Hepatocellular Carcinoma, Including the Viral Genome and Glycosylated Proteins. Int. J. Mol. Sci. 2021, 22, 11051. [Google Scholar] [CrossRef] [PubMed]
- Bellan, M.; Castello, L.M.; Pirisi, M. Candidate Biomarkers of Liver Fibrosis: A Concise, Pathophysiology-oriented Review. J. Clin. Transl. Hepatol. 2018, 6, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Miyake, T.; Kuno, A.; Imai, Y.; Sawai, Y.; Hino, K.; Hara, Y.; Hige, S.; Sakamoto, M.; Yamada, G.; et al. Association between Wisteria floribunda agglutinin-positive Mac-2 binding protein and the fibrosis stage of non-alcoholic fatty liver disease. J. Gastroenterol. 2015, 50, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Dohi, Y.; Takase, H.; Yamashita, S.; Murai, S.; Tsuzuki, Y.; Ogawa, S.; Tanaka, Y.; Ohte, N. Serum levels of Mac-2 binding protein increase with cardiovascular risk and reflect silent atherosclerosis. Atherosclerosis 2016, 251, 192–196. [Google Scholar] [CrossRef]
- Sugiura, T.; Dohi, Y.; Takase, H.; Yamashita, S.; Tsuzuki, Y.; Ogawa, S.; Tanaka, Y.; Ohte, N. Factors associated with longitudinal changes in serum concentrations of Mac-2 binding protein: A prospective 3-year observational study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 1337–1344. [Google Scholar] [CrossRef]
- Arai, T.; Atsukawa, M.; Tsubota, A.; Kawano, T.; Koeda, M.; Yoshida, Y.; Tanabe, T.; Okubo, T.; Hayama, K.; Iwashita, A.; et al. Factors influencing subclinical atherosclerosis in patients with biopsy-proven nonalcoholic fatty liver disease. PLoS ONE 2019, 14, e0224184. [Google Scholar] [CrossRef]
- Oh, S.; Tsujimoto, T.; Kim, B.; Uchida, F.; Suzuki, H.; Iizumi, S.; Isobe, T.; Sakae, T.; Tanaka, K.; Shoda, J. Weight-loss-independent benefits of exercise on liver steatosis and stiffness in Japanese men with NAFLD. JHEP Rep. Innov. Hepatol. 2021, 3, 100253. [Google Scholar] [CrossRef]
- Ban, L.A.; Shackel, N.A.; McLennan, S.V. Extracellular Vesicles: A New Frontier in Biomarker Discovery for Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 376. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Alen, R.; Rada, P.; Valverde, A.M. Insights Into Extracellular Vesicles as Biomarker of NAFLD Pathogenesis. Front. Med. 2020, 7, 395. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Masyuk, T.V.; Larusso, N.F. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J. Hepatol. 2013, 59, 621–625. [Google Scholar] [CrossRef]
- Szabo, G.; Momen-Heravi, F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Moratti, E.; Vezzalini, M.; Tomasello, L.; Giavarina, D.; Sorio, C. Identification of protein tyrosine phosphatase receptor gamma extracellular domain (sPTPRG) as a natural soluble protein in plasma. PLoS ONE 2015, 10, e0119110. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles from Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Tomita, K.; Bronk, S.F.; Werneburg, N.W.; Harrison, S.A.; Goodfellow, V.S.; Malhi, H.; Gores, G.J. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016, 63, 731–744. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, M.F.; Jiang, S.; Wu, J.; Liu, J.; Yuan, X.W.; Shen, D.; Zhang, J.Z.; Zhou, N.; He, J.; et al. Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat. Commun. 2020, 11, 719. [Google Scholar] [CrossRef]
- Ore, A.; Akinloye, O.A. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef]
- Korish, A.A.; Arafah, M.M. Camel milk ameliorates steatohepatitis, insulin resistance and lipid peroxidation in experimental non-alcoholic fatty liver disease. BMC Complement. Altern. Med. 2013, 13, 264. [Google Scholar] [CrossRef]
- Schröder, T.; Kucharczyk, D.; Bär, F.; Pagel, R.; Derer, S.; Jendrek, S.T.; Sünderhauf, A.; Brethack, A.K.; Hirose, M.; Möller, S.; et al. Mitochondrial gene polymorphisms alter hepatic cellular energy metabolism and aggravate diet-induced non-alcoholic steatohepatitis. Mol. Metab. 2016, 5, 283–295. [Google Scholar] [CrossRef]
- Elshazly, S.M. Ameliorative effect of nicorandil on high fat diet induced non-alcoholic fatty liver disease in rats. Eur. J. Pharmacol. 2015, 748, 123–132. [Google Scholar] [CrossRef]
- Sugatani, J.; Wada, T.; Osabe, M.; Yamakawa, K.; Yoshinari, K.; Miwa, M. Dietary inulin alleviates hepatic steatosis and xenobiotics-induced liver injury in rats fed a high-fat and high-sucrose diet: Association with the suppression of hepatic cytochrome P450 and hepatocyte nuclear factor 4alpha expression. Drug Metab. Dispos. 2006, 34, 1677–1687. [Google Scholar] [CrossRef]
- Li, W.; Lu, Y. Hepatoprotective Effects of Sophoricoside against Fructose-Induced Liver Injury via Regulating Lipid Metabolism, Oxidation, and Inflammation in Mice. J. Food Sci. 2018, 83, 552–558. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Rodríguez-Juan, C.; Díaz-Sanjuan, T.; Del Hoyo, P.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591. [Google Scholar] [CrossRef]
- Verbeek, J.; Spincemaille, P.; Vanhorebeek, I.; Van Den Berghe, G.; Vander Elst, I.; Windmolders, P.; Van Pelt, J.; Van Der Merwe, S.; Bedossa, P.; Nevens, F.; et al. Dietary intervention, but not losartan, completely reverses non-alcoholic steatohepatitis in obese and insulin resistant mice. Lipids Health Dis. 2017, 16, 46. [Google Scholar] [CrossRef]
- Ciapaite, J.; Bakker, S.J.L.; Van Eikenhorst, G.; Wagner, M.J.; Teerlink, T.; Schalkwijk, C.G.; Fodor, M.; Ouwens, D.M.; Diamant, M.; Heine, R.J.; et al. Functioning of oxidative phosphorylation in liver mitochondria of high-fat diet fed rats. Biochim. Biophys. Acta 2007, 1772, 307–316. [Google Scholar] [CrossRef]
- Janevski, M.; Antonas, K.N.; Sullivan-Gunn, M.J.; McGlynn, M.A.; Lewandowski, P.A. The effect of cocoa supplementation on hepatic steatosis, reactive oxygen species and LFABP in a rat model of NASH. Comp. Hepatol. 2011, 10, 10. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Fernández-Moreira, D.; Solís-Muñoz, P.; Rodríguez-Juan, C.; Díaz-Sanjuán, T.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Mitochondrial complex I subunits are decreased in murine nonalcoholic fatty liver disease: Implication of peroxynitrite. J. Proteome Res. 2010, 9, 2450–2459. [Google Scholar] [CrossRef]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A.; Mak, K.M.; Xu, Y.; Cao, Q.; Ren, C.; Ponomarenko, A.; DeCarli, L.M. Model of nonalcoholic steatohepatitis. Am. J. Clin. Nutr. 2004, 79, 502–509. [Google Scholar] [CrossRef]
- Cardoso, A.R.; Kakimoto, P.A.H.B.; Kowaltowski, A.J. Diet-sensitive sources of reactive oxygen species in liver mitochondria: Role of very long chain acyl-CoA dehydrogenases. PLoS ONE 2013, 8, e77088. [Google Scholar] [CrossRef]
- Kathirvel, E.; Chen, P.; Morgan, K.; French, S.W.; Morgan, T.R. Oxidative stress and regulation of anti-oxidant enzymes in cytochrome P4502E1 transgenic mouse model of non-alcoholic fatty liver. J. Gastroenterol. Hepatol. 2010, 25, 1136–1143. [Google Scholar] [CrossRef]
- Yoshioka, S.; Hamada, A.; Jobu, K.; Yokota, J.; Onogawa, M.; Kyotani, S.; Miyamura, M.; Saibara, T.; Onishi, S.; Nishioka, Y. Effects of Eriobotrya japonica seed extract on oxidative stress in rats with non-alcoholic steatohepatitis. J. Pharm. Pharmacol. 2010, 62, 241–246. [Google Scholar] [CrossRef]
- Chung, M.Y.; Park, H.J.; Manautou, J.E.; Koo, S.I.; Bruno, R.S. Green tea extract protects against nonalcoholic steatohepatitis in ob/ob mice by decreasing oxidative and nitrative stress responses induced by proinflammatory enzymes. J. Nutr. Biochem. 2012, 23, 361–367. [Google Scholar] [CrossRef]
- Song, L.; Qu, D.; Zhang, Q.; Jiang, J.; Zhou, H.; Jiang, R.; Li, Y.; Zhang, Y.; Yan, H. Phytosterol esters attenuate hepatic steatosis in rats with non-alcoholic fatty liver disease rats fed a high-fat diet. Sci. Rep. 2017, 7, 41604. [Google Scholar] [CrossRef]
- Stiuso, P.; Scognamiglio, I.; Murolo, M.; Ferranti, P.; De Simone, C.; Rizzo, M.R.; Tuccillo, C.; Caraglia, M.; Loguercio, C.; Federico, A. Serum oxidative stress markers and lipidomic profile to detect NASH patients responsive to an antioxidant treatment: A pilot study. Oxid. Med. Cell. Longev. 2014, 2014, 169216. [Google Scholar] [CrossRef]
- Leghi, G.E.; Domenici, F.A.; Vannucchi, H. Influence of oxidative stress and obesity in patients with nonalcoholic steatohepatitis. Arq. Gastroenterol. 2015, 52, 228–233. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef]
- Loguercio, C.; De Girolamo, V.; De Sio, I.; Tuccillo, C.; Ascione, A.; Baldi, F.; Budillon, G.; Cimino, L.; Di Carlo, A.; Pia Di Marino, M.; et al. Non-alcoholic fatty liver disease in an area of southern Italy: Main clinical, histological, and pathophysiological aspects. J. Hepatol. 2001, 35, 568–574. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Oliveira, C.P.M.S.; Faintuch, J.; Rascovski, A.; Furuya, C.K.; Do Socorro Bastos, M.; Matsuda, M.; Della Nina, B.I.; Yahnosi, K.; Abdala, D.S.P.; Vezozzo, D.C.P.; et al. Lipid peroxidation in bariatric candidates with nonalcoholic fatty liver disease (NAFLD)—Preliminary findings. Obes. Surg. 2005, 15, 502–505. [Google Scholar] [CrossRef]
- Dasarathy, S.; Yang, Y.; McCullough, A.J.; Marczewski, S.; Bennett, C.; Kalhan, S.C. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2011, 23, 382–388. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, X.; Jiao, Y.; Xiong, X.; Wang, E.; Wang, X.; Zhang, Z.; Zhang, H.; Pan, L.; Guan, Y.; et al. Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARα. J. Clin. Investig. 2014, 124, 3501–3513. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Z.; Zhu, H.T.; Dai, Y.N.; Li, C.X.; Fang, Z.Y.; Zhao, D.J.; Wan, X.Y.; Wang, Y.M.; Wang, F.; Yu, C.H.; et al. Serum periostin is a potential biomarker for non-alcoholic fatty liver disease: A case-control study. Endocrine 2016, 51, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, H.; Niu, Y.; Zhang, W.; Zhu, L.; Li, X.; Lu, S.; Fan, J.; Li, X.; Ning, G.; et al. Circulating periostin in relation to insulin resistance and nonalcoholic fatty liver disease among overweight and obese subjects. Sci. Rep. 2016, 6, 37886. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Qu, H.; Wang, H.; Wei, H.; Wu, J.; Duan, Y.; Liu, D.; Deng, H. Plasma Periostin Levels Are Increased in Chinese Subjects with Obesity and Type 2 Diabetes and Are Positively Correlated with Glucose and Lipid Parameters. Mediators Inflamm. 2016, 2016, 6423637. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Anstee, Q.; Valenti, L. Genetic predisposition in NAFLD and NASH: Impact on severity of liver disease and response to treatment. Curr. Pharm. Des. 2013, 19, 5219–5238. [Google Scholar] [CrossRef]
- Salomone, F.; Li Volti, G.; Rosso, C.; Grosso, G.; Bugianesi, E. Unconjugated bilirubin, a potent endogenous antioxidant, is decreased in patients with non-alcoholic steatohepatitis and advanced fibrosis. J. Gastroenterol. Hepatol. 2013, 28, 1202–1208. [Google Scholar] [CrossRef]
- Chtioui, H.; Semela, D.; Ledermann, M.; Zimmermann, A.; Dufour, J.F. Expression and activity of the cytochrome P450 2E1 in patients with nonalcoholic steatosis and steatohepatitis. Liver Int. 2007, 27, 764–771. [Google Scholar] [CrossRef]
- Marcolin, É.; Forgiarini, L.F.; Tieppo, J.; Dias, A.S.; de Freitas, L.A.R.; Marroni, N.P. Methionine- and choline-deficient diet induces hepatic changes characteristic of non-alcoholic steatohepatitis. Arq. Gastroenterol. 2011, 48, 72–79. [Google Scholar] [CrossRef]
- Nosrati, N.; Aghazadeh, S.; Yazdanparast, R. Effects of Teucrium polium on Insulin Resistance in Nonalcoholic Steatohepatitis. J. Acupunct. Meridian Stud. 2010, 3, 104–110. [Google Scholar] [CrossRef]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef]
- Marcolin, E.; San-Miguel, B.; Vallejo, D.; Tieppo, J.; Marroni, N.; González-Gallego, J.; Tuñón, M.J. Quercetin treatment ameliorates inflammation and fibrosis in mice with nonalcoholic steatohepatitis. J. Nutr. 2012, 142, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, A.; Duseja, A.; Das, A.; Dhiman, R.K.; Chawla, Y.K.; Kohli, K.K.; Bhansali, A. Patients with Nonalcoholic Fatty Liver Disease (NAFLD) have Higher Oxidative Stress in Comparison to Chronic Viral Hepatitis. J. Clin. Exp. Hepatol. 2013, 3, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.K.M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar] [PubMed]
- Sumida, Y.; Nakashima, T.; Yoh, T.; Furutani, M.; Hirohama, A.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Okanoue, T.; et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J. Hepatol. 2003, 38, 32–38. [Google Scholar] [CrossRef]
- Köroǧlu, E.; Canbakan, B.; Atay, K.; Hatemi, I.; Tuncer, M.; Dobrucali, A.; Sonsuz, A.; Gültepe, I.; Şentürk, H. Role of oxidative stress and insulin resistance in disease severity of non-alcoholic fatty liver disease. Turk. J. Gastroenterol. 2016, 27, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Osterreicher, C.H.; Brenner, D.A. The genetics of nonalcoholic fatty liver disease. Ann Hepatol. 2007, 6, 83–88. [Google Scholar] [CrossRef]
- Willner, I.R.; Waters, B.; Patil, S.R.; Reuben, A.; Morelli, J.; Riely, C.A. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am. J. Gastroenterol. 2001, 96, 2957–2961. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Bezsonov, E.E.; Baig, M.S.; Popkova, T.V.; Nedosugova, L.V.; Starodubova, A.V.; Orekhov, A.N. Mitochondrial Mutations and Genetic Factors Determining NAFLD Risk. Int. J. Mol. Sci. 2021, 22, 4459. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.P.; Stefano, J.T. Genetic polymorphisms and oxidative stress in non-alcoholic steatohepatitis (NASH): A mini review. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (Suppl. 1), S35–S40. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Malaguarnera, L. Genetic variants in candidate genes influencing NAFLD progression. J. Mol. Med. 2012, 90, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Taliento, A.E.; Dallio, M.; Federico, A.; Prati, D.; Valenti, L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2755. [Google Scholar] [CrossRef] [PubMed]
- Wispe’, J.R.; Clark, J.C.; Burhans, M.S.; Kropp, K.E.; Korfhagen, T.R.; Whitsett, J.A. Synthesis and processing of the precursor for human mangano-superoxide dismutase. Biochim. Biophys. Acta 1989, 994, 30–36. [Google Scholar] [CrossRef]
- Sutton, A.; Imbert, A.; Igoudjil, A.; Descatoire, V.; Cazanave, S.; Pessayre, D.; Degoul, F. The manganese superoxide dismutase Ala16Val dimorphism modulates both mitochondrial import and mRNA stability. Pharmacogenet. Genomics 2005, 15, 311–319. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Dongiovanni, P. Genetic and metabolic factors: The perfect combination to treat metabolic associated fatty liver disease. Explor. Med. 2020, 1, 218–243. [Google Scholar] [CrossRef]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.S.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef]
- Namikawa, C.; Shu-Ping, Z.; Vyselaar, J.R.; Nozaki, Y.; Nemoto, Y.; Ono, M.; Akisawa, N.; Saibara, T.; Hiroi, M.; Enzan, H.; et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatol. 2004, 40, 781–786. [Google Scholar] [CrossRef]
- Schrauwen, P.; Xia, J.; Walder, K.; Snitker, S.; Ravussin, E. A novel polymorphism in the proximal UCP3 promoter region: Effect on skeletal muscle UCP3 mRNA expression and obesity in male non-diabetic Pima Indians. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 1242–1245. [Google Scholar] [CrossRef]
- Aller, R.; De Luis, D.A.; Izaola, O.; González Sagrado, M.; Conde, R.; Alvarez, T.; Pacheco, D.V.M. Role of -55CT polymorphism of UCP3 gene on non alcoholic fatty liver disease and insulin resistance in patients with obesity. Nutr. Hosp. 2010, 25, 572–576. [Google Scholar]
- Fares, R.; Petta, S.; Lombardi, R.; Grimaudo, S.; Dongiovanni, P.; Pipitone, R.; Rametta, R.; Fracanzani, A.L.; Mozzi, E.; Craxì, A.; et al. The UCP2-866 G>A promoter region polymorphism is associated with nonalcoholic steatohepatitis. Liver Int. 2015, 35, 1574–1580. [Google Scholar] [CrossRef]
- Dong, C.; Della-Morte, D.; Wang, L.; Cabral, D.; Beecham, A.; McClendon, M.S.; Luca, C.C.; Blanton, S.H.; Sacco, R.L.; Rundek, T. Association of the sirtuin and mitochondrial uncoupling protein genes with carotid plaque. PLoS ONE 2011, 6, e27157. [Google Scholar] [CrossRef]
- Oliveira, C.P.M.S.; Stefano, J.T.; Cavaleiro, A.M.; Zanella Fortes, M.A.H.; Vieira, S.M.; Rodrigues Lima, V.M.; Santos, T.E.; Santos, V.N.; De Azevedo Salgado, A.L.F.; Parise, É.R.; et al. Association of polymorphisms of glutamate-cystein ligase and microsomal triglyceride transfer protein genes in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2010, 25, 357–361. [Google Scholar] [CrossRef]
- Hashemi, M.; Hoseini, H.; Yaghmaei, P.; Moazeni-Roodi, A.; Bahari, A.; Hashemzehi, N.; Shafieipour, S. Association of polymorphisms in glutamate-cysteine ligase catalytic subunit and microsomal triglyceride transfer protein genes with nonalcoholic fatty liver disease. DNA Cell Biol. 2011, 30, 569–575. [Google Scholar] [CrossRef]
- Emdin, C.A.; Haas, M.E.; Khera, A.V.; Aragam, K.; Chaffin, M.; Klarin, D.; Hindy, G.; Jiang, L.; Wei, W.Q.; Feng, Q.; et al. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020, 16, e1008629. [Google Scholar] [CrossRef]
- Sparacino-Watkins, C.E.; Tejero, J.; Sun, B.; Gauthier, M.C.; Thomas, J.; Ragireddy, V.; Merchan, B.A.; Wang, J.; Azarov, I.; Basu, P.; et al. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J. Biol. Chem. 2014, 289, 10345–10358. [Google Scholar] [CrossRef]
- Schneider, J.; Girreser, U.; Havemeyer, A.; Bittner, F.; Clement, B. Detoxification of Trimethylamine N-Oxide by the Mitochondrial Amidoxime Reducing Component mARC. Chem. Res. Toxicol. 2018, 31, 447–453. [Google Scholar] [CrossRef]
- Wang, C.; Gong, J.; Wu, H. Development of gene polymorphisms in meditators of nonalcoholic fatty liver disease. Biomed. Rep. 2017, 7, 95–104. [Google Scholar] [CrossRef]
- Lin, Y.C.; Wu, C.C.; Ni, Y.H. New Perspectives on Genetic Prediction for Pediatric Metabolic Associated Fatty Liver Disease. Front. Pediatr. 2020, 8, 603654. [Google Scholar] [CrossRef]
- Nobili, V.; Carpino, G.; Alisi, A.; De Vito, R.; Franchitto, A.; Alpini, G.; Onori, P.; Gaudio, E. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e88005. [Google Scholar] [CrossRef]
- Lin, Y.C.; Chang, P.F.; Hu, F.C.; Chang, M.H.; Ni, Y.H. Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics 2009, 124, e1221–e1227. [Google Scholar] [CrossRef]
- Puri, K.; Nobili, V.; Melville, K.; Corte, C.D.; Sartorelli, M.R.; Lopez, R.; Feldstein, A.E.; Alkhouri, N. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 114–118. [Google Scholar] [CrossRef]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 overexpression and underexpression: Clinical implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef]
- Soyal, S.; Krempler, F.; Oberkofler, H.; Patsch, W. PGC-1alpha: A potent transcriptional cofactor involved in the pathogenesis of type 2 diabetes. Diabetologia 2006, 49, 1477–1488. [Google Scholar] [CrossRef]
- Lin, Y.C.; Chang, P.F.; Chang, M.H.; Ni, Y.H. A common variant in the peroxisome proliferator-activated receptor-γ coactivator-1α gene is associated with nonalcoholic fatty liver disease in obese children. Am. J. Clin. Nutr. 2013, 97, 326–331. [Google Scholar] [CrossRef]
- Ladero, J.M.; Martínez, C.; García-Martin, E.; Fernández-Arquero, M.; López-Alonso, G.; De La Concha, E.G.; Díaz-Rubio, M.; Agúndez, J.A.G. Polymorphisms of the glutathione S-transferases mu-1 (GSTM1) and theta-1 (GSTT1) and the risk of advanced alcoholic liver disease. Scand. J. Gastroenterol. 2005, 40, 348–353. [Google Scholar] [CrossRef]
- Stickel, F.; Österreicher, C.H.; Datz, C.; Ferenci, P.; Wölfel, M.; Norgauer, W.; Kraus, M.R.; Wrba, F.; Hellerbrand, C.; Schuppan, D. Prediction of progression to cirrhosis by a glutathione S-transferase P1 polymorphism in subjects with hereditary hemochromatosis. Arch. Intern. Med. 2005, 165, 1835–1840. [Google Scholar] [CrossRef][Green Version]
- Reynolds, W.F.; Patel, K.; Pianko, S.; Blatt, L.M.; Nicholas, J.J.; McHutchison, J.G. A genotypic association implicates myeloperoxidase in the progression of hepatic fibrosis in chronic hepatitis C virus infection. Genes Immun. 2002, 3, 345–349. [Google Scholar] [CrossRef]
- Österreicher, C.H.; Datz, C.; Stickel, F.; Hellerbrand, C.; Penz, M.; Hofer, H.; Wrba, F.; Penner, E.; Schuppan, D.; Ferenci, P. Association of myeloperoxidase promotor polymorphism with cirrhosis in patients with hereditary hemochromatosis. J. Hepatol. 2005, 42, 914–919. [Google Scholar] [CrossRef]
- Sonzogni, L.; Silvestri, L.; De Silvestri, A.; Gritti, C.; Foti, L.; Zavaglia, C.; Bottelli, R.; Mondelli, M.U.; Civardi, E.; Silini, E.M. Polymorphisms of microsomal epoxide hydrolase gene and severity of HCV-related liver disease. Hepatology 2002, 36, 195–201. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Colletti, A.; Bellentani, S. Nutraceutical Approach to Non-Alcoholic Fatty Liver Disease (NAFLD): The Available Clinical Evidence. Nutrients 2018, 10, 1153. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Cheung, F.; Hong, M.; Feng, Y. The Potential and Action Mechanism of Polyphenols in the Treatment of Liver Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 8394818. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary Polyphenols Protect Against Oleic Acid-Induced Steatosis in an in Vitro Model of NAFLD by Modulating Lipid Metabolism and Improving Mitochondrial Function. Nutrients 2019, 11, 541. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef]
- Hodges, J.K.; Sasaki, G.Y.; Bruno, R.S. Anti-inflammatory activities of green tea catechins along the gut-liver axis in nonalcoholic fatty liver disease: Lessons learned from preclinical and human studies. J. Nutr. Biochem. 2020, 85, 108478. [Google Scholar] [CrossRef]
- Xia, H.M.; Wang, J.; Xie, X.J.; Xu, L.J.; Tang, S.Q. Green tea polyphenols attenuate hepatic steatosis, and reduce insulin resistance and inflammation in high-fat diet-induced rats. Int. J. Mol. Med. 2019, 44, 1523–1530. [Google Scholar] [CrossRef]
- Yang, J.P.; Shin, J.H.; Seo, S.H.; Kim, S.G.; Lee, S.H.; Shin, E.H. Effects of Antioxidants in Reducing Accumulation of Fat in Hepatocyte. Int. J. Mol. Sci. 2018, 19, 2563. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Ivancovsky-Wajcman, D.; Fliss-Isakov, N.; Salomone, F.; Webb, M.; Shibolet, O.; Kariv, R.; Zelber-Sagi, S. Dietary vitamin E and C intake is inversely associated with the severity of nonalcoholic fatty liver disease. Dig. Liver Dis. 2019, 51, 1698–1705. [Google Scholar] [CrossRef]
- Perumpail, B.; Li, A.; John, N.; Sallam, S.; Shah, N.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Vuppalanchi, R.; Gawrieh, S.; Ghabril, M.; Saxena, R.; Cummings, O.W.; Chalasani, N. Vitamin E Improves Transplant-Free Survival and Hepatic Decompensation Among Patients with Nonalcoholic Steatohepatitis and Advanced Fibrosis. Hepatology 2020, 71, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Raso, G.M.; Esposito, E.; Iacono, A.; Pacilio, M.; Cuzzocrea, S.; Canani, R.B.; Calignano, A.; Meli, R. Comparative therapeutic effects of metformin and vitamin E in a model of non-alcoholic steatohepatitis in the young rat. Eur. J. Pharmacol. 2009, 604, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Podszun, M.C.; Frank, J. Impact of vitamin E on redox biomarkers in non-alcoholic fatty liver disease. Redox Biol. 2021, 42, 101937. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Paital, B.; Jena, S.; Swain, S.S.; Kumar, S.; Yadav, M.K.; Chainy, G.B.N.; Samanta, L. Possible activation of NRF2 by Vitamin E/Curcumin against altered thyroid hormone induced oxidative stress via NFĸB/AKT/mTOR/KEAP1 signalling in rat heart. Sci. Rep. 2019, 9, 7408. [Google Scholar] [CrossRef]
- He, W.; Xu, Y.; Ren, X.; Xiang, D.; Lei, K.; Zhang, C.; Liu, D. Vitamin E Ameliorates Lipid Metabolism in Mice with Nonalcoholic Fatty Liver Disease via Nrf2/CES1 Signaling Pathway. Dig. Dis. Sci. 2019, 64, 3182–3191. [Google Scholar] [CrossRef]
- Podszun, M.C.; Alawad, A.S.; Lingala, S.; Morris, N.; Huang, W.C.A.; Yang, S.; Schoenfeld, M.; Rolt, A.; Ouwerkerk, R.; Valdez, K.; et al. Vitamin E treatment in NAFLD patients demonstrates that oxidative stress drives steatosis through upregulation of de-novo lipogenesis. Redox Biol. 2020, 37, 101710. [Google Scholar] [CrossRef]
- Wei, J.; Lei, G.H.; Fu, L.; Zeng, C.; Yang, T.; Peng, S.F. Association between Dietary Vitamin C Intake and Non-Alcoholic Fatty Liver Disease: A Cross-Sectional Study among Middle-Aged and Older Adults. PLoS ONE 2016, 11, e0147985. [Google Scholar] [CrossRef]
- Oliveira, C.P.M.S.; Da Costa Gayotto, L.C.; Tatai, C.; Della Nina, B.I.; Lima, E.S.; Abdalla, D.S.P.; Lopasso, F.P.; Laurindo, F.R.M.; Carrilho, F.J. Vitamin C and Vitamin E in Prevention of Nonalcoholic Fatty Liver Disease (NAFLD) in Choline Deficient Diet Fed Rats. Nutr. J. 2003, 2, 9. [Google Scholar] [CrossRef]
- Lee, H.; Ahn, J.; Shin, S.S.; Yoon, M. Ascorbic acid inhibits visceral obesity and nonalcoholic fatty liver disease by activating peroxisome proliferator-activated receptor α in high-fat-diet-fed C57BL/6J mice. Int. J. Obes. 2019, 43, 1620–1630. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Martin, S. Health Effects of Coffee: Mechanism Unraveled? Nutrients 2020, 12, 1842. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Thongprayoon, C.; Ungprasert, P. Coffee consumption and risk of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2017, 29, e8–e12. [Google Scholar] [CrossRef] [PubMed]
- Boettler, U.; Volz, N.; Teller, N.; Haupt, L.M.; Bakuradze, T.; Eisenbrand, G.; Bytof, G.; Lantz, I.; Griffiths, L.R.; Marko, D. Induction of antioxidative Nrf2 gene transcription by coffee in humans: Depending on genotype? Mol. Biol. Rep. 2012, 39, 7155–7162. [Google Scholar] [CrossRef] [PubMed]
- Priftis, A.; Angeli-Terzidou, A.E.; Veskoukis, A.S.; Spandidos, D.A.; Kouretas, D. Cell-specific and roasting-dependent regulation of the Keap1/Nrf2 pathway by coffee extracts. Mol. Med. Rep. 2018, 17, 8325–8331. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Huang, Q.; Guan, Y.; Lv, M.; He, X.; Fang, C.; Wang, X.; Sheng, J. Caffeine promotes conversion of palmitic acid to palmitoleic acid by inducing expression of fat-5 in Caenorhabditis elegans and scd1 in mice. Front. Pharmacol. 2018, 9, 321. [Google Scholar] [CrossRef]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef]
- Wang, Q.; Dai, X.; Yang, W.; Wang, H.; Zhao, H.; Yang, F.; Yang, Y.; Li, J.; Lv, X. Caffeine protects against alcohol-induced liver fibrosis by dampening the cAMP/PKA/CREB pathway in rat hepatic stellate cells. Int. Immunopharmacol. 2015, 25, 340–352. [Google Scholar] [CrossRef]
- Bhandarkar, N.S.; Brown, L.; Panchal, S.K. Chlorogenic acid attenuates high-carbohydrate, high-fat diet-induced cardiovascular, liver, and metabolic changes in rats. Nutr. Res. 2019, 62, 78–88. [Google Scholar] [CrossRef]
- Mazza, A.; Fruci, B.; Garinis, G.A.; Giuliano, S.; Malaguarnera, R.; Belfiore, A. The role of metformin in the management of NAFLD. Exp. Diabetes Res. 2012, 2012, 716404. [Google Scholar] [CrossRef]
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 6668. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, Y.H.; Kim, J.W.; Ham, D.S.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef]
- Rouabhia, S.; Milic, N.; Abenavoli, L. Metformin in the treatment of non-alcoholic fatty liver disease: Safety, efficacy and mechanism. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 343–349. [Google Scholar] [CrossRef]
- Parhiz, H.; Roohbakhsh, A.; Soltani, F.; Rezaee, R.; Iranshahi, M. Antioxidant and anti-inflammatory properties of the citrus flavonoids hesperidin and hesperetin: An updated review of their molecular mechanisms and experimental models. Phytother. Res. 2015, 29, 323–331. [Google Scholar] [CrossRef]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-κB Signaling in an Aβ Mouse Model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef]
- Geng, Y.; Wu, Z.; Buist-Homan, M.; Blokzijl, H.; Moshage, H. Hesperetin protects against palmitate-induced cellular toxicity via induction of GRP78 in hepatocytes. Toxicol. Appl. Pharmacol. 2020, 404, 115183. [Google Scholar] [CrossRef]
- Wah Kheong, C.; Nik Mustapha, N.R.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 1940–1949.e8. [Google Scholar] [CrossRef]
- Herrero, M.; Cifuentes, A.; Ibañez, E. Sub- and supercritical fluid extraction of functional ingredients from different natural sources: Plants, food-by-products, algae and microalgae: A review. Food Chem. 2006, 98, 136–148. [Google Scholar] [CrossRef]
- Zhong, S.; Fan, Y.; Yan, Q.; Fan, X.; Wu, B.; Han, Y.; Zhang, Y.; Chen, Y.; Zhang, H.; Niu, J. The therapeutic effect of silymarin in the treatment of nonalcoholic fatty disease: A meta-analysis (PRISMA) of randomized control trials. Medicine 2017, 96, e9061. [Google Scholar] [CrossRef]
- Taleb, A.; Ahmad, K.A.; Ihsan, A.U.; Qu, J.; Lin, N.; Hezam, K.; Koju, N.; Hui, L.; Qilong, D. Antioxidant effects and mechanism of silymarin in oxidative stress induced cardiovascular diseases. Biomed. Pharmacother. 2018, 102, 689–698. [Google Scholar] [CrossRef]
- Navarro, V.J.; Belle, S.H.; D’Amato, M.; Adfhal, N.; Brunt, E.M.; Fried, M.W.; Rajender Reddy, K.; Wahed, A.S.; Harrison, S. Silymarin in non-cirrhotics with non-alcoholic steatohepatitis: A randomized, double-blind, placebo controlled trial. PLoS ONE 2019, 14, e0221683. [Google Scholar] [CrossRef]
- Tang, J.T.; Mao, Y.M. Development of new drugs for the treatment of nonalcoholic steatohepatitis. J. Dig. Dis. 2020, 21, 3–11. [Google Scholar] [CrossRef]



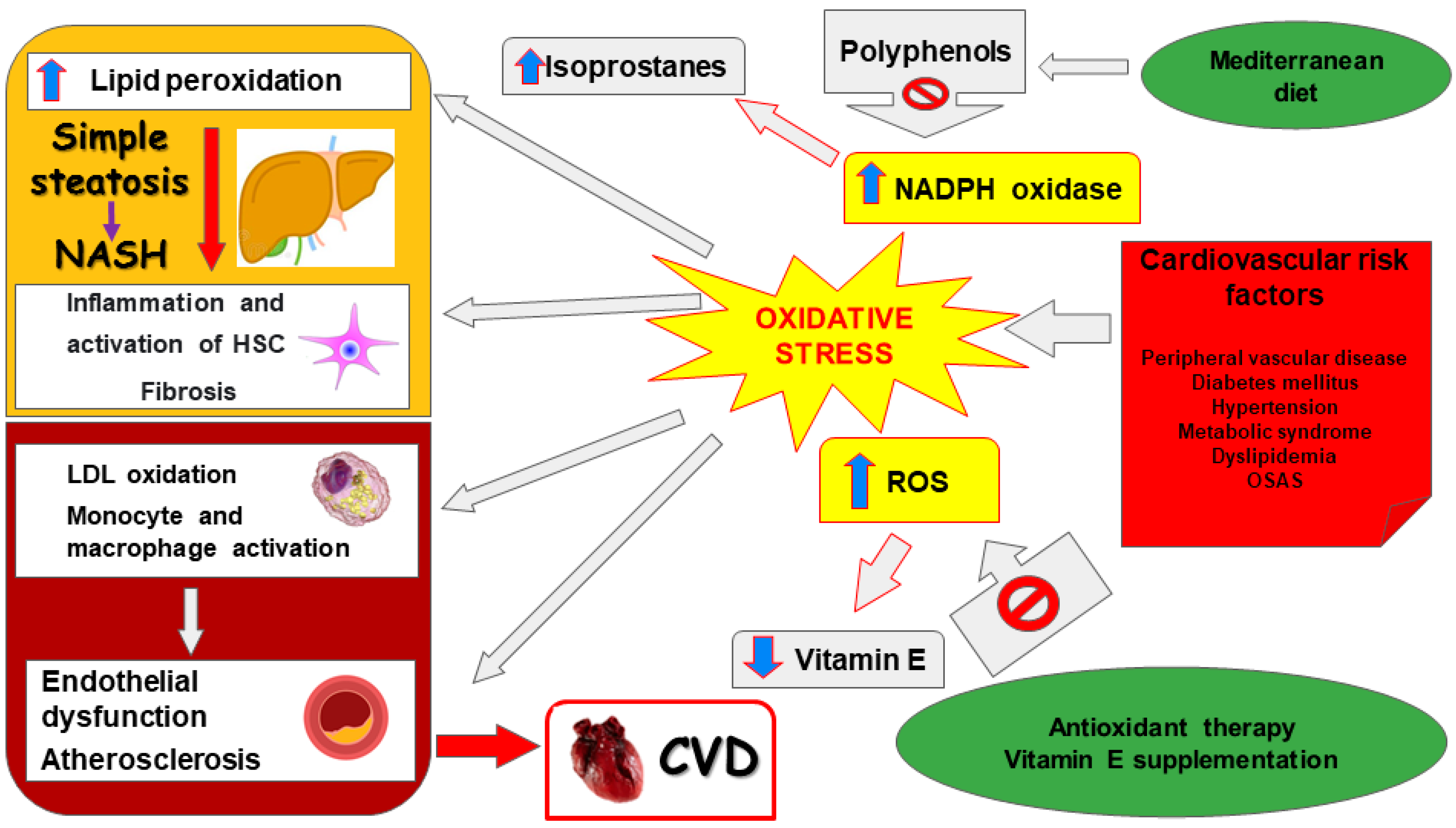{kind=link}
{kind=link}
| Oxidative Stress Markers | Disease Status | Experimental Model and Species | Sample | Changes in Concentration/ Activity/Expression | Reference(s) |
|---|---|---|---|---|---|
| TBARS/MDA | Steatosis, NASH | HFD, HF-HSD, MCD, ob/ob mice, CDHF diet | Liver | ↑ | [310,311] |
| HCD | ↓ | ||||
| HFD, MCD | NS | ||||
| 4-HNE | Steatosis, NASH | HFD, MCD, HF-HSD, CDHF diet | Liver | ↑ | [312,313] |
| 8-OH-dG | NASH | MCD, ob/ob mice HFMCD | Liver | ↑ ↓ | [314] |
| 8-isoprostane | NASH | HFMCD | Liver | ↑ | [315] |
| Protein carbonyl | Steatosis, NASH | HFD, MCD HFD | Liver | ↑ NS | [316] |
| Nitrotyrosine | NASH | HFD, ob/ob mice | Liver | ↑ | [317] |
| Periostin | NASH | HFD, ob/ob mice | Serum, liver | ↑ | [318] |
| CYP2E1 | Steatosis, NASH | HFD, HF-HSD, CDHF | Liver | ↑ | [311,319] |
| Dityrosine | Steatosis | HFD | Liver | ↑ | [320] |
| Hydroxyproline | NASH | MCD/WD | Liver | ↑ | [321] |
| H2O2 | NASH | MCD | Liver | ↑ | [309] |
| Lipid peroxide | NASH | MCD | Liver | ↑ | [322] |
| NADPH oxidase | NASH | ob/ob mice | Liver | ↑ | [323] |
| Xanthine oxidase | Steatosis | HFD | Liver | ↑ | [324] |
| Oxidative Stress Markers | Disease Status | Sample | Changes in Concentration/ Activity/Expression | Reference(s) |
|---|---|---|---|---|
| TBARS/MDA | Steatosis, NASH | Serum, liver, blood | ↑ | [87,117,328] |
| Serum | NS | |||
| 4-HNE | NASH | Liver | ↑ | [329] |
| Hydroperoxides | NASH | Liver | ↑ | [330] |
| 8-OH-dG | Steatosis, NASH | Liver, plasma Liver | ↑ NS | [331] |
| 8-isoprostane | NASH | Plasma | NS | [326] |
| Protein carbonyl | Steatosis, NASH | Liver | ↑ | [327] |
| Nitrotyrosine | Steatosis, NASH | Liver | ↑ | [25] |
| Blood | NS | |||
| Periostin | Steatosis, NASH | Serum, plasma, liver Serum, plasma | ↑ | [332,333,334,335] |
| NS | ||||
| Nitric oxide | Steatosis, NASH | Serum, blood | ↑ | [336,337] |
| CYP2E1 | Steatosis, NASH | Liver | ↑ | [338] |
| NS |
| Antioxidant Marker | Disease Status | Experimental Model and Species | Sample | Changes in Concentration/ Activity/Expression | Reference(s) |
|---|---|---|---|---|---|
| SOD | Steatosis, NASH | HF, HFD, OLETF rats, MCD | Liver | ↓ | [344,346] |
| HFD, MCD | ↑ | ||||
| HFD | NS | ||||
| Catalase | Steatosis, NASH | HFD, MCD, HCD | Liver | ↓ | [117,343] |
| MCD | ↑ | ||||
| GPx | Steatosis, NASH | HFD, MCD, HF | Liver | ↓ | [326,327] |
| MCD | ↑ | ||||
| HFD | NS | ||||
| GSH | Steatosis, NASH | HFD, HCD, MCD, ob/ob mice | Liver | ↓ | [346] |
| OLETF rats, HF MCD | ↑ | ||||
| GR | NASH | MCD | Liver | ↓ | [340] |
| Antioxidant Marker | Disease Status | Sample | Changes in Concentration/ Activity/Expression | Reference(s) |
|---|---|---|---|---|
| SOD | Steatosis, NASH | Serum, plasma, liver | ↓ | [344,346] |
| Blood, serum | ↑ | |||
| Serum | NS | |||
| Catalase | Steatosis, NASH | Plasma, blood, liver | ↓ | [117,343] |
| Serum | NS | |||
| GPx | Steatosis, NASH | Liver, serum | ↓ | [326,327] |
| Blood | ↑ | |||
| Serum | NS | |||
| GSH | Steatosis, NASH | Liver, blood | ↓ | [346] |
| Serum | ↑ | |||
| GR | Steatosis, NASH | Serum, blood | ↑ | [344] |
| TRX | Steatosis | Serum | ↑ | [345] |
| α-Tocopherol | Steatosis, NASH | Serum | ↓ | [333] |
| NS | ||||
| Ubiquinone | Steatosis | Serum | ↓ | [347] |
| Bilirubin | Steatosis, NASH | Serum | ↓ | [337] |
| Ascorbic acid | Steatosis, NASH | Serum | ↓ | [346] |
| NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirne, C.; Croce, E.; Di Benedetto, D.; Cantaluppi, V.; Comi, C.; Sainaghi, P.P.; Minisini, R.; Grossini, E.; Pirisi, M. Oxidative Stress in Non-Alcoholic Fatty Liver Disease. Livers 2022, 2, 30-76. https://doi.org/10.3390/livers2010003
Smirne C, Croce E, Di Benedetto D, Cantaluppi V, Comi C, Sainaghi PP, Minisini R, Grossini E, Pirisi M. Oxidative Stress in Non-Alcoholic Fatty Liver Disease. Livers. 2022; 2(1):30-76. https://doi.org/10.3390/livers2010003
Chicago/Turabian StyleSmirne, Carlo, Eleonora Croce, Davide Di Benedetto, Vincenzo Cantaluppi, Cristoforo Comi, Pier Paolo Sainaghi, Rosalba Minisini, Elena Grossini, and Mario Pirisi. 2022. "Oxidative Stress in Non-Alcoholic Fatty Liver Disease" Livers 2, no. 1: 30-76. https://doi.org/10.3390/livers2010003
APA StyleSmirne, C., Croce, E., Di Benedetto, D., Cantaluppi, V., Comi, C., Sainaghi, P. P., Minisini, R., Grossini, E., & Pirisi, M. (2022). Oxidative Stress in Non-Alcoholic Fatty Liver Disease. Livers, 2(1), 30-76. https://doi.org/10.3390/livers2010003