
Targeting Gut–Liver Axis for Treatment of Liver Fibrosis and Portal Hypertension


Abstract
:1. Introduction
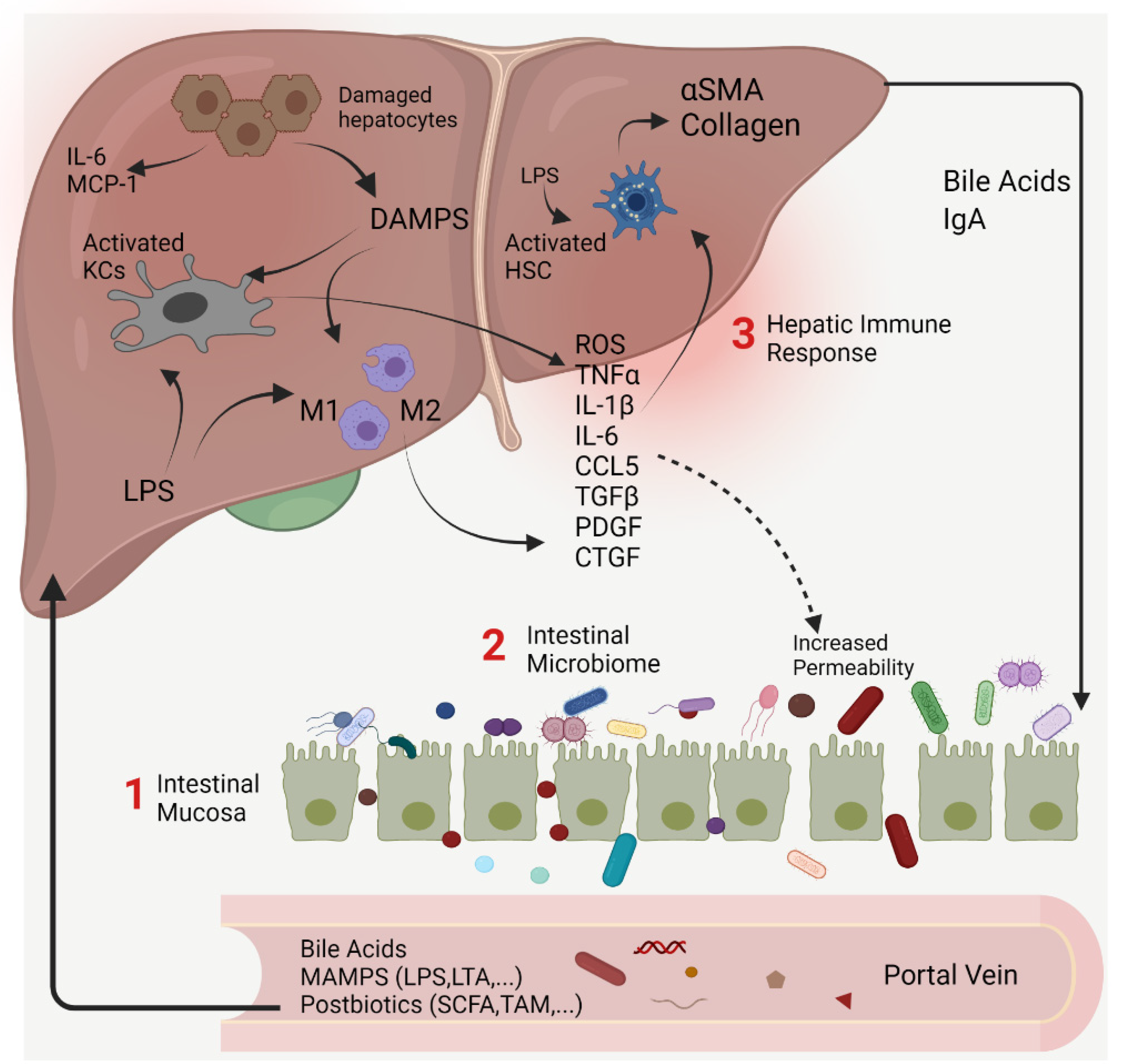
2. The Gut–Liver Axis at the Frontier of Host–Microbial Interactions
Intestinal Permeability
3. Therapies Targeting the Gut–Liver Axis to Improve Liver Fibrosis and Portal Hypertension
3.1. Interventions Targeting the Intestinal Mucosa
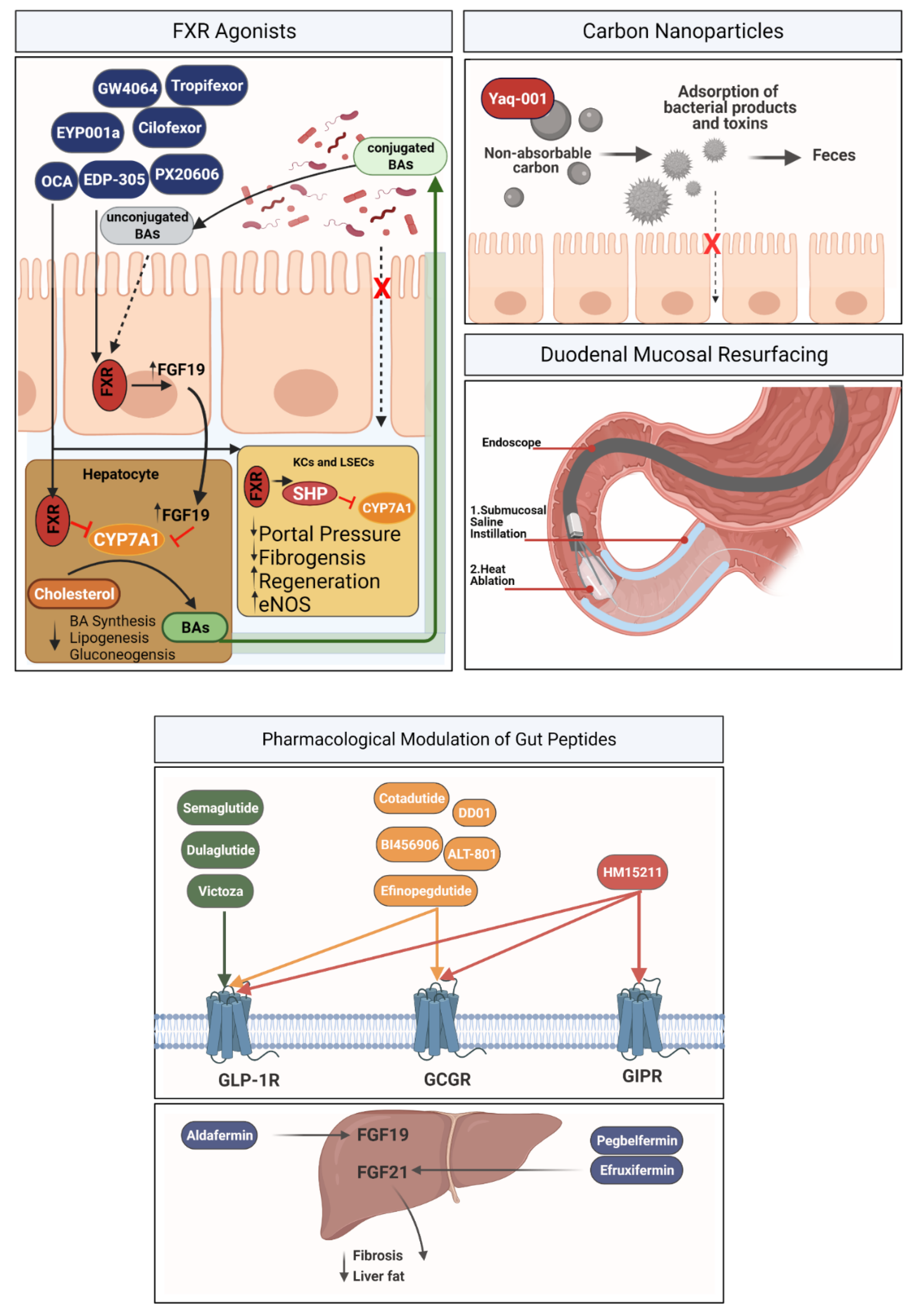
3.1.1. FXR Agonists
3.1.2. Carbon Nanoparticles
3.1.3. Duodenal Mucosal Resurfacing
3.1.4. Pharmacological Modulation of Gut Peptides
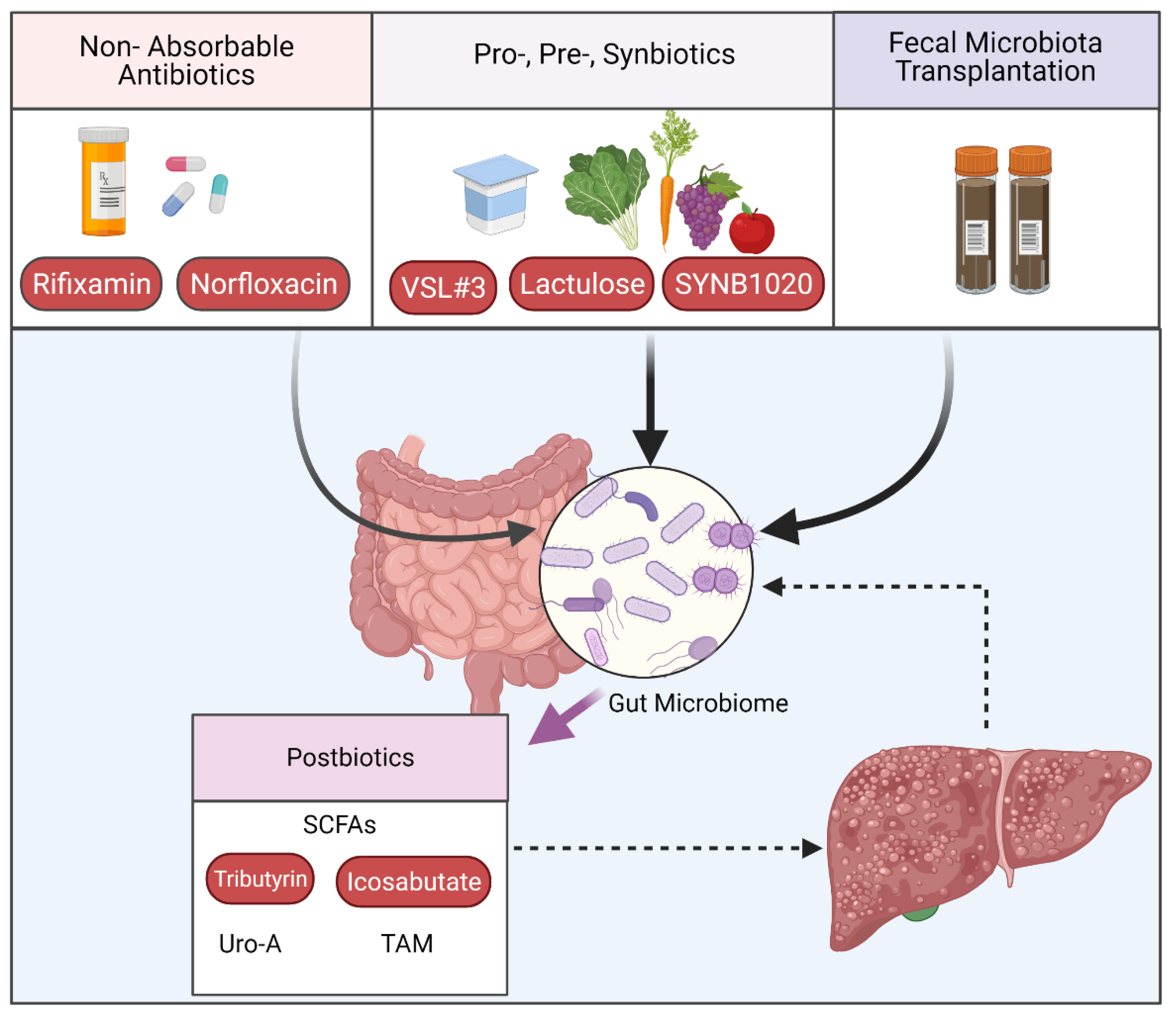
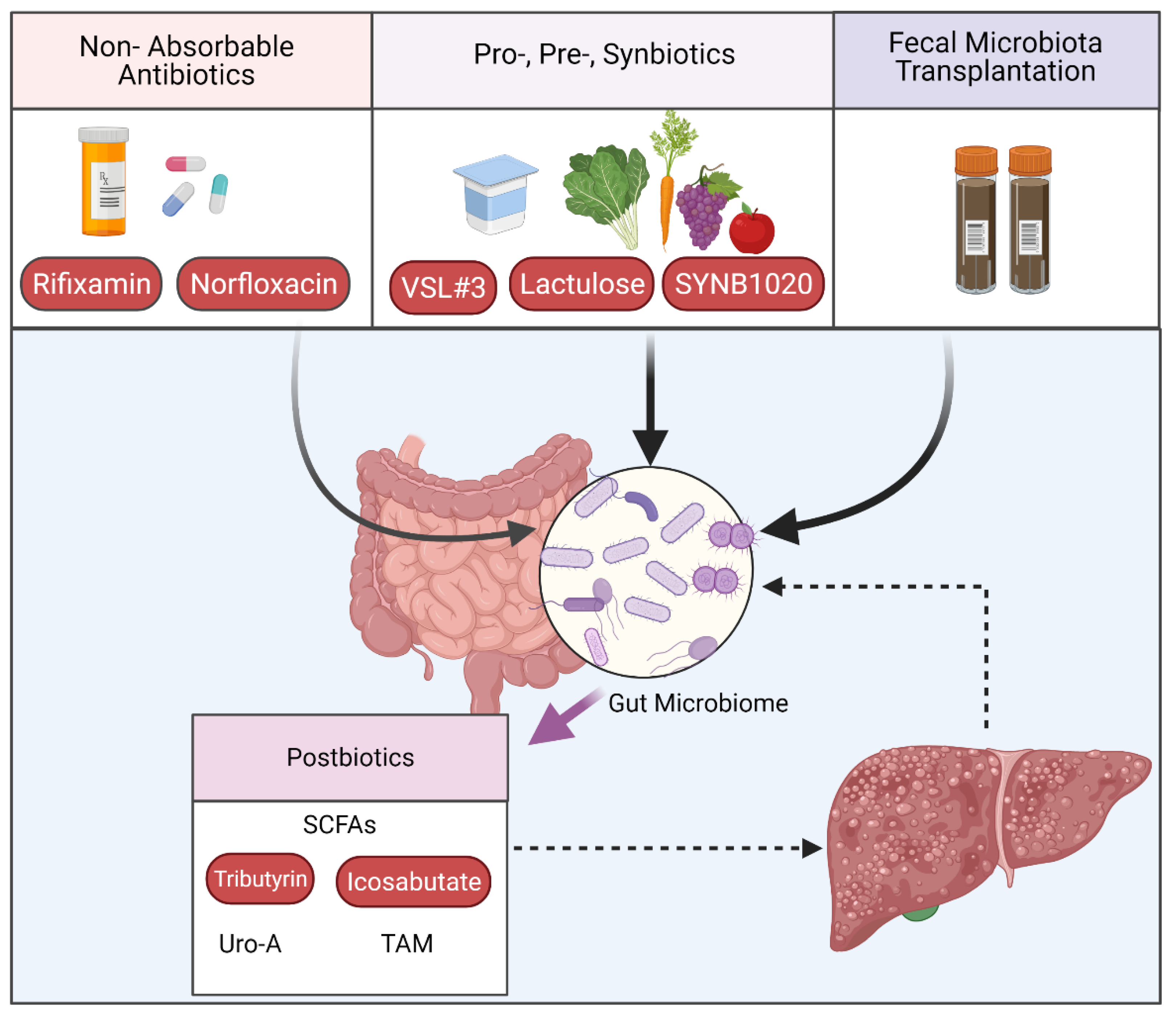
3.2. Interventions Targeting the Intestinal Microbiome
3.2.1. Targeting Microbiome Composition
3.2.2. Postbiotics
4. Interventions Targeting Hepatic Immune Response
4.1. Targeting Pattern Recognition Receptors
4.1.1. Key Toll-like Receptors in Liver Fibrosis
4.1.2. NLRP3
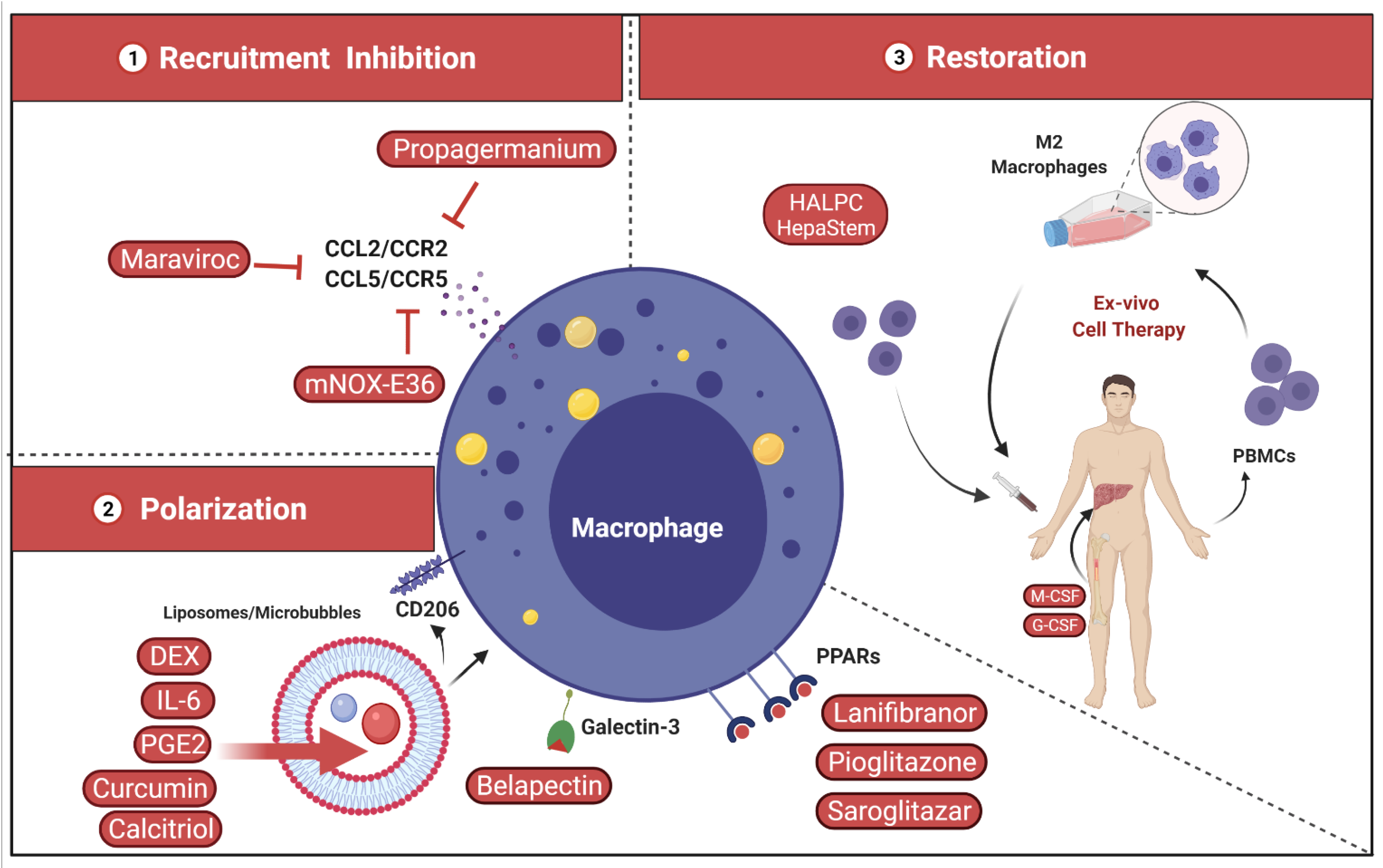
4.2. Targeting Liver Macrophages
4.2.1. Inhibition of Inflammatory Monocyte Recruitment
4.2.2. Shape and Polarization of Hepatic Macrophage Function
4.2.3. Restoration of Hepatic Macrophage Count and Function
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations
| AASLD | American Association for the Study of Liver Diseases |
| ACLF | Acute-on-chronic liver failure |
| ALD | Alcoholic liver disease |
| ASH | Alcoholic steatohepatitis |
| BAs | Bile acids |
| BT | Bacterial translocation |
| CCL2/MCP-1 | Chemokine (C-C motif) ligand 2/ Monocyte chemoattractant protein-1 |
| CCL5 | Chemokine (C-C motif) ligand 5 |
| CCl4 | Carbon tetrachloride |
| CCR5 | C-C chemokine receptor type 5 |
| CLD | Chronic liver disease |
| CTGF/CCN2 | Connective tissue growth factor |
| CVC | Cenicriviroc |
| DAMPs | Damage-associated molecular patterns |
| DMR | Duodenal mucosal resurfacing |
| DEX | Dexamethasone |
| ECM | Extracellular matrix |
| ESAL | European Association for the Study of the Liver |
| FGF19 | Fibroblast growth factor 19 |
| FGF21 | Fibroblast growth factor 21 |
| FMT | Fecal microbiota transplantation |
| FXR | Farnesoid X receptor |
| Gal-3 | Galectin-3 |
| G-CSF | Granulocyte colony-stimulating factor |
| GGG | Glucagon/GIP/GLP-1 |
| GIP | Glucose-dependent insulinotropic peptide |
| GLP-1 | Glucagon like peptide-1 |
| GLP-1R | Glucagon-like peptide-1 receptor |
| HE | Hepatic encephalopathy |
| HMGB1 | High-mobility group box 1 |
| HSCs | Hepatic stellate cells |
| HVPG | Hepatic venous pressure gradient |
| IL-1β | Interleukin one beta |
| IL-6 | Interleukin six |
| ISAPP | International Scientific Association for Probiotics and Prebiotics |
| KCs | Kupffer cells |
| LPS | Lipopolysaccharide |
| LBP | Lipopolysaccharide binding protein |
| LSECs | Liver sinusoidal endothelial cells |
| mAb | Monoclonal antibody |
| MAFLD | Metabolic associated fatty liver disease |
| MAMPS | Microbial -associated molecular patterns |
| M-CSF | Macrophage-colony stimulating factor |
| MCD | Methionine Choline- deficient |
| MELD | Model for end-stage liver disease |
| mtDNA | Mitochondrial DNA |
| NAS | NAFLD activity score |
| NASH | Non-alcoholic steatohepatitis |
| NF-κB | Nuclear factor-kappa B |
| NK | Natural killer |
| NSBBs | Nonselective beta blockers |
| OCA | Obeticholic acid |
| PAMPS | Pathogen-associated molecular patterns |
| PDGF | Platelet-derived growth factor |
| PPARs | Peroxisome proliferator-activated receptors |
| PRRs | Pattern recognition receptors |
| PTH | Portal hypertension |
| RCT | Randomized control trial |
| RIG-1 | Retinoic acid-inducible gene I |
| ROS | Reactive oxygen species |
| SBP | Spontaneous bacterial peritonitis |
| SCFA(s) | Short-chain fatty acids |
| SSD | Soluble solid dispersion |
| STING | Stimulator of interferon genes |
| TAA | Thioacetamide |
| TGF-β | Transforming growth factor beta |
| TGR5 | Takeda G-protein-coupled receptor 5 |
| TLRs | Toll-like receptors |
| TMA | Trimethylamine |
| TMAO | Trimethylamine-N-oxide |
| TNF-α | Tumor necrosis factor alpha |
| Uro A | Urolithin A |
| VH | Variceal hemorrhage |
| ZO-1 | Zonula occludens-1 |
| α-SMA | Alpha Smooth muscle actin |
References
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Zhai, M.; Liu, Z.; Long, J.; Zhou, Q.; Yang, L.; Zhou, Q.; Liu, S.; Dai, Y. The incidence trends of liver cirrhosis caused by nonalcoholic steatohepatitis via the GBD study 2017. Sci. Rep. 2021, 11, 5195. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases From 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef]
- Adams, L.A.; Roberts, S.K.; Strasser, S.I.; Mahady, S.E.; Powell, E.; Estes, C.; Razavi, H.; George, J. Nonalcoholic fatty liver disease burden: Australia, 2019–2030. J. Gastroenterol. Hepatol. 2020, 35, 1628–1635. [Google Scholar] [CrossRef] [Green Version]
- Collaborators, G.B.D.C. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef] [Green Version]
- Australian Institute of Health and Welfare. Leading Cause of Premature Mortality in Australia Fact Sheet: Liver Disease, Cat. no. PHE 199; AIHW: Canberra, Australia, 2015. [Google Scholar]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Hernaez, R.; Kramer, J.R.; Liu, Y.; Tansel, A.; Natarajan, Y.; Hussain, K.B.; Gines, P.; Sola, E.; Moreau, R.; Gerbes, A.; et al. Prevalence and short-term mortality of acute-on-chronic liver failure: A national cohort study from the USA. J. Hepatol. 2019, 70, 639–647. [Google Scholar] [CrossRef] [PubMed]
- WHO Global Health Observatory data repository. Liver Cirrhosis (15+), Age-Standardized Death Rates by Country for 2016. Available online: https://apps.who.int/gho/data/view.main.53420 (accessed on 10 July 2021).
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Bosch, J.; Abraldes, J.G.; Berzigotti, A.; Garcia-Pagan, J.C. The clinical use of HVPG measurements in chronic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, C.; Groszmann, R.J.; Garcia-Tsao, G.; Bosch, J.; Grace, N.; Burroughs, A.; Planas, R.; Escorsell, A.; Garcia-Pagan, J.C.; Makuch, R.; et al. Hepatic venous pressure gradient predicts development of hepatocellular carcinoma independently of severity of cirrhosis. J. Hepatol. 2009, 50, 923–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, V.; Moreau, R.; Kamath, P.S.; Jalan, R.; Gines, P.; Nevens, F.; Fernandez, J.; To, U.; Garcia-Tsao, G.; Schnabl, B. Acute-on-chronic liver failure in cirrhosis. Nat. Rev. Dis. Primers 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, R.; Jalan, R.; Gines, P.; Pavesi, M.; Angeli, P.; Cordoba, J.; Durand, F.; Gustot, T.; Saliba, F.; Domenicali, M.; et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013, 144, 1426–1437. [Google Scholar] [CrossRef]
- Gustot, T.; Stadlbauer, V.; Laleman, W.; Alessandria, C.; Thursz, M. Transition to decompensation and acute-on-chronic liver failure: Role of predisposing factors and precipitating events. J. Hepatol. 2021, 75, S36–S48. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The microbiota in cirrhosis and its role in hepatic decompensation. J. Hepatol. 2021, 75, S67–S81. [Google Scholar] [CrossRef]
- Reiberger, T.; Ferlitsch, A.; Payer, B.A.; Mandorfer, M.; Heinisch, B.B.; Hayden, H.; Lammert, F.; Trauner, M.; Peck-Radosavljevic, M.; Vogelsang, H.; et al. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J. Hepatol. 2013, 58, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.G.; Mendoza, Y.P.; Bosch, J. Beta-blockers in cirrhosis: Evidence-based indications and limitations. JHEP Rep. 2020, 2, 100063. [Google Scholar] [CrossRef] [Green Version]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Rep. 2019, 1, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. Publisher Correction: The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 785. [Google Scholar] [CrossRef]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Baffy, G. Potential mechanisms linking gut microbiota and portal hypertension. Liver Int. 2019, 39, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, G.; Gustot, T.; Mookerjee, R.P.; Garcia-Pagan, J.C.; Fallon, M.B.; Shah, V.H.; Moreau, R.; Jalan, R. Inflammation and portal hypertension—the undiscovered country. J. Hepatol. 2014, 61, 155–163. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Grander, C.; Moschen, A.R. How does the microbiome affect liver disease? Clin. Liver Dis. 2016, 8, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Davis, B.C.; Bajaj, J.S. The Human Gut Microbiome in Liver Diseases. Semin. Liver Dis. 2017, 37, 128–140. [Google Scholar] [CrossRef]
- Cho, I.; Yamanishi, S.; Cox, L.; Methe, B.A.; Zavadil, J.; Li, K.; Gao, Z.; Mahana, D.; Raju, K.; Teitler, I.; et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 2012, 488, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Eksteen, B.; Curbishley, S.M. Immunology of the gut and liver: A love/hate relationship. Gut 2008, 57, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A. Metabolic liver disease—what’s in a name? Nat. Rev. Endocrinol. 2021, 17, 79–80. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.L.; Chen, H.; Wang, C.L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Goel, A.; Gupta, M.; Aggarwal, R. Gut microbiota and liver disease. J. Gastroenterol. Hepatol. 2014, 29, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebicka, J.; Reiberger, T.; Laleman, W. Gut-Liver Axis Links Portal Hypertension to Acute-on-Chronic Liver Failure. Visc. Med. 2018, 34, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Halilbasic, E.; Fuchs, C.; Traussnigg, S.; Trauner, M. Farnesoid X Receptor Agonists and Other Bile Acid Signaling Strategies for Treatment of Liver Disease. Dig. Dis. 2016, 34, 580–588. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Schwabl, P.; Reiberger, T.; Trauner, M. Liver Capsule: FXR agonists against liver disease. Hepatology 2016, 64, 1773. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Farre, R.; Trebicka, J.; Komuta, M.; Roskams, T.; Klein, S.; Elst, I.V.; Windmolders, P.; Vanuytsel, T.; Nevens, F.; et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 2014, 59, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kuruba, R.; Wilson, A.; Gao, X.; Zhang, Y.; Li, S. Inhibition of endothelin-1-mediated contraction of hepatic stellate cells by FXR ligand. PLoS ONE 2010, 5, e13955. [Google Scholar] [CrossRef] [Green Version]
- Ubeda, M.; Lario, M.; Munoz, L.; Borrero, M.J.; Rodriguez-Serrano, M.; Sanchez-Diaz, A.M.; Del Campo, R.; Lledo, L.; Pastor, O.; Garcia-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Farre, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am. J. Pathol. 2015, 185, 409–419. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Mookerjee, R.; Rosselli, M.; Pieri, G.; Beecher-Jones, T.; Hooshmand-Rad, R.; Chouhan, M.; Mehta, G.; Jalan, R.; Shapiro, D. Effects of the FXR agonist obeticholic acid on hepatic venous pressure gradient (HVPG) in alcoholic cirrhosis: A proof of concept phase 2a study. J. Hepatol. 2014, 60, S7–S8. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Pares, A.; Mason, A.; et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, P.; Wei, G.; Huang, P.; Li, W.; Qi, X.; Lin, Y.; Vaid, K.A.; Wang, J.; Zhang, S.; Li, Y.; et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 2020, 40, 1655–1669. [Google Scholar] [CrossRef]
- ENYO PHARMA. Vonafexor (EYP001) in NASH. 2019. Available online: http://www.enyopharma.com/pipeline/vonafexor-in-nash/ (accessed on 15 July 2021).
- Schwabl, P.; Hambruch, E.; Seeland, B.A.; Hayden, H.; Wagner, M.; Garnys, L.; Strobel, B.; Schubert, T.L.; Riedl, F.; Mitteregger, D.; et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J. Hepatol. 2017, 66, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef]
- Schwabl, P.; Hambruch, E.; Budas, G.R.; Supper, P.; Burnet, M.; Liles, J.T.; Birkel, M.; Brusilovskaya, K.; Konigshofer, P.; Peck-Radosavljevic, M.; et al. The Non-Steroidal FXR Agonist Cilofexor Improves Portal Hypertension and Reduces Hepatic Fibrosis in a Rat NASH Model. Biomedicines 2021, 9, 60. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Y.; Liu, Y.; Wang, W.; Tian, X.; Chen, S.; Lu, Y.; Du, J.; Cai, W. A nonbile acid farnesoid X receptor agonist tropifexor potently inhibits cholestatic liver injury and fibrosis by modulating the gut-liver axis. Liver Int. 2021. [Google Scholar] [CrossRef]
- Lucas, K.J.; Lopez, P.; Lawitz, E.; Sheikh, A.; Aizenberg, D.; Hsia, S.; Boon Bee, G.G.; Vierling, J.; Frias, J.; White, J. Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig. Liver Dis. 2021, 52, e38. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated With the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Renga, B.; Cipriani, S.; Carino, A.; Simonetti, M.; Zampella, A.; Fiorucci, S. Reversal of Endothelial Dysfunction by GPBAR1 Agonism in Portal Hypertension Involves a AKT/FOXOA1 Dependent Regulation of H2S Generation and Endothelin-1. PLoS ONE 2015, 10, e0141082. [Google Scholar] [CrossRef] [Green Version]
- Klindt, C.; Reich, M.; Hellwig, B.; Stindt, J.; Rahnenfuhrer, J.; Hengstler, J.G.; Kohrer, K.; Schoonjans, K.; Haussinger, D.; Keitel, V. The G Protein-Coupled Bile Acid Receptor TGR5 (Gpbar1) Modulates Endothelin-1 Signaling in Liver. Cells 2019, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Macnaughtan, J.; Ranchal, I.; Soeda, J.; Sawhney, R.; Oben, J.; Davies, N.; Mookerjee, R.; Marchesi, J.; Cox, J.; Jalan, R. O091: Oral therapy with non-absorbable carbons of controlled porosity (YAQ-001) selectively modulates stool microbiome and its function and this is associated with restoration of immune function and inflammasome activation. J. Hepatol. 2015, 62, S240. [Google Scholar] [CrossRef]
- Haidry, R.J.; van Baar, A.C.; Galvao Neto, M.P.; Rajagopalan, H.; Caplan, J.; Levin, P.S.; Bergman, J.J.; Rodriguez, L.; Deviere, J.; Thompson, C.C. Duodenal mucosal resurfacing: Proof-of-concept, procedural development, and initial implementation in the clinical setting. Gastrointest. Endosc. 2019, 90, 673–681. [Google Scholar] [CrossRef]
- Van Baar, A.C.G.; Holleman, F.; Crenier, L.; Haidry, R.; Magee, C.; Hopkins, D.; Rodriguez Grunert, L.; Galvao Neto, M.; Vignolo, P.; Hayee, B.; et al. Endoscopic duodenal mucosal resurfacing for the treatment of type 2 diabetes mellitus: One year results from the first international, open-label, prospective, multicentre study. Gut 2020, 69, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology 2002, 122, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribble, F.M.; Reimann, F. Metabolic Messengers: Glucagon-like peptide 1. Nat. Metab. 2021, 3, 142–148. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A.; Clausen, W.H.; Elbrond, B.; Gough, S.C.; Tomlinson, J.W.; Newsome, P.N. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: Individual patient data meta-analysis of the LEAD program. Aliment. Pharmacol. Ther. 2013, 37, 234–242. [Google Scholar] [CrossRef]
- Nahra, R.; Wang, T.; Gadde, K.M.; Oscarsson, J.; Stumvoll, M.; Jermutus, L.; Hirshberg, B.; Ambery, P. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults With Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care 2021. [Google Scholar] [CrossRef]
- Choi, J.; Kim, J.K.; Lee, S.M.; Kwon, H.; Lee, J.; Bae, S.; Kim, D.; Choi, I.Y. 1830-P:Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in CDHFD-Induced NASH and Fibrosis Mice. Am. Diabetes Assoc. Diabetes 2020, 69 (Suppl. 1). [Google Scholar] [CrossRef]
- Abdelmalek, M.; Choi, J.; Kim, Y.; Seo, K.; Hompesch, M.; Baek, S. LBP03-HM15211, a novel GLP-1/GIP/Glucagon triple-receptor co-agonist significantly reduces liver fat and body weight in obese subjects with non-alcoholic fatty liver disease: A Phase 1b/2a, multi-center, randomized, placebo-controlled trial. J. Hepatol. 2020, 73, S124. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; Investigators, N.N. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Choudhary, N.S.; Singh, M.K.; Wasir, J.S.; Kaur, P.; Gill, H.K.; Bano, T.; Farooqui, K.J.; et al. Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: Randomised controlled trial (D-LIFT trial). Diabetologia 2020, 63, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.R.; Johnston, I.M.; Nolan, J.D.; Vassie, C.; Pruzanski, M.E.; Shapiro, D.A. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment. Pharmacol. Ther. 2015, 41, 54–64. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled, phase 2a trial. Nat. Med. 2021, 27, 1262–1271. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2019, 30, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clement, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci. Rep. 2016, 6, 34055. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de la Hera, A.; Gonzalez, M.; Moya, J.L.; Calleja, J.L.; Monserrat, J.; Ruiz-del-Arbol, L.; Alvarez-Mon, M. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology 2003, 37, 208–217. [Google Scholar] [CrossRef]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taura, P.; Fuster, J.; Garcia-Valdecasas, J.C.; Lacy, A.; Suarez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar] [CrossRef]
- Wiest, R.; Garcia-Tsao, G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005, 41, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Saxinger, L.; Ma, M.; Prado, V.; Fernandez, J.; Kumar, D.; Gonzalez-Abraldes, J.; Keough, A.; Bastiampillai, R.; Carbonneau, M.; et al. Bacterial infections in acute variceal hemorrhage despite antibiotics-a multicenter study of predictors and clinical impact. United Eur. Gastroenterol. J. 2017, 5, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadamrad, S.; McCoy, K.D.; Geuking, M.B.; Sagesser, H.; Kirundi, J.; Macpherson, A.J.; De Gottardi, A. Attenuated portal hypertension in germ-free mice: Function of bacterial flora on the development of mesenteric lymphatic and blood vessels. Hepatology 2015, 61, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calanni, F.; Renzulli, C.; Barbanti, M.; Viscomi, G.C. Rifaximin: Beyond the traditional antibiotic activity. J. Antibiot. 2014, 67, 667–670. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. Electronic address and L. European Association for the Study of the, L. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460. [Google Scholar] [CrossRef] [Green Version]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Takaya, H.; Saikawa, S.; Furukawa, M.; Sato, S.; Kawaratani, H.; Kitade, M.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Rifaximin ameliorates hepatic encephalopathy and endotoxemia without affecting the gut microbiome diversity. World J. Gastroenterol. 2017, 23, 8355–8366. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heimanson, Z.; Israel, R.; Sanyal, A. SAT-014-Efficacy of rifaximin soluble solid dispersion in patients with early decompensated cirrhosis and a Conn score of 0: A post hoc analysis of a randomized, double-blind, placebo-controlled trial. J. Hepatol. 2019, 70, e631. [Google Scholar] [CrossRef]
- Zhu, Q.; Zou, L.; Jagavelu, K.; Simonetto, D.A.; Huebert, R.C.; Jiang, Z.D.; DuPont, H.L.; Shah, V.H. Intestinal decontamination inhibits TLR4 dependent fibronectin-mediated cross-talk between stellate cells and endothelial cells in liver fibrosis in mice. J. Hepatol. 2012, 56, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachogiannakos, J.; Saveriadis, A.S.; Viazis, N.; Theodoropoulos, I.; Foudoulis, K.; Manolakopoulos, S.; Raptis, S.; Karamanolis, D.G. Intestinal decontamination improves liver haemodynamics in patients with alcohol-related decompensated cirrhosis. Aliment. Pharmacol. Ther. 2009, 29, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Kimer, N.; Pedersen, J.S.; Busk, T.M.; Gluud, L.L.; Hobolth, L.; Krag, A.; Moller, S.; Bendtsen, F.; Copenhagen Rifaximin Study, G. Rifaximin has no effect on hemodynamics in decompensated cirrhosis: A randomized, double-blind, placebo-controlled trial. Hepatology 2017, 65, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.L.; Kim, M.Y.; Jang, Y.O.; Baik, S.K.; Kwon, S.O. Rifaximin and Propranolol Combination Therapy Is More Effective than Propranolol Monotherapy for the Reduction of Portal Pressure: An Open Randomized Controlled Pilot Study. Gut Liver 2017, 11, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, Y.; Kawaratani, H.; Kaya, D.; Tsuji, Y.; Ozutsumi, T.; Furukawa, M.; Kitagawa, K.; Sato, S.; Nishimura, N.; Sawada, Y.; et al. Effective Combination Therapy of Angiotensin-II Receptor Blocker and Rifaximin for Hepatic Fibrosis in Rat Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5589. [Google Scholar] [CrossRef] [PubMed]
- Hennenberg, M.; Trebicka, J.; Buecher, D.; Heller, J.; Sauerbruch, T. Lack of effect of norfloxacin on hyperdynamic circulation in bile duct-ligated rats despite reduction of endothelial nitric oxide synthase function: Result of unchanged vascular Rho-kinase? Liver Int. 2009, 29, 933–941. [Google Scholar] [CrossRef]
- Kemp, W.; Colman, J.; Thompson, K.; Madan, A.; Vincent, M.; Chin-Dusting, J.; Kompa, A.; Krum, H.; Roberts, S. Norfloxacin treatment for clinically significant portal hypertension: Results of a randomised double-blind placebo-controlled crossover trial. Liver Int. 2009, 29, 427–433. [Google Scholar] [CrossRef]
- Moreau, R.; Elkrief, L.; Bureau, C.; Perarnau, J.M.; Thevenot, T.; Saliba, F.; Louvet, A.; Nahon, P.; Lannes, A.; Anty, R.; et al. Effects of Long-term Norfloxacin Therapy in Patients With Advanced Cirrhosis. Gastroenterology 2018, 155, 1816–1827. [Google Scholar] [CrossRef] [Green Version]
- Rasaratnam, B.; Kaye, D.; Jennings, G.; Dudley, F.; Chin-Dusting, J. The effect of selective intestinal decontamination on the hyperdynamic circulatory state in cirrhosis. A randomized trial. Ann. Intern. Med. 2003, 139, 186–193. [Google Scholar] [CrossRef]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhiman, R.K.; Rana, B.; Agrawal, S.; Garg, A.; Chopra, M.; Thumburu, K.K.; Khattri, A.; Malhotra, S.; Duseja, A.; Chawla, Y.K. Probiotic VSL#3 reduces liver disease severity and hospitalization in patients with cirrhosis: A randomized, controlled trial. Gastroenterology 2014, 147, 1327–1337 e1323. [Google Scholar] [CrossRef] [PubMed]
- Duseja, A.; Acharya, S.K.; Mehta, M.; Chhabra, S.; Rana, S.; Das, A.; Dattagupta, S.; Dhiman, R.K.; Chawla, Y.K. High potency multistrain probiotic improves liver histology in non-alcoholic fatty liver disease (NAFLD): A randomised, double-blind, proof of concept study. BMJ Open Gastroenterol. 2019, 6, e000315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincon, D.; Vaquero, J.; Hernando, A.; Galindo, E.; Ripoll, C.; Puerto, M.; Salcedo, M.; Frances, R.; Matilla, A.; Catalina, M.V.; et al. Oral probiotic VSL#3 attenuates the circulatory disturbances of patients with cirrhosis and ascites. Liver Int. 2014, 34, 1504–1512. [Google Scholar] [CrossRef]
- Jayakumar, S.; Carbonneau, M.; Hotte, N.; Befus, A.D.; St Laurent, C.; Owen, R.; McCarthy, M.; Madsen, K.; Bailey, R.J.; Ma, M.; et al. VSL#3 (R) probiotic therapy does not reduce portal pressures in patients with decompensated cirrhosis. Liver Int. 2013, 33, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Moncrief, K.; Madsen, K.; Arrieta, M.C.; Owen, R.J.; Bain, V.G.; Wong, W.W.; Ma, M.M. Effects of probiotic therapy on portal pressure in patients with cirrhosis: A pilot study. Liver Int. 2009, 29, 1110–1115. [Google Scholar] [CrossRef]
- Gupta, N.; Kumar, A.; Sharma, P.; Garg, V.; Sharma, B.C.; Sarin, S.K. Effects of the adjunctive probiotic VSL#3 on portal haemodynamics in patients with cirrhosis and large varices: A randomized trial. Liver Int. 2013, 33, 1148–1157. [Google Scholar] [CrossRef]
- Kurtz, C.B.; Millet, Y.A.; Puurunen, M.K.; Perreault, M.; Charbonneau, M.R.; Isabella, V.M.; Kotula, J.W.; Antipov, E.; Dagon, Y.; Denney, W.S.; et al. An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Rose, C.F.; Amodio, P.; Bajaj, J.S.; Dhiman, R.K.; Montagnese, S.; Taylor-Robinson, S.D.; Vilstrup, H.; Jalan, R. Hepatic encephalopathy: Novel insights into classification, pathophysiology and therapy. J. Hepatol. 2020, 73, 1526–1547. [Google Scholar] [CrossRef]
- Sharma, B.C.; Sharma, P.; Agrawal, A.; Sarin, S.K. Secondary prophylaxis of hepatic encephalopathy: An open-label randomized controlled trial of lactulose versus placebo. Gastroenterology 2009, 137, 885–891. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients With Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065. [Google Scholar] [CrossRef]
- Garcia-Lezana, T.; Raurell, I.; Bravo, M.; Torres-Arauz, M.; Salcedo, M.T.; Santiago, A.; Schoenenberger, A.; Manichanh, C.; Genesca, J.; Martell, M.; et al. Restoration of a healthy intestinal microbiota normalizes portal hypertension in a rat model of nonalcoholic steatohepatitis. Hepatology 2018, 67, 1485–1498. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Gavis, E.A.; Fagan, A.; Wade, J.B.; Thacker, L.R.; Fuchs, M.; Patel, S.; Davis, B.; Meador, J.; Puri, P.; et al. A Randomized Clinical Trial of Fecal Microbiota Transplant for Alcohol Use Disorder. Hepatology 2021, 73, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B.; Kunze, W.A.; Clerc, N. Nutrient tasting and signaling mechanisms in the gut. II. The intestine as a sensory organ: Neural, endocrine, and immune responses. Am. J. Physiol. 1999, 277, G922–G928. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeiro, M.H.; Ramirez, M.J.; Milagro, F.I.; Martinez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [Green Version]
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2015, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, H.; Li, B.; Lee, C.; Alganabi, M.; Zheng, S.; Pierro, A. Beneficial effects of butyrate in intestinal injury. J. Pediatr. Surg. 2020, 55, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Juanola, O.; Ferrusquia-Acosta, J.; Garcia-Villalba, R.; Zapater, P.; Magaz, M.; Marin, A.; Olivas, P.; Baiges, A.; Bellot, P.; Turon, F.; et al. Circulating levels of butyrate are inversely related to portal hypertension, endotoxemia, and systemic inflammation in patients with cirrhosis. FASEB J. 2019, 33, 11595–11605. [Google Scholar] [CrossRef] [PubMed]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Sanyal, A. Abstract: Icosabutate, A Novel Structurally Engineered Fatty Acid, Significantly Reduces Relevant Markers of NASH and Fibrosis in 16 Weeks: Interim Analysis Results of the ICONA Trial. Available online: https://easl.eu/press-release/treatment-advances-for-non-alcoholic-fatty-liver-disease-nafld-announced-at-ilc-2021/ (accessed on 15 July 2021).
- Singh, R.; Chandrashekharappa, S.; Bodduluri, S.R.; Baby, B.V.; Hegde, B.; Kotla, N.G.; Hiwale, A.A.; Saiyed, T.; Patel, P.; Vijay-Kumar, M.; et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun. 2019, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Jala, V.R.; Singh, R.; Chandrashekharappa, S.; Joshi-Barve, S.; McClain, C.; Bodduluri, B.; Vemula, P.K. Gut microbial metabolites as therapeutics to treat of alcoholic liver disease. J. Immunol. 2020, 204, 83.17. [Google Scholar]
- Schon, H.T.; Bartneck, M.; Borkham-Kamphorst, E.; Nattermann, J.; Lammers, T.; Tacke, F.; Weiskirchen, R. Pharmacological Intervention in Hepatic Stellate Cell Activation and Hepatic Fibrosis. Front. Pharmacol. 2016, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Le, T.H.; Shahidipour, H.; Read, S.A.; Ahlenstiel, G. The Role of Gut-Derived Microbial Antigens on Liver Fibrosis Initiation and Progression. Cells 2019, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Zeromski, J.; Kierepa, A.; Brzezicha, B.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Pattern Recognition Receptors: Significance of Expression in the Liver. Arch. Immunol. Ther. Exp. 2020, 68, 29. [Google Scholar] [CrossRef]
- Engelmann, C.; Sheikh, M.; Sharma, S.; Kondo, T.; Loeffler-Wirth, H.; Zheng, Y.B.; Novelli, S.; Hall, A.; Kerbert, A.J.C.; Macnaughtan, J.; et al. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. J. Hepatol. 2020, 73, 102–112. [Google Scholar] [CrossRef]
- Liaunardy-Jopeace, A.; Gay, N.J. Molecular and cellular regulation of toll-like receptor-4 activity induced by lipopolysaccharide ligands. Front. Immunol. 2014, 5, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Liu, M.; Sehrawat, T.S.; Shah, V.H. Regulation and functional roles of chemokines in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Aoyama, T.; Paik, Y.H.; Seki, E. Toll-like receptor signaling and liver fibrosis. Gastroenterol Res. Pract. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Feng, J.; Du, J.; Zhuo, Z.; Yang, S.; Zhang, W.; Wang, W.; Zhang, S.; Iwakura, Y.; Meng, G.; et al. Macrophage-derived IL-1alpha promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell Mol. Immunol. 2018, 15, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazawa, T.; Tsujimoto, T.; Kawaratani, H.; Fukui, H. Therapeutic approach to regulate innate immune response by Toll-like receptor 4 antagonist E5564 in rats with D-galactosamine-induced acute severe liver injury. J. Gastroenterol. Hepatol. 2009, 24, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, T.; Tsujimoto, T.; Kawaratani, H.; Fukui, H. Salvage effect of E5564, Toll-like receptor 4 antagonist on d-galactosamine and lipopolysaccharide-induced acute liver failure in rats. J. Gastroenterol. Hepatol. 2010, 25, 1009–1012. [Google Scholar] [CrossRef]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Monnet, E.; Lapeyre, G.; Poelgeest, E.V.; Jacqmin, P.; Graaf, K.; Reijers, J.; Moerland, M.; Burggraaf, J.; Min, C. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin. Pharmacol. Ther. 2017, 101, 200–208. [Google Scholar] [CrossRef]
- Bennett, R.G.; Simpson, R.L.; Hamel, F.G. Serelaxin increases the antifibrotic action of rosiglitazone in a model of hepatic fibrosis. World J. Gastroenterol. 2017, 23, 3999–4006. [Google Scholar] [CrossRef]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef]
- Vergis, N.; Atkinson, S.R.; Knapp, S.; Maurice, J.; Allison, M.; Austin, A.; Forrest, E.H.; Masson, S.; McCune, A.; Patch, D.; et al. In Patients With Severe Alcoholic Hepatitis, Prednisolone Increases Susceptibility to Infection and Infection-Related Mortality, and Is Associated With High Circulating Levels of Bacterial DNA. Gastroenterology 2017, 152, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, S.; Marcos, M.; Gattu, A.; Catalano, D.; Szabo, G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS ONE 2014, 9, e96864. [Google Scholar] [CrossRef]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Gabele, E.; Muhlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Scholmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Farooq, A.; Malik, A.; Trawick, B.N.; Berberich, D.W.; McClurg, J.P.; Galen, K.P.; Mehal, W. A novel small-molecule enantiomeric analogue of traditional (-)-morphinans has specific TLR9 antagonist properties and reduces sterile inflammation-induced organ damage. J. Immunol. 2013, 190, 4297–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaker, M.E.; Trawick, B.N.; Mehal, W.Z. The novel TLR9 antagonist COV08-0064 protects from ischemia/reperfusion injury in non-steatotic and steatotic mice livers. Biochem. Pharmacol. 2016, 112, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Fukui, R.; Motoi, Y.; Shibata, T.; Saitoh, S.I.; Sato, R.; Miyake, K. The protective effect of the anti-Toll-like receptor 9 antibody against acute cytokine storm caused by immunostimulatory DNA. Sci. Rep. 2017, 7, 44042. [Google Scholar] [CrossRef]
- Gilboa-Geffen, A.; Wolf, Y.; Hanin, G.; Melamed-Book, N.; Pick, M.; Bennett, E.R.; Greenberg, D.S.; Lester, S.; Rischmueller, M.; Soreq, H. Activation of the alternative NFkappaB pathway improves disease symptoms in a model of Sjogren’s syndrome. PLoS ONE 2011, 6, e28727. [Google Scholar] [CrossRef]
- Kiripolsky, J.; Kramer, J.M. Current and Emerging Evidence for Toll-Like Receptor Activation in Sjogren’s Syndrome. J. Immunol. Res. 2018, 2018, 1246818. [Google Scholar] [CrossRef]
- Byun, J.S.; Suh, Y.G.; Yi, H.S.; Lee, Y.S.; Jeong, W.I. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in mice. J. Hepatol. 2013, 58, 342–349. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Song, H.; Li, H.; Cui, A.; Liu, Y.; Su, L.; Crispe, I.N.; Tu, Z. Activated NK cells kill hepatic stellate cells via p38/PI3K signaling in a TRAIL-involved degranulation manner. J. Leukoc. Biol. 2019, 105, 695–704. [Google Scholar] [CrossRef]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.S.; Lee, Y.S.; Park, S.H.; Jang, M.J.; Jo, E.; Kim, S.C.; Han, Y.M.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.-Y.; Liu, C.; Li, Z.-R.; Niu, C.; Wu, J. NLRP3 deficiency did not attenuate NASH development under high fat calorie diet plus high fructose and glucose in drinking water. Lab. Investig. 2021, 101, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Qu, J.; Yuan, Z.; Pan, R.; Li, K. Calcipotriol Inhibits NLRP3 Signal Through YAP1 Activation to Alleviate Cholestatic Liver Injury and Fibrosis. Front. Pharmacol. 2020, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- NodThera is Unlocking the Significant Therapeutic Potential of NLRP3 Inflammasome Activation Inhibitors Through Our Novel Drug Discovery Platform. Available online: https://www.nodthera.com/approach-progress/ (accessed on 27 August 2021).
- Advancing Our Pipeline. Available online: https://www.ifmthera.com/pipeline/ (accessed on 27 August 2021).
- Xu, R.; Zhang, Z.; Wang, F.S. Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell Mol. Immunol. 2012, 9, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Zimmermann, H.W.; Roderburg, C.; Gassler, N.; Wasmuth, H.E.; Luedde, T.; Trautwein, C.; Tacke, F. The fractalkine receptor CX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology 2010, 52, 1769–1782. [Google Scholar] [CrossRef]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Mockel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. [Google Scholar] [CrossRef]
- Mulder, P.; van den Hoek, A.M.; Kleemann, R. The CCR2 Inhibitor Propagermanium Attenuates Diet-Induced Insulin Resistance, Adipose Tissue Inflammation and Non-Alcoholic Steatohepatitis. PLoS ONE 2017, 12, e0169740. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambade, A.; Lowe, P.; Kodys, K.; Catalano, D.; Gyongyosi, B.; Cho, Y.; Iracheta-Vellve, A.; Adejumo, A.; Saha, B.; Calenda, C.; et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology 2019, 69, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.J.; Fuchs, B.C.; Masia, R.; Holmes, J.A.; Salloum, S.; Sojoodi, M.; Ferreira, D.S.; Rutledge, S.M.; Caravan, P.; Alatrakchi, N.; et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol. Commun. 2018, 2, 529–545. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [Green Version]
- NIH, U.S. National Library of Medicine Clinicaltrials.gov. AURORA: Phase 3 Study for the Efficacy and Safety of CVC for the Treatment of Liver Fibrosis in Adults With NASH, ClinicalTrials.gov Identifier, NCT03028740. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03028740 (accessed on 10 July 2021).
- Perez-Martinez, L.; Perez-Matute, P.; Aguilera-Lizarraga, J.; Rubio-Mediavilla, S.; Narro, J.; Recio, E.; Ochoa-Callejero, L.; Oteo, J.A.; Blanco, J.R. Maraviroc, a CCR5 antagonist, ameliorates the development of hepatic steatosis in a mouse model of non-alcoholic fatty liver disease (NAFLD). J. Antimicrob. Chemother. 2014, 69, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, D.; Gilleece, Y.; Verma, S.; Abramowicz, I.; Bremner, S.; Perry, N. Protocol for a phase IV, open-label feasibility study investigating non-invasive markers of hepatic fibrosis in people living with HIV-1 and non-alcoholic fatty liver disease randomised to receiving optimised background therapy (OBT) plus maraviroc or OBT alone. BMJ Open 2020, 10, e035596. [Google Scholar] [CrossRef]
- Colino, C.I.; Lanao, J.M.; Gutierrez-Millan, C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 2020, 11, 218. [Google Scholar] [CrossRef] [Green Version]
- Ergen, C.; Heymann, F.; Al Rawashdeh, W.; Gremse, F.; Bartneck, M.; Panzer, U.; Pola, R.; Pechar, M.; Storm, G.; Mohr, N.; et al. Targeting distinct myeloid cell populations in vivo using polymers, liposomes and microbubbles. Biomaterials 2017, 114, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Scheyda, K.M.; Warzecha, K.T.; Rizzo, L.Y.; Hittatiya, K.; Luedde, T.; Storm, G.; Trautwein, C.; Lammers, T.; Tacke, F. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 2015, 37, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Bygd, H.C.; Ma, L.; Bratlie, K.M. Physicochemical properties of liposomal modifiers that shift macrophage phenotype. Mater. Sci. Eng. C. Mater. Biol. Appl. 2017, 79, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Maradana, M.R.; Yekollu, S.K.; Zeng, B.; Ellis, J.; Clouston, A.; Miller, G.; Talekar, M.; Bhuyan, Z.A.; Mahadevaiah, S.; Powell, E.E.; et al. Immunomodulatory liposomes targeting liver macrophages arrest progression of nonalcoholic steatohepatitis. Metabolism 2018, 78, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Iacobini, C.; Menini, S.; Ricci, C.; Blasetti Fantauzzi, C.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Traber, P.G.; Chou, H.; Zomer, E.; Hong, F.; Klyosov, A.; Fiel, M.I.; Friedman, S.L. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 2013, 8, e75361. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [Green Version]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andres-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernandez-Iglesias, A.; Broqua, P.; Junien, J.L.; Wettstein, G.; Bosch, J.; et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Marra, F.; Efsen, E.; Romanelli, R.G.; Caligiuri, A.; Pastacaldi, S.; Batignani, G.; Bonacchi, A.; Caporale, R.; Laffi, G.; Pinzani, M.; et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology 2000, 119, 466–478. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrene, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Francque, S.; Bedossa, P.; Ratziu, V.; Anstee, Q.; Bugianesi, E.; Sanyal, A.; Loomba, R.; Harrison, S.A.; Balabanska, R.I.; Mateva, L.; et al. The panPPAR agonist lanifibranor induces both resolution of NASH and regression of fibrosis after 24 weeks of treatment in non-cirrhotic nash: Results of the NATIVE phase 2b trial. Hepatology 2020, 72, 9A. [Google Scholar]
- Sven, M.F.; Pierre, B.; Manal, F.A.; Quentin, M.A.; Elisabetta, B.; Vlad, R.; Philippe, H.M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar] [CrossRef]
- Goyal, O.; Nohria, S.; Goyal, P.; Kaur, J.; Sharma, S.; Sood, A.; Chhina, R.S. Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: A prospective, observational, real world study. Sci. Rep. 2020, 10, 21117. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-alpha/gamma Agonist, for Treatment of Nonalcoholic Fatty Liver Disease: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, C.; Dupuis, T.; Jouvion, G.; Eberl, G.; Disson, O.; Lecuit, M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 2015, 42, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [Green Version]
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat. Med. 2019, 25, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Stutchfield, B.M.; Antoine, D.J.; Mackinnon, A.C.; Gow, D.J.; Bain, C.C.; Hawley, C.A.; Hughes, M.J.; Francis, B.; Wojtacha, D.; Man, T.Y.; et al. CSF1 Restores Innate Immunity After Liver Injury in Mice and Serum Levels Indicate Outcomes of Patients With Acute Liver Failure. Gastroenterology 2015, 149, 1896–1909. [Google Scholar] [CrossRef] [Green Version]
- Kedarisetty, C.K.; Anand, L.; Bhardwaj, A.; Bhadoria, A.S.; Kumar, G.; Vyas, A.K.; David, P.; Trehanpati, N.; Rastogi, A.; Bihari, C.; et al. Combination of granulocyte colony-stimulating factor and erythropoietin improves outcomes of patients with decompensated cirrhosis. Gastroenterology 2015, 148, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Promethera Biosciences. HepaStem: Toward an Alternative to Liver Transplantation. Available online: https://www.nature.com/articles/d43747-020-00724-x (accessed on 10 July 2021).
- Nevens, F.; Gustot, T.; Laterre, P.F.; Lasser, L.L.; Haralampiev, L.E.; Vargas, V.; Lyubomirova, D.; Albillos, A.; Najimi, M.; Michel, S.; et al. A phase II study of human allogeneic liver-derived progenitor cell therapy for acute-on-chronic liver failure and acute decompensation. JHEP Rep. 2021, 3, 100291. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ClinicalTrials.gov Identifier and Sponsor | Study | Estimated Completion |
|---|---|---|
| NCT02642172 Kaplan Medical Center, Israel | Evaluating whether prebiotics—ITF (Inulin/OFS 75/25) are effective in treating patients with NFALD. | 2023 |
| NCT0256860 University of Calgary, Canada | Effect of prebiotic fiber oligofructose-enriched inulin (Synergy1) supplementation, in conjunction with diet-induced weight loss, on reduction of liver fat and injury. | 2022 |
| NCT03467282 Hospital de Clinicas de Porto Alegre, Brazil | Probiotic supplementation (Lactobacillus acidophilus, Bifidobacterium lactis, Lactobacillus rhamnosus and Lactobacillus paracasei) in nonalcoholic steatohepatitis patients (PROBILIVER trail). | 2021 |
| NCT03863730 Odense University Hospital, Denmark | Prevention of progression in alcoholic liver disease by modulating dysbiotic microbiota by Profermin Plus, FSMP (food for special medical purposes), probiotics (based on fermented oats, Lactobacillus plantarum 299v, barley malt and Lecithin plus Thiamin) (SYN-ALD). | 2021 |
| NCT04175392 William Beaumont Hospitals, USA | Effect of probiotics (Align) in non-alcoholic fatty liver disease and steatohepatitis measured by transient elastography (PRONE Study). | 2023 |
| NCT04671186 Northwell Health, USA | Role of probiotics (Culturelle (Lactobacillus rhamnosus strain GG)) in treatment of pediatric nonalcoholic fatty liver disease (NAFLD) patients by assessing with fibroscan. | 2021 |
| NCT03749070, Camila Ribeiro de Avelar, Brazil | Effect of Silymarin (dietary supplement) on clinical evolution and nutritional variables of patients with non-alcoholic fatty liver disease. | 2021 |
| NCT04871360, Universidad de Guanajuato, Mexico | Effect of oral L-Citrulline supplementation on liver function and non-alcoholic fatty liver disease in adolescents with obesity. | 2021 |
| NCT04198805, Naga P. Chalasani, Indiana University School of Medicine, Indiana USA | The effect of Vitamin E and Docosahexaenoic Acid Ethyl Ester on non-alcoholic fatty liver disease (NAFLD). | 2022 |
| NCT04823676 Hospital General Dr. Manuel Gea GonzalezMexico city, Mexico | Efficacy and safety of a probiotic composition (mixture of two Lactoplantibacillus plantarum strains (formerly Lactobacillus plantarum) and one Levilactobacillus brevis strain (formerly Lactobacillus brevis), in a maltodextrin carrier (E1400)) as adjunct treatment in the comprehensive management of metabolism-associated hepatic steatosis in adults. | 2022 |
| NCT04781933 Mativa-Tech SA, France | “Combo” (a combination of dietary supplements including probiotics (Lactobacillus rhamnosus GG, Bifidobacterium breve BR03, Lactobacillus plantarum) and Glutamine, Quercetin, Vitamin E, Curcumin, Silybin, Pectin) in NASH improvement (ICAN). | 2022 |
| NCT03897218, 1. Medical University of Vienna, Austria 2. University Hospital RWTH Aachen, Germany 3. Sahlgrenska University Hospital, Sweden | Dietary modulation of intestinal microbiota as trigger of liver health: Role of Bile acids—“A Diet for Liver Health” (ADLH) using oatmeal flakes with prebiotic food supplements. | 2022 |
| NCT04465032, Leiden University Medical Center, The Netherlands | The effect of consecutive fecal microbiota transplantation on non-alcoholic fatty liver disease (NAFLD). Fecal transplantation will be performed via gastroduodenal endoscopy of autologous vs allogenic (lean donor) at 3 and 6 weeks (NAFTx). | 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalo, E.; Read, S.; Ahlenstiel, G. Targeting Gut–Liver Axis for Treatment of Liver Fibrosis and Portal Hypertension. Livers 2021, 1, 147-179. https://doi.org/10.3390/livers1030014
Kalo E, Read S, Ahlenstiel G. Targeting Gut–Liver Axis for Treatment of Liver Fibrosis and Portal Hypertension. Livers. 2021; 1(3):147-179. https://doi.org/10.3390/livers1030014
Chicago/Turabian StyleKalo, Eric, Scott Read, and Golo Ahlenstiel. 2021. "Targeting Gut–Liver Axis for Treatment of Liver Fibrosis and Portal Hypertension" Livers 1, no. 3: 147-179. https://doi.org/10.3390/livers1030014
APA StyleKalo, E., Read, S., & Ahlenstiel, G. (2021). Targeting Gut–Liver Axis for Treatment of Liver Fibrosis and Portal Hypertension. Livers, 1(3), 147-179. https://doi.org/10.3390/livers1030014