Abstract
Antifibrotic therapies for the treatment of liver fibrosis represent an unconquered area of drug development. The significant involvement of the gut microbiota as a driving force in a multitude of liver disease, be it pathogenesis or fibrotic progression, suggest that targeting the gut–liver axis, relevant signaling pathways, and/or manipulation of the gut’s commensal microbial composition and its metabolites may offer opportunities for biomarker discovery, novel therapies and personalized medicine development. Here, we review potential links between bacterial translocation and deficits of host-microbiome compartmentalization and liver fibrosis that occur in settings of advanced chronic liver disease. We discuss established and emerging therapeutic strategies, translated from our current knowledge of the gut–liver axis, targeted at restoring intestinal eubiosis, ameliorating hepatic fibrosis and rising portal hypertension that characterize and define the course of decompensated cirrhosis.
1. Introduction
In contrast to acute inflammatory reactions, which are characterized by rapidly resolving vascular changes, edema and neutrophilic inflammation, fibrosis is an intrinsic response to chronic, non-resolving injury and inflammation. The latter triggers a wound healing process that mitigates inflammatory tissue destruction and excessive scarring. Orchestrated by a spectrum of activated extracellular matrix (ECM)-producing cells, protracted injuries often progress towards remodeling and replacement of organ parenchyma by acellular scar tissue accompanied by severe architectural and vascular distortion. Fibrosis is intimately linked to wound healing, serving to maintain organ integrity when tissue disassembly occurs during inflammation, apoptosis, necrosis, and release of lytic enzymes.
The buildup of scar tissue is a hallmark of chronic liver disease (CLD) progression. In the liver, fibrosis is the common endpoint of a plethora of conditions such as chronic viral hepatitis B or C, autoimmune and biliary diseases, alcoholic steatohepatitis (ASH), and a worsening trend of non-alcoholic steatohepatitis (NASH) [1,2,3,4]. While mild fibrosis remains largely asymptomatic and usually reversible within days to weeks, its progression towards cirrhosis is a major cause of liver related morbidity and mortality [5,6,7]. Acute-on-chronic liver disease (ACLD) represents the most advanced stage of liver cirrhosis characterized by acute decompensation of chronic liver disease that can result in multi-system organ failure and a significant short-term mortality [8]. Consequently, cirrhosis and CLD pose a substantial health burden on many countries that has increased at the global scale since the 1990s [5]. In Australia, the age-standardized death rate of cirrhosis in 2016 per 100,000 is 9.6 [9], and the main etiology of decompensated cirrhosis is alcoholic liver disease (ALD) [6]. With this rapid rise in the burden of liver cirrhosis, there is an immense need to understand the mechanisms of disease pathogenesis and specific targets to reverse or cease fibrosis progression.
Clinically, cirrhosis is associated with progressive liver failure, the risk of hepatocellular carcinoma, and is often accompanied by the development of portal hypertension (PTH). Clinically significant portal hypertension is characterized by hepatic venous pressure gradient (HVPG) ≥ 10 mm Hg. PTH triggers many complications, including secondary splanchnic vasodilation and extrahepatic shunt formation resulting in the development of portosystemic collaterals (varices) with significant risk of gastrointestinal bleeding, hyperdynamic syndrome, ascites and hepatic encephalopathy (HE) [10,11,12]. Severe consequences of PTH can predispose patients to the development of acute decompensation and acute-on-chronic liver failure (ACLF) that is associated with high short-term mortality [13,14,15,16].
Currently, there are few therapeutic measures that can prevent progression of clinically significant portal hypertension. Notably, such treatments do not target the main underlying mechanisms and consist of extrahepatic vasoconstrictors (i.e., nonselective beta blockers [NSBBs] [17,18], vasopressin analogues and somatostatin analogues) aimed at ameliorating PTH, or therapies focused on the prevention of PTH-derived complications. Effective artificial liver support remains a major unmet need in patients with end-stage liver disease, with liver transplantation being the only available curative option to date. Organ shortage remains one of the major challenges in liver transplantation and ultimately leads to mortality for those caught on the waiting list.
The bidirectional relationship between the gut and the liver implicates the gastrointestinal microbiome in the development and progression of chronic liver disease [19,20,21,22]. While liver-derived bile acids and antimicrobial molecules help shape the gastrointestinal microbiome, the portal vein delivers gut-derived metabolites and microbial products into the liver. Alcohol and diet, two of the main drivers of chronic liver disease, cause significant “local” damage in the liver but are major contributors of microbial dysbiosis in the gut as well as intestinal permeability resulting in microbial translocation into the portal venous system. These factors are exacerbated in end-stage liver disease where bacterial translocation is worsened, combined with impaired hepatic microbial clearance [23]. Conversely, the gut communicates with the liver via close links through the biliary tract, portal vein and systemic circulation.
Growing evidence for the role of gastrointestinal dysfunction in liver disorders is supported by an abundance of evidence from clinical trials demonstrating that liver fibrosis and rising portal hypertension can be efficiently ameliorated by targeting the gut–liver axis [24,25]. Current therapies include pre- and probiotics, and antibiotics to modulate gut microbial composition and intestinal barrier integrity, as well as inhibition of antigen recognition in the liver to limit the local response to microbial products delivered from the gut. This review explores upcoming and state of the art therapeutic strategies for the management of liver fibrosis and portal hypertension translated from our advances in knowledge of the gut–liver axis.
2. The Gut–Liver Axis at the Frontier of Host–Microbial Interactions
The human gastrointestinal tract is the largest barrier surface in contact with the external environment. Therein, the gut microbiota represents a massive microbial ecosystem, harboring upwards of 4 × 1013 microbial cells, with a pool of genetic material over one hundred times larger than the human genome, and a metabolic capacity akin to the liver [26,27,28]. This interdependency that has developed over more than a billion years of mammalian–microbial coevolution has resulted in the entrenchment of our microbiota in every one of our biological systems: the maturation and continued education of the host immune response, selective exclusion of pathogens, regulation of intestinal endocrine functions, neurologic signaling, provision of metabolically available energy sources, vitamins and neurotransmitters, metabolism of bile salts, toxins and drugs and the bidirectional communication between the gut and other organ systems [29]. This bidirectional crosstalk is best exemplified by the gut–liver axis.
The liver communicates with the intestinal tract through the biliary system and systemic circulation mostly via bile acids (BAs), bioactive mediators and immunoglobulin A antibodies. BAs are amphipathic molecules synthesized from cholesterol in the pericentral hepatocytes. These are conjugated to glycine or taurine and released in the biliary tract. On reaching the small intestine through the duodenum, BAs, together with other biliary components, facilitate emulsification and absorption of dietary fats, cholesterol, and fat-soluble vitamins. About 95% of the BAs are actively reabsorbed in the terminal ileum and transported back to the liver [30,31]. The remaining five percent are deconjugated, dehydrogenated and dehydroxylated by the intestinal microbiota to form secondary bile acids, which reach the liver via passive absorption into the portal circulation. Due to their amphipathic nature, bile acids are toxic for bacterial cells and, thus, exert a strong selective pressure on the microbial populations inhabiting the human gut; they additionally promote the synthesis and secretion of antimicrobial molecules by the intestinal epithelium. This effect helps maintain gut eubiosis and its pool size regulates the microbiome at the highest taxonomic levels.
The term gut–liver axis was coined to highlight the close functional and bidirectional relationship between both these organs resulting from the integration of dietary, metabolic and environmental factors, among others. The present understanding of the many etiologies of liver diseases is underpinned by intestinal dysbiosis and impaired intestinal permeability termed leaky gut [32]. Considerable changes to our diet, alcohol intake, and lifestyle, accompanied with the sanitation revolution and excessive use of antimicrobial agents and medications have drastically accelerated our microbial dysbiosis and augmented systemic inflammation in ways we do not yet comprehend [33].
Intestinal Permeability
The liver receives 70% of its blood supply directly from the gut, where it sits at the crossroad between the portal blood flow coming from the intestinal circulation and peripheral organs. This close anatomical position offers continuous exposure to gastrointestinal antigens, particularly in the context of CLD. These include translocated microbes, microbial products and translocated microbial/pathogen-associated molecular patterns (MAMPs/PAMPS) such as microbial DNA and endotoxins (lipopolysaccharide, flagellin, lipoteichoic acid and peptidoglycan) [34]. Cell death in the gut and liver also generates damage-associated molecular patterns (DAMPs) including adenosine triphosphate (ATP), and intracellular proteins such as heat shock proteins and chromatin associated high-mobility group box 1 (HMGB1) [35].
Liver cell populations including Kupffer cells, hepatic stellate cells (HSCs), sinusoidal cells, biliary epithelial cells, and hepatocytes express innate immune receptors known as pattern recognition receptors (PRRs) that respond to the constant influx of microbial-derived ligands from the gut. When translocated MAMPs/PAMPs reach the liver, they bind PRRs including Toll-like receptors (TLRs) to activate immunomodulatory and inflammatory cascades mediated primarily by signal transducer and activator of transcription (STAT) and nuclear factor-kappa B (NF-κB) transcription factors. These effects are beneficial in the short term by limiting pathogen infection and dispersion, yet detrimental over longer periods of activation by stimulating excessive inflammation, fibrosis and organ damage. The many etiologies of CLD affect gastrointestinal homeostasis by causing changes in the microbiome, innate immune defenses and intestinal permeability. Gastrointestinal dysbiosis, particularly in the context of poor diet or excessive alcohol, inevitably exacerbates chronic liver damage, and is a key target to limit inflammatory and fibrotic progression in CLD patients.
In alcohol-induced liver disease, ethanol impairs intestinal epithelial barrier, elicits intestinal bacterial overgrowth [36] and profoundly disrupts the composition of the microbiome and its metabolome [37]. These effects cumulatively lead to elevated bacterial LPS in portal circulation and can result in rapid systemic endotoxemia. LPS activates Kupffer cell NF-κB signaling leading to induction of reactive oxygen species (ROS), tumor necrosis factor alpha (TNF-α) and transforming growth factor beta (TGF-β) production. Sustained TNF-α drives mitochondrial dysfunction and neutrophilic infiltration, subsequently triggering inflammation and stimulating apoptosis of hepatocytes. Chronic hepatocyte injury causes release of DAMPS and apoptotic bodies, leading to activation of resident HSCs into myofibroblasts to produce matrix proteins faster than they are degraded. Moreover, the major ethanol metabolite, acetaldehyde, is fibrogenic and causes the release of ROS resulting in paracrine stimulation of HSCs.
In the context of metabolic associated fatty liver disease (MAFLD) [38,39], a two hit [40] theory has been historically adopted to explain the resulting pathogenesis: This theory suggests that hyperglycemia and insulin resistance stimulate the development of hepatic steatosis. The second ‘hit’ is mediated by lipid-induced cellular stresses such as oxidative stress, apoptosis and gut-derived lipopolysaccharide (LPS) that are required for the development of NASH. More recently, this theory has been considered overly simplistic by ignoring the systemic effects of obesity and has been replaced with the “substrate-overload liver injury model/multi-hit theory [41,42]”: Here, surplus fatty acids that develop in MAFLD overwhelm the liver’s metabolic capacity and serve as substrates for the generation of lipotoxic species that provoke endoplasmic reticulum stress and hepatocellular injury leading to a pro-fibrogenic response and genomic instability. In recent years, a line of evidence has suggested a close link between intestinal dysbiosis and the pathogenesis of NAFLD [43,44,45] (e.g., increased production of intestinal ethanol, bacterial translocation and small intestinal bacterial overgrowth [SIBO]).
Portal pressure is strongly linked to intestinal permeability; venous congestion and splanchnic neoangiogenesis, due to chronic rise of pressure in portal vein, induce phlebectasia, mucosal hypoperfusion and lead to increased permeability in the gut [46]. Abnormal intestinal permeability and bacterial translocation in cirrhotic patients are common and correlated with the degree of portal hypertension.
3. Therapies Targeting the Gut–Liver Axis to Improve Liver Fibrosis and Portal Hypertension
Several interventions targeting the gut–liver axis have been developed in recent years or are otherwise undergoing clinical trials. Here, we will outline current treatments based on their primary target: (1) the intestinal mucosa, (2) the intestinal microbiome, or (3) the hepatic immune response (Figure 1).
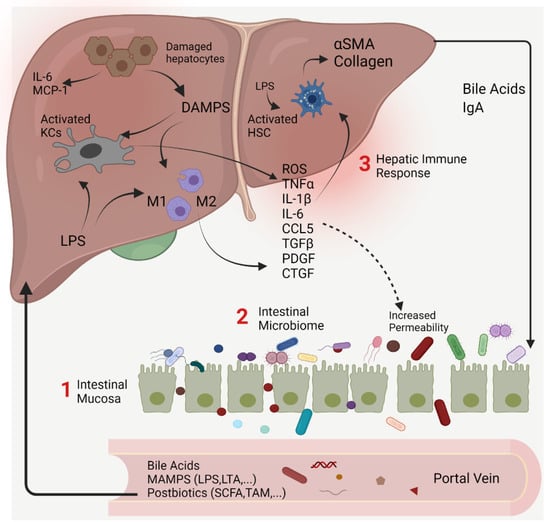
Figure 1.
The gut–liver axis and its intersection with the intestinal microbiome as a potential therapeutic target for the treatment of liver fibrosis and portal hypertension. The bidirectional relationship between the gut, its microbiome and liver is established via the portal vein which transports immunogenic antigens from the gut. Conversely, the liver feedback route is via bile and antibody secretion in the gut. Our current understanding of the many etiologies of liver diseases is underpinned by intestinal dysbiosis, functional impairment of intestinal barrier, and systemic dissemination of gut MAMPs that trigger an abnormal immune-inflammatory cascade in the liver. Activation of HSCs into proliferative, fibrogenic myofibroblasts is well established as the central driver of hepatic fibrosis. Therapeutic interventions developed or undergoing clinical trials target elements of gut- liver interaction primarily the (1) intestinal mucosa, (2) microbiome and (3) diverse repertoire of immune cell populations in the liver and their sensors. α-SMA, alpha smooth muscle actin; CTGF/CCN2, connective tissue growth factor; DAMPs, damage associated molecular patterns; HSCs, hepatic stellate cells; IgA, immunoglobulin A; IL-1β, interleukin one beta; IL-6, interleukin six; CCL5, chemokine (C-C motif) ligand 5; HSCs, hepatic stellate cells; KCs, Kupffer cells; LPS, lipopolysaccharides; LTA, lipoteichoic acid; M1, macrophage type1; M2, macrophage type 2; MAMPs, microbe-associated molecular patterns; CCL2/MCP-1, chemokine (C-C motif) ligand 2/monocyte chemoattractant protein-1; PDGF, platelet-derived growth factor; ROS, reactive oxygen species; SCFA, short-chain fatty acids; TMA, trimethylamine; TGFβ, transforming growth factor beta; TNF-α, tumor necrotizing factor alpha.
3.1. Interventions Targeting the Intestinal Mucosa
3.1.1. FXR Agonists
The regulatory effects of primary BAs have been best studied via their interaction with nuclear receptors such as farnesoid X receptor (FXR) and Takeda G-protein-coupled receptor 5 (TGR5), which modulate hepatic bile acid synthesis, metabolic regulation, inflammation, hepatic fibrosis and vascular homeostasis [47,48]. Obeticholic acid (Ocaliva) (OCA) is a selective semi-synthetic FXR agonist that has been shown to reduce hepatic resistance and portal pressure without systemic effects via increased intrahepatic endothelial nitric oxide synthase activity [49]. Furthermore, OCA has anti-inflammatory properties in vitro, inhibiting pro-inflammatory NF-κB activation in Kupffer cells (KCs) and liver sinusoidal endothelial cells (LSECs) [50]. Moreover, these anti-inflammatory properties reduced HSC activation in rat model of thioacetamide (TAA) fibrosis, as demonstrated by a significant decrease in hepatic alpha-smooth muscle actin (α-SMA) [50]. This data is supported by studies using the synthetic FXR agonist GW4064, which inhibits contraction of HSCs mediated by endothelin-1 [51]. In addition, OCA has been shown to reduce bacterial translocation and attenuate intestinal inflammation in cirrhotic rats by improving the ileal gut-vascular barrier function via antimicrobial peptide induction, improved tight junction expression and reduced loss of fecal albumin [52,53].
OCA has recently been examined in a multicenter, double-blind, placebo-controlled, randomized clinical trial (RCT), FLINT (Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment), in patients with non-cirrhotic, non-alcoholic steatohepatitis. When given orally for 72 weeks, OCA improved the histological features of non-alcoholic steatohepatitis and improved liver fibrosis in 45% of patients compared with 23% of patients in the placebo group [54]. Even after one week of treatment, another study by Mookerjee et al. reported that nine out of 16 patients with alcoholic cirrhosis receiving OCA responded with a mean HVPG reduction of 28% [55].
Beyond the clinical potential of the first generation of FXR agonists, OCA therapy has been associated with several side effects including high incidences of drug-induced pruritus in both NASH and primary biliary cirrhosis trials [54,56], increased in low density lipoprotein cholesterol levels and elevated risk of gallstone formation. Most of these side effects are related to its steroidal BA-like chemical structure that enhances some of the TGR5-related side effects. In this context, novel FXR agonists are devoid of TGR5 cross reactivity thus avoiding off-target side effects. EDP-305 is an example of a non-bile acid derivative endowed with FXR agonism/GPBAR1 antagonism that can profoundly inhibit perisinusoidal fibrosis, with over 80% reduction in collagen deposition in methionine choline-deficient (MCD) diet fed mice [57]. EYP001a (Vonafexor, PXL007) is another non-bile acid FXR agonist that is currently being assessed in phase 2 clinical trials for NASH [58] (Enyo Pharma, NCT03812029).
The novel non-steroidal FXR agonist PX20606 has also been shown to improve portal pressure by reducing vascular remodeling while limiting hepatic fibrosis progression, angiogenesis and endothelial dysfunction in rodents [59]. A reduction of bacterial translocation was confirmed by a significant decrease in mesenteric lymph node bacterial count, as well as serum concentrations of lipopolysaccharide binding protein (LBP), TNF and interleukin 6 (IL-6). In cirrhotic animals, PX20606 reduced intestinal fluorescein isothiocyanate (FITC)-dextran uptake (a marker of intestinal permeability) and demonstrated a tendency towards increased ileal zonula occludens 1 (ZO-1) expression, indicating an improvement in gut barrier function due to a reduction in portal hypertensive enteropathy. Another recent study demonstrated that FXR activation stimulates TGR5 expression in the intestinal L cells and drives gut microbiome remodeling to change bile acid composition [60]. This resulted in increased levels of lithocholic acid and taurolithocholic acid which are potent endogenous agonists for TGR5 (GPBAR1). OCA was found to stabilize the gut–vascular barrier, whereas both FXR agonists abrogated gut–liver translocation of E. coli, highlighting its ability to block microbial transit into the liver [61].
Another novel FXR agonist, Cilofexor (GS-9674 or PX-201), exerts dose-dependent antifibrotic effects and ameliorates portal hypertension in cirrhotic NASH rats [62]. The combination of GS-9674 with the beta-blocker propranolol appeared safe and resulted in an additional decrease of mesenteric hyper-perfusion [62]. Tropifexor (LJN452) is another highly potent FXR agonist, producing robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy based on results from the Novartis, FLIGHT- FXR phase 2b study [63,64] (Novartis Pharmaceuticals, NCT02855164). In two preclinical distinct rodent models, Tropifexor mediated abrogation of steatohepatitis and fibrosis and induced transcriptome signatures associated with reduction of oxidative stress, fibrogenesis and inflammation [65].
In rats, treatment with the synthetic TGR5 agonist BAR501 for 6 days prior to cannulation of the portal vein reduced the norepinephrine-mediated rise in portal perfusion pressure. Furthermore, administration of the TGR5 agonist inhibited portal hypertension in mice treated for 9 weeks with carbon tetrachloride (CCl4), while it did not affect fibrosis progression [66]. It was postulated that TGR5 activation promotes the generation and secretion of vasodilatory agents, hydrogen sulfide, and nitric oxide, and inhibits the expression and secretion of the potent vasoconstrictor endothelin-1 from LSECs [67].
3.1.2. Carbon Nanoparticles
Non-absorbable carbon nanoparticles exhibit a high adsorptive capacity for bacterial fragments and represent a novel tool to counteract dysbiosis and translocation of bacterial-derived products. Experimental evidence from a bile-duct ligated cirrhotic rodent model showed that oral therapy with non-absorbable carbon nanoparticles of controlled porosity (Yaq-001) was associated with a significant increase in Firmicutes, particularly Clostridia, and a decrease in Bacteroidetes in stool samples. In addition, this treatment attenuated LPS-induced ROS production and inflammasome activation by monocytes and neutrophils in bile duct-ligated rats [68].
3.1.3. Duodenal Mucosal Resurfacing
Duodenal mucosal resurfacing (DMR) is a safe, minimally invasive endoscopic procedure that involves a single cycle of circumferential hydrothermal ablation of at least 10 cm of the post-papillary duodenal mucosa [69]. The precise mechanism of action of DMR remains to be determined; however, a recent study by Van Baar et al. demonstrated that DMR can improve liver aminotransferases, decrease hepatocyte mitochondrial and reduce fibrosis-4 scores at 6 months post DMR. These effects were sustained at 12 and 24 months post procedure [70].
3.1.4. Pharmacological Modulation of Gut Peptides
Gut peptides play an important role in relaying signals of nutritional and energy status from the gut. They are released in response to dietary nutrients as well as microbial products and metabolites. Pharmacological modulation of gut peptides holds promise to re-establish metabolic homeostasis in NAFLD and hepatic fibrosis [71,72]. Glucagon-like peptide-1 (GLP-1) is a potent incretin hormone produced and stored by the enteroendocrine L cells of the distal ileum and colon. GLP-1 regulates energy metabolism by promoting glucose-dependent insulin secretion, improving peripheral insulin sensitivity, suppressing glucagon secretion, inhibiting gastric emptying, and promoting satiety. Importantly, several in vitro studies have demonstrated that GLP-1 analogues improve the ability of hepatocytes to handle excess non-esterified fatty acids and lipid production [73].
The LEAN trial (Liraglutide Efficacy and Action in NASH) was the first RCT to document on the efficacy of 48-week treatment period of human glucagon-like peptide-1 receptor (GLP-1R) analogue (Victoza) in adults with biopsy-proven NASH [74,75]. Despite the relatively short duration of the trial, the long-acting GLP-1 receptor agonist Liraglutide histologically reduced active steatohepatitis and lobular inflammation with no worsening of fibrosis from baseline based on Kleiner Fibrosis stage. Recently, Cotadutide (MEDI0382), a dual GLP-1 and glucagon receptor GCGR agonist, has been reported to exert multifactorial reductions in NAFLD activity score, pro-peptide of type III collagen level, fibrosis-4 index to an extent more pronounced than the GLP-1 mono agonist Liraglutide [76] (AstraZeneca, NCT03235050).
The success of the various GLP-1/GCG and GLP-1/GIP (glucose-dependent insulinotropic peptide) dual agonists inspired research towards the development of unimolecular multifunctional peptides with improved plasma half-life and potency that combine agonism for two or more G protein-coupled receptors. The (Glucagon/GIP/GLP-1) GGG tri-agonist (HM15211/LAPS) is a long acting, monomeric peptide triple agonist that is conjugated to the human aglycosylate Fc fragment to extend its circulating half-life. This tri-agonist synergistically reduces liver fat, oxidative stress, and HSC activation (reduced TGF-β and α-SMA gene expression), resulting in greater NAFLD activity score (NAS) reduction than GLP-1RA, apoptosis signal-regulating kinase 1 inhibitor, or a FXR agonist in an MCD mouse model [77]. In line with these results, the GGG tri-agonist HM15211 is currently being evaluated for NASH and fibrosis as part of a Phase 1b/2a clinical trial. Preliminary data have demonstrated improvement of hepatic steatosis, inflammation and fibrosis [78]. Phase 1 clinical trials using ALT-801 (Altimmune, Inc., NCT04561245) and DD01 (Neuraly, Inc. NCT04812262) as well as Phase 2 clinical for trials of Efinopegdutide (JNJ 64565111/HM12525A) (Merck Sharp & Dohme Corp., NCT04944992) and BI456906 (Boehringer Ingelheim, NCT04771273), all dual GLP-1/Glucagon receptor agonists, are underway in patients with histologically proven NASH, underlining the utility of this approach.
Other GLP-1 analogs were similarly effective in the treatment of NASH but failed to resolve fibrosis. Phase 2 trials using the long-lasting GLP-1 analog Semaglutide (Ozempic) for 72-week in patients with biopsy-confirmed NASH and liver fibrosis of stage F1, F2, or F3, resulted in NASH resolution but unexpected lack of improvement of fibrosis stage [79] (Novo Nordisk A/S, NCT02970942). Similarly, results from (D-LIFT trial [80]) demonstrated that Dulaglutide significantly reduced liver fat content and improved GGT levels in participants with NAFLD but was unable to reduce liver stiffness and transaminases.
Fibroblast growth factor 19 (FGF19) is a hormone produced by enterocytes of the terminal ileum in response to bile acid-mediated FXR activation to regulate bile acid synthesis. Therefore, FXR agonists such as OCA possess potent dual activity by acting directly on the liver and indirectly via FGF19 [81]. From a clinical standpoint, it is challenging to identify whether the multi-faceted and anti-fibrotic effects of FXR agonists are due to FXR activation in the liver or the effects of FGF19 from the gut. In a proof-of-concept study, an engineered FGF19 analog (Aldafermin) was administered for up to 24 weeks in 53 patients with histologically confirmed NASH. It reduced liver fat and improved liver fibrosis (≥1-stage decrease in fibrosis score) in 38% of patients versus 18% in the placebo group [82] (NGM Biopharmaceuticals, Inc., NCT02443116).
Another member of the FGF subfamily is fibroblast growth factor 21 (FGF21). FGF21 is a pleiotropic hormone produced mainly by hepatocytes in a PPAR-alpha regulated fashion, acting as regulator of energy balance, glucose and lipid homeostasis via a heterodimeric receptor consisting of FGF receptor 1 (FGFR1) and β-klotho [83]. FGF21 represents an intriguing target for modulation of NASH and progression to advanced fibrosis. Data obtained from the phase 2 clinical trial using the PEGylated human FGF21 analogue, Pegbelfermin (BMS-986036) administered for 16 weeks for patients with non-alcoholic steatohepatitis (fibrosis stage 1–3) resulted in significant reduction in hepatic fat as measured by Magnetic Resonance Imaging Proton Density Fat Fraction—MRI-PDFF, PRO-C3 (a fibrosis biomarker), and improvement of metabolic parameters (adiponectin and lipid concentrations) [84] (Bristol-Myers Squibb, NCT02413372). In view of this, a handful of drugs using the enterokine pathway are in the clinical pipeline, including Efruxifermin (AKR-001), another FGF21 mimetic. In Phase 2 studies in non-alcoholic steatohepatitis (NASH) patients with F1-F3 fibrosis, Efruxifermin demonstrated effective reduction of liver fat content with 48% of treatment responders achieving an improvement in fibrosis stage [85] (Akero Therapeutics, Inc. NCT03976401, NCT04767529).
In summary, there are a variety of therapeutic interventions targeting the intestinal mucosa for the treatment of advanced liver disease. Figure 2 summarizes several of these interventions that aim to restore homeostasis within the gut–liver axis.
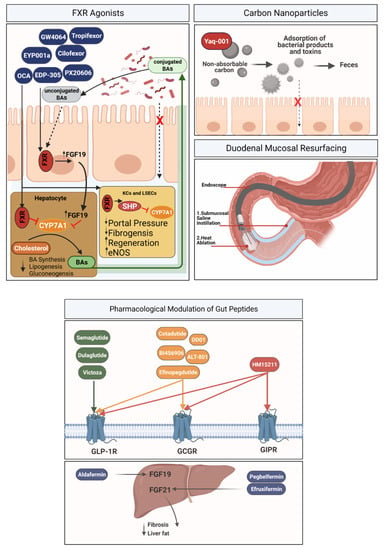
Figure 2.
Interventions targeting intestinal mucosa. FXR agonists—In Kupffer cells (KCs) and liver sinusoidal endothelial cells (LSECs), FXR induction leads to the expression of short heterodimeric partner (SHP), and further downregulation of cholesterol 7a-hydroxylase CYP71A. FXR agonists: Cilofexor, CW4064, EDP-305, EYP001a, OCA (Obeticholic acid), PX20606, Tropifexor. Carbon nanoparticles—Non-absorbable carbon particles exhibit a high adsorptive capacity for bacterial-derived products, counteracting bacterial translocation. Duodenal mucosal resurfacing (DMR) is a minimally invasive upper endoscopic procedure that involves circumferential mucosal lifting followed by hydrothermal ablation of duodenal mucosa. Pharmacological modulation of gut peptide agonists of mucosal gut receptor, including GLP-1 agonists, GLP-1/GCG agonists and tri-agonists (GLP1/GCG/GIP), are all intriguing drugs for modulation of fibrosis. Aldafermin (an engineered FGF19 analog), Pegbelfermin (PEGylated human FGF21 analogue) and Efruxifermin (FGF21 mimetic) represent promising targets for modulation of liver fibrosis. GLP-1R, glucagon-like peptide-1 receptor; GCGR, glucagon receptor; GIPR, glucagon-like peptide-1 receptor.
3.2. Interventions Targeting the Intestinal Microbiome
Individuals with liver fibrosis have a markedly altered microbial diversity, characterized by a decline in microbial gene richness and function [86,87]. Perturbations in bacterial metagenomic and metabolomic signatures and their association with liver disease suggests that manipulation of commensal microbial composition or function is essential to restore homeostasis [88,89,90]. Therapies that aim to achieve restoration of the intestinal microbiome include selected combinations of metabolites (postbiotics) produced by the microbiome that are generated from dietary components, as well as probiotics and prebiotics that are used to stimulate the growth of “good bacteria”. Alternatively, antibiotics and fecal microbiota transplantation are used to broadly remove/replace the majority of the microbial ecosystem and are often used in combination with gentler approaches (pre-/probiotics) to recolonize the gut (Figure 3).
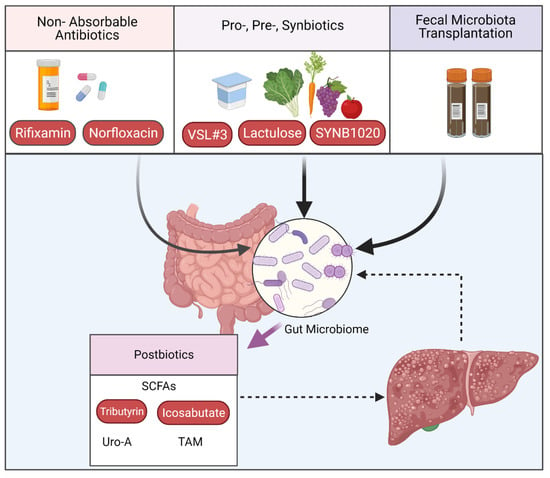
Figure 3.
Interventions targeting the intestinal microbiome. Advances in our knowledge of the gut–liver axis are driving the development of therapeutic tools based on microbiota composition and its metabolites (postbiotics). Modifying intestinal content with non-absorbable antibiotics (Rifixamin, Norfloxacin) or specific pro- pre- or synbiotics (VSL#1, lactulose, SYNB1020) or target fecal microbiota transplantation are increasingly recognized in clinical trials as effective interventions targeting the microbiome to effectively treat liver disease. SCFAs, short-chain fatty acids; TAM, trimethylamine; Uro-A, Urilothlin-A.
3.2.1. Targeting Microbiome Composition
(a) Non-absorbable Antibiotics. Patients with cirrhosis are predisposed to intestinal dysmotility, bacterial overgrowth, and increased intestinal permeability, all leading to an increase in bacterial translocation (BT) and increased endotoxemia. In cirrhosis, there is an increased relative abundance of bacterial taxa belonging to Enterobacteriaceae (Gram-negative (-) rods such as Escherichia coli (E. coli) and Klebsiella), Enterococcaceae (Enterococcus faecalis and E. faecium), and Streptococcaceae, combined with a lower abundance of potentially beneficial autochthonous taxa such as Lachnospiraceae Ruminococcaceae, and Clostridiales XIV in advanced cirrhosis [86]. The invasion of oral bacteria (such as Prevotella or Veillonella) into the distal intestine is also observed in cirrhotic patients [91,92].
In addition to changes in microbiome composition, PTH damages the intestinal barrier and thus increases microbial translocation into the portal system. A surrogate marker of BT, LBP, was observed to be increased in 42% of cirrhotic patients [93]. In addition, up to 30.8% of patients with Child-Pugh C cirrhosis have positive bacterial cultures of mesenteric lymph nodes compared to 8.6% of non-cirrhotics [94]. BT has also been associated with other portal hypertension related complications such as HE and spontaneous bacterial peritonitis (SBP) [95]. In recent years, an association between bacterial infection and portal hypertension in cirrhosis has been established. Bacterial infection is an independent predictor of the occurrence of variceal hemorrhage (VH) and is also the strongest independent factor associated with failure to control VH, earlier re-bleeding, coagulation abnormalities and mortality [96]. Hence, current guidelines recommend continuous prophylaxis with antibiotics to protect against the development of decompensation events such as SBP, either as primary prophylaxis in specific conditions or as secondary prophylaxis after an episode of SBP. Meanwhile, third-generation cephalosporins and fluoroquinolones, are recommended for prophylaxis of variceal bleeding. A study by Moghadamrad et al. showed that wild type mice colonized with intestinal microbiota presented with significantly higher portal pressure levels after partial portal vein ligation, when compared to germ free mice [97]. Consequently, the effect of antibiotic therapy on portal pressure has become heavily investigated in human trials.
The administration of antibiotics can eliminate dysbiosis and pathobionts, and additionally reduces enteric production of inflammatory cytokines, stabilizes the gut barrier and decreases the production of harmful secondary bile acids [98]. Rifaximin is a non-absorbable entero-selective broad-spectrum antibiotic that remains relevant in this context, even since its approval over 30 years ago in 1987 [99]. Rifaximin is beneficial for HE in cirrhosis and is currently recommended by the European Association for the Study of Liver [100] and the American Association for the Study of Liver Diseases (AASLD) [101] as one of the first-line drug for PTH therapy and prophylaxis. Kaji et al. demonstrated that Rifaximin ameliorates HE and lowers endotoxemia with minimal change in microbiome composition [102]. Moreover, Rifaximin is recommended as add-on therapy to lactulose for prevention of overt HE according to AASLD and EASL guidelines.
Unfortunately, the hydrophobic nature of Rifaximin makes it largely insoluble in water, and it requires BA for adequate solubilization. A newer formulation termed Rifaximin soluble solid dispersion (SSD) is water soluble and is of therapeutic benefit for patients with advanced liver disease who have lower intestinal BA concentration. A phase 2 study concluded that oral Rifaximin SSD treatment in patients with early decompensated cirrhosis could reduce all-cause hospitalization or mortality [103] (Bausch Health Americas, Inc. NCT01904409).
Rifaximin is believed to reduce hepatic fibrosis progression by improving intestinal permeability by increasing intestinal ZO-1 expression. In a murine model of bile duct ligation-induced liver fibrosis, Zhu et al. demonstrated that Rifaximin reduced fibrosis, angiogenesis and portal hypertension via inhibition of TLR4 pathway activation [104]. As expected, both aerobic and anaerobic fecal bacteria counts, which were increased after bile duct ligation, were significantly reduced in animals receiving Rifaximin.
A small cohort study by Vlachogiannakos et al. in 2009 demonstrated that HVPG values decreased significantly after intestinal decontamination with Rifaximin for 28 days in patients with alcohol-related decompensated cirrhosis [105]. Furthermore, long-term use of Rifaximin reduced the risk of developing complications of PTH and improved survival. On the contrary, a recent randomized, double blinded, placebo-controlled trial investigating the hemodynamic effect of Rifaximin in 54 patients with cirrhotic ascites without signs of overt HE observed no difference in HVPG compared to placebo [106]. A possible explanation for this discrepancy is due to Rifaximin’s effect on the gut microbiome. It was hypothesized that Rifaximin limits HE development by stimulating the growth of colonic microbes that produce less ROS and amino acids (Copenhagen University Hospital, Hvidovre, NCT01769040).
Rifaximin has also proven useful when combined with non-selective beta-blockers. NSSBs function to prevent rebleeding by decreasing cardiac output, as well as inducing splanchnic arterial vasoconstriction and therefore reducing splanchnic blood flow. They have additionally been shown to improve intestinal permeability in cirrhosis and consequently decreased bacterial translocation. A large clinical trial investigating the hemodynamic response of Rifaximin and propanol combination therapy versus propanol monotherapy on complications of decompensated cirrhosis and portal hypertension showed that Rifaximin combination therapy with propanol has an additive effect in improving PTH [107]. A recent study in a rat model of NASH by Fujinaga et al. showed that the combination of an angiotensin-II receptor blocker (ARB) and Rifaximin showed a stronger inhibitory effect compared to that conferred by a single agent [108].
Norfloxacin is a synthetic broad-spectrum antibiotic, and poorly absorbed fluoroquinolone, that has been used to achieve selective intestinal decontamination in cirrhotic patients. Treatment with Norfloxacin has been shown to reverse the hyperdynamic state, albeit without an effect on HVPG [109,110]. Norfloxacin nonetheless seems to improve survival in cirrhotic patients with reduced ascitic fluid protein concentrations and decrease risk of AD and ACLF [111]. In a small RCT, selective intestinal decontamination with Norfloxacin partially reversed the hyperdynamic circulatory state in cirrhotic patients with a reduction of serum LPS [112]. Furthermore, a RCT has shown that Norfloxacin, when combined with standard medical therapy improved survival in patients with decompensated alcoholic cirrhosis and liver failure (Assistance Publique—Hôpitaux de Paris, NCT01037959).
(b) Probiotics, prebiotics and synbiotics. Antibiotic regimens cause a lasting disruption to the composition of the gut microbiome, opening the doors to antibiotic resistance. Conseqently, the use of pre-, pro- and/or synbiotics has long been advocated for restoration of intestinal microbial diversity. Prebiotics are substrates that are selectively used by host microorganisms conferring a health benefit [113] (International Scientific Association for Probiotics and Prebiotics-ISAPP consensus 2016), while probiotics are “live microorganisms that, when administered in adequate amounts confer a health benefit on the host [114] (Food and Agriculture Organization of the United Nations (FAO)/World Health Organization (WHO)-ISAPP 2013). Synbiotics are a synergistic combination of probiotics and prebiotics, which serve to improve the therapeutic benefits of probiotics by combining them with prebiotics to enhance their growth in the colon. The therapeutic and prophylactic effects of probiotics can be predetermined by modifying bacteria to produce biotherapeutic metabolites, and immune-modulating compounds to enhance host immunity and barrier integrity in the form of post-biotics. The beneficial effects induced by pre-, pro-, synbiotics are largely individual, and dependent on host genetic background, diet and gut microbial milieu.
In randomized control trial VSL#3, a live formulation of eight bacterial species (four strains of Lactobacillus, three strains of Bifidobacterium (Bifidobacterium breve, longum, and infantis), and one strain of Streptococcus) reduced the risk of hospitalization for HE in patients with cirrhosis [115]. VSL#3 treatment stimulated an increase in plasma albumin and hemoglobin, which can lead to lower MELD scores in patients with decompensated liver cirrhosis. In addition, a long-term investigation of 39 patients with biopsy-proven NAFLD demonstrated that VSL#3 (12 strains, 675 billion colony forming units (CFU)/day) administered for one year significantly improved NAS, and resulted in significant improvement in hepatocyte ballooning and hepatic fibrosis [116].
Upon examining all available clinical evidence, the impact of VSL#3 on HVPG remains uncertain and has led to reservations on use of probiotics in management of portal hypertension. One study of 12 patients demonstrated that administration of the probiotic mixture VSL#3 improved the hepatic and systemic hemodynamics and serum sodium levels in patients with cirrhosis [117], while two additional studies of a similar size showed that VSL#3 did not impact HVPG in both compensated and decompensated patients with cirrhosis [118,119]. When combined with the beta blocker propranolol, the VSL#3 probiotic mixture was safe and well tolerated in patients with cirrhosis and improved the response rate of propranolol with respect to HVPG [120].
Apart from their production of beneficial metabolites, probiotics can also be used to consume harmful bacterial products. SYNB1020, an engineered Escherichia Coli Nissle strain has been designed to consume colonic ammonia in patients with cirrhosis [121] (Synlogic, NCT03447730). Capturing part of gut the ammonia can attenuate clinical symptoms of hyperammonemia in conditions like urea cycle disorders and HE. Its development has unfortunately been discontinued given the negative trial data from an interim analysis of a placebo-controlled phase 1b/2a.
The benefits of prebiotics have been known since many years ago. Prebiotics, such as inulin, were associated with an increase in short-chain fatty acids such as propionate in the colon and portal vein. Lactulose, a non-absorbable disaccharide effectively reduces ammonia absorption in the gut and is an effective treatment for HE [101,122,123]. Despite its widespread use as a laxative and prebiotic, its influence on gut microbiota composition remains undefined. Lactulose acidifies the colonic contents resulting in decreased passive non-ionic diffusion of ammonia into the systemic concentration, as well as reduced formation of toxic SCFAs. Furthermore, lactulose prompts the growth of non-urease-producing bacteria such as lactobacillus and bifidobacteria.
(c) Fecal microbiota transplantation. Fecal microbiota transplantation (FMT) is used to replenish a healthy gut microbial environment and restore physiological colonization by transfer of microbial flora from a healthy donor. It represents a more robust method of manipulating the gut microbiota as compared to dietary/probiotic treatments and is now an accepted therapy for recurrent or refractory Clostridium difficile infection. A phase 1 study showed that FMT with oral capsules, following antibiotic pre-treatment with Rifaximin, was well tolerated and safe long term, and was associated with a reduction in serum LBP, and improved mucosal barrier integrity and EncephalApp performance in patients with cirrhosis and recurrent HE with MELD < 17 [124]. This is consistent with a RCT of 21 NAFLD patients provided with allogenic FMT, demonstrating improved intestinal barrier function, albeit no change in steatosis or insulin resistance [125]. Additional beneficial effects of FMT on the liver have been demonstrated in rats. In a model of non-alcoholic steatohepatitis with portal hypertension, transplantation of stool from healthy animals significantly reduced HVPG by 31% and restored the sensitivity to insulin via the hepatic protein kinase B-dependent endothelial nitric oxide synthase signaling pathway [126].
Surprisingly, FMT has also demonstrated promise as a measure for limiting alcoholic cirrhosis progression. In a phase 1 RCT of 20 patients, FMT enema from a donor enriched in Ruminococcaceae and short-chain fatty acid-producing taxa Lachnospiraceae was associated with short-term reduction in alcohol craving and consumption in patients with alcohol-associated cirrhosis (Hunter Holmes Mcguire Veteran Affairs Medical Center, NCT03416751). These data hint at a particularly potent effect of FMT in restoring microbiota composition and functionality in the course of alcoholic liver disease. FMT success is likely to be dependent on functionality of particular microbial consortia. Indeed, FMT has increased relative abundance of butyrate-producing genera such as Roseburia and Odoribacter, which are typically reduced during alcoholic cirrhosis [127]. It is thought that SCFA modulation along with an increase in beneficial taxa engages the gut–brain axis and hence could explain the reduction in alcohol craving.
Despite the clinically evident success and safety of FMT, it remains a second-line treatment owing to the risk of disease transmission between the donor and recipient, undesirable side effects, sustainability of the post-FMT microbiota, and the unclear effects on the recipient’s immune system. Further rigorous clinical studies are warranted to determine the utility of FMT in liver fibrosis. Table 1 summarizes ongoing pro-, pre-, synbiotic and FMT clinical tails for treatment of liver disease.

Table 1.
Summary of ongoing pre-, pro, synbiotics and FMT clinical trials for treatment of liver disease.
3.2.2. Postbiotics
The gut is the residence for up to 80% of immune cells in the body [128], where they respond to bacterial metabolites (postbiotics) responsible for immune system ontogeny, modulation of immune signaling and intestinal mucosal integrity. Postbiotics potentiate the morphological structures of the intestinal barrier by increasing the expression of tight junction proteins ZO-1 and intestinal mucin levels and increasing the secretion of anti-inflammatory cytokines such as IL-10 [129]. This protective role extends to the liver, as demonstrated by increased susceptibility to liver fibrosis in germ-free mice [130].
Gut commensal microbes produce a myriad of metabolites that modulate their environment. These include short-chain fatty acids (SCFAs) such as acetate, propionate and butyrate, which are end products of bacterial fermentation of dietary fibers, proteins with immunomodulatory activities (e.g., bacterial p40, HM0539), biosurfactants, bacteriocins, polysaccharides, and vitamins, to name a few. Given the versatility of postbiotics, meticulous clinical trials are required to support their use in diseases of gut-barrier dysfunction. Nonetheless, there remains a significant amount of evidence demonstrating the efficacy of bacterial metabolites as a treatment, as well as their dietary precursors and metabolizing microbes.
(a) Choline. Choline is an essential macronutrient with many functions, ranging from lipid metabolism and neurotransmitter synthesis to cell structure and DNA methylation [131]. The gut microbiota metabolizes dietary choline into trimethylamine (TMA), which enters into the portal circulation where it is oxidized by hepatic flavin-containing monooxygenases in the liver, forming trimethylamine-N-oxide (TMAO) [132]. This conversion of choline into methylamines results in deficiency of phosphatidylcholine, one of the major cytoprotective components of hepatocyte membranes against bile salts. Furthermore, the microbial TMA metabolite TMAO has been strongly associated with the presence and severity of NAFLD [133]. TMAO is thought to aggravate liver steatosis via modulation of the bile acid pool and suppression BA-mediated hepatic FXR signaling. Metagenomic analysis of stool microbiome of pediatric NAFLD patients revealed increased abundance of Gammaproteobacteria and Prevotella in comparison with the microbiota of obese children without NAFLD [134]. The class Gammaproteobacteria is known to harbor high concentrations of choline-metabolizing enzymes, which can influence susceptibility to and progression of hepatic steatosis.
(b) Short-chain fatty acids. Short-chain fatty acids (SCFAs) such as acetate, propionate, and butyrate are anaerobic fermentation products generated by cecal and colonic microbiota from non-digestible carbohydrates such as non-starch polysaccharides, resistant starch, and miscellaneous low-digestible saccharide prebiotics. Research has predominantly been focused on the least abundant SCFA, which perhaps possesses the most important biological roles—butyrate. Butyrate is a primary enterocyte energy source that dynamically promotes the maintenance of the colonic barrier via induction of tight junction proteins and mucins [135,136]. Moreover, butyrate exerts an anti-inflammatory effect and can suppress colonic and hepatic LPS-induced production of pro-inflammatory cytokines via inhibition of NF-κB activation. Studies suggest that butyrate produced by intestinal microbiota can modulate the pathogenesis of liver fibrosis. Butyric acid has been shown to be inversely correlated with the model for end-stage liver disease (MELD) score and was further reduced in patients with history of ascites, HE, and SBP [137]. Of note, the fraction of SCFA-producing bacterial phyla such as Firmicutes and Bacteroidetes are diminished during advanced stages of liver cirrhosis. Moreover, chronic alcohol intake induces skewing of intestinal SCFA concentrations, increasing the luminal acetate: butyrate ratio. Butyrate when supplemented in the form of rapidly absorbing prodrug, Tributyrin, to mice on chronic binge alcohol exposure, altered alcohol-induced intestinal permeability, inflammatory cytokine expression and liver transaminases when administered to mice following chronic alcohol exposure [138]. Icosabutate is a structurally engineered fatty acid that selectively targets the liver through the portal vein. Preliminary data presented at the International Liver Congress 2021 from an ongoing phase 2 study of patients with biopsy-confirmed NASH are encouraging. A 4-month treatment with Icosabutate caused significant dose-dependent decreases in alanine transaminase, aspartate transaminase, gamma-glutamyltransferase, and alkaline phosphatase combined with significant reductions in fibrosis PRO-C3 and Enhanced Liver Fibrosis (ELF) scores [139] (NorthSea Therapeutics B.V., NCT04052516).
(c) Urolithlin A. Among the metabolites of hydrolysable tannins that are produced in the gut microbiome, urolithin A (UroA) has received enormous attention recently as a novel candidate with anti-inflammatory and antioxidant effects in vitro and in vivo. UroA is believed to enhance gut barrier function by inducing tight junction proteins (Occludin, Claudin-4 and ZO-1) via activation of the aryl hydrocarbon receptor [140]. UroA demonstrates potent anti-inflammatory activity, reducing LPS-mediated IL-6 and TNF production via NF-κB suppression. In a recent study, UroA has been shown to attenuate ALD pathogenesis in both acute and chronic experimental mouse models by reducing alcohol induced barrier permeability, systemic endotoxin levels and inflammatory mediators [141].
4. Interventions Targeting Hepatic Immune Response
The progression of chronic liver disease is mediated primarily by the hepatic insult that triggers chronic immune activation, inflammation and fibrosis. The etiologies responsible for CLD and its related pathologies (e.g., portal hypertension and biliary dysfunction) also stimulate gastrointestinal dysfunction and significant alterations in the microbiome, allowing antigens to transit into the liver where they can exacerbate inflammatory conditions. Consequently, numerous therapeutic strategies are currently under development targeting the diverse repertoire of immune cell populations in the liver and their sensors responsible for antigen recognition and immune cell activation. The abundance of cell-surface, cytoplasmic and nuclear molecules that contribute to HSC activation provide fertile ground for novel antifibrotic therapies, several of which are undergoing drug development and clinical trials. A detailed cataloguing of these approaches is beyond the scope of this review and can be reviewed elsewhere [142]. This section will outline the mechanisms by which gastrointestinal and hepatic antigens exacerbate the progression of CLD, and current treatments aimed to quench the hepatic immune response. We pay special attention to liver macrophage populations due to their central role in the initiation and exacerbation of chronic liver disease and HSC activation.
4.1. Targeting Pattern Recognition Receptors
PRRs are a diverse group of sensors capable of recognizing molecular patterns conserved among microbial species, termed PAMPs [143]. In addition, they recognize an ever-growing list of endogenous molecules released following cellular damage/death called DAMPs. The first-discovered and best-characterized PRR families are TLRs. TLRs are evolutionarily conserved type I transmembrane proteins, expressed in many internal organs including the liver. At present, 13 human TLRs have been identified that recognize diverse intracellular and extracellular microbial antigens ranging from DNA and RNA to bacterial membrane and fungal wall components. In addition to TLRs, a variety of PRRs have been characterized, including cell surface c-type lectin receptors (CLRs), as well as intracellular receptors such as the family of nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I), stimulator of interferon genes (STING).
As outlined above, a compromised gut barrier in CLD allows influx of gut-derived antigenic loads via the portal vein, triggering chronic breakdown in TLR tolerance against endogenous ligands and further transcriptional expression of pro-inflammatory/anti-inflammatory mediators and interferons. This inflammatory milieu/micro-environment in the liver results in activation of quiescent HSCs to initiate the production of several extracellular matrix proteins including collagen. In liver injury and hepatic fibrogenesis TLR3, TLR4 and TLR9 have been best characterized with respect to inflammation and fibrosis resulting from gut-derived PAMPs and host-derived DAMPs. Innate immune sensing of gut-derived microbial products by PRRs and their impact on chronic liver disease have been recently reviewed elsewhere [144,145,146]. Herein, we will focus on the PRRs for which antifibrotic treatments are in development, including TLR3, 4, and 9, as well as the NLRP3 inflammasome.
4.1.1. Key Toll-like Receptors in Liver Fibrosis
(a) Toll-like Receptor 4. TLR4 in combination with its co-receptors MD2 and CD14 recognize potent inflammatory PAMPs (flagellin and LPS) and endogenous DAMPS such as calprotectin, S100A8/9 HMGB1 and HSP70 [147]. In the liver, TLR4 is ubiquitously expressed by both hepatocytes and non-parenchymal cells, including LSECs and KCs. Importantly, the activation of TLR4 is directly linked to circulating LPS, hepatic inflammation and fibrosis development. Activated Kupffer cells secrete pro-inflammatory cytokines (TNF, IL-1β, IL-6) and fibrogenic stimuli (TGF- β, platelet-derived growth factor [PDGF]) to stimulate HSC differentiation into extracellular matrix producing myofibroblasts [148,149]. TLR4 stimulation additionally leads to upregulation of inflammatory cytokines (TNF, IL-1β, IL-6) and chemokines (such as MCP-1, MIP-1β, and RANTES) in HSCs, further recruiting monocytes and KCs [150]. During chronic LPS stimulation this positive feedback loop is potentiated by NFκB mediated repression of Bambi transcriptional activity which contributes to TLR4-mediated enhancement of TGF-β signaling in HSCs [149,151].
Recent studies in murine models have demonstrated that TLR4 deficiency reduces pro-inflammatory cytokine production of IL-1α, IL-1β and IL-6 as well as liver injury in acetaminophen-induced liver injury and ALD [152]. In addition, endotoxin-resistant TLR4 mutant mice fed the (MCD) NASH diet possessed significantly reduced hepatic inflammation and injury markers supporting the pro-inflammatory role of TLR4 [153]. Consequently, the prevention of excessive activation and inhibition of TLR4 have become attractive pharmacological strategies to inhibit fibrogenesis.
Although many TLR4 antagonists have been examined, very few have progressed into clinical trials due to worries regarding potential effects on systemic immunity. Nonetheless, both animal models and in vitro studies have demonstrated a clear benefit of TLR4 antagonism and in multiple etiologies of CLD. The most well-known TLR4 antagonist to enter clinical trials was Eritoran, followed by TAK-242 and NI-0101. Eritoran tetrasodium (E5564) is a synthetic Lipid A mimic that binds to the MD2-TLR4 interface. It has been reported to decrease LPS-induced acute severe liver injury in rats by decreasing activation of MAP kinases and TNF gene expression [154,155]. Inhibition of TLR4 with the antagonist, E5564 was tested in humans with severe sepsis in the ACCESS trial, showing no effect on 28-day all cause mortality [156].TAK-242 (Resatorvid) is another small molecule inhibitor of TLR4 that reduces LPS-induced cytokine secretion and cell death in hepatocytes and monocytes in vitro. Importantly, TAK-242 reduced the severity of inflammation, hepatocyte death and improved organ function in two animal models of ACLF (bile duct ligation + LPS; CCl4 + LPS) and one model of acute liver failure (Galactosamine + LPS) [146]. Monoclonal antibodies have also proven effective, as exemplified by the NI-0101 humanized monoclonal antibody (mAb) that interferes with TLR4 dimerization and activation. In a randomized phase 1 dose escalation study of healthy volunteers receiving LPS, NI-010 was shown to possess durable anti-inflammatory properties, suppressing the production of IL-6, TNFα, CXCL10, and IFNβ [157].
The use of TLR4 antagonists in combination with other treatments, may provide additional therapeutic benefits. Using a murine model of CCl4-induced fibrosis, the TLR4 inhibitor Serelaxin (RLX030), when combined with the PPARγ agonist rosiglitazone, has been shown to amplify the beneficial effects of rosiglitazone and simultaneously reduce hepatic collagen content and HSC activation [158]. In addition, the small molecule Ibudilast, a phosphodiesterase-4 inhibitor, is currently being assessed for the treatment of extrahepatic conditions [159] in combination with other TLR4 antagonists such as TAP2, TLR4-IN-C34 and M62812, which are clinically effective for the management of chronic inflammatory conditions and sepsis. Regardless of how promising TLR4 antagonists are in the treatment of liver fibrosis, there are still challenges in bioavailability and delivery. Nonetheless, anti-TLR4 therapies may represent an alternative strategy for future treatments for liver fibrosis.
(b) Toll-like Receptor 9. TLR9 binds double-stranded CpG unmethylated DNA from bacteria, fungi and viruses, as well as host-derived DNA derived from apoptotic cells. In line with changes observed in intestinal permeability, an increase in circulating bacterial DNA is often an early event in many experimental models of alcoholic liver disease and fatty liver, even preceding hepatic fibrosis [160,161]. Activation of TLR9 from host-derived apoptotic hepatocyte DNA can exacerbate fibrogenic signaling by retaining HSCs at sites of cellular apoptosis, where they become activated and up-regulate collagen production [162]. TLR9 and STING have been shown to synergistically trigger a macrophage pro-inflammatory response to self-mitochondrial DNA (mtDNA) during the development of NASH [163]. This has been confirmed in murine models of diet-induced NASH, where TLR9 deletion or pharmacological antagonism resulted in an attenuated response to bacterial DNA and mtDNA, leading to reduced IL-1β production, steatosis and liver injury [164]. These findings are supported by TLR9 knockout (KO) models, where mice lacking TLR9 develop less severe steatohepatitis and liver fibrosis when compared to wild-type mice on a choline-deficient l-amino acid-defined diet [165,166].
Compared to TLR4, fewer therapies are available to inhibit TLR9 activation. The novel TLR9 antagonist COV08-0064, a small-molecule inhibitor with greater specificity for TLR9 than oligo-based antagonists, has shown promise in damping hepatocellular death in animal models of sterile liver inflammation [167,168]. A newly developed TLR9 mAb, clone NaR9, has also shown promise, rescuing mice from fulminant hepatitis caused by administering the TLR9 ligand CpGB and D-(+)-galactosamine [169]. Human studies using TLR9 agonism have been exploited in an attempt to improve antiviral responses against chronic infections as well as cancer therapies. TLR9 inhibition, however, is considerably less common, having been employed to limit excessive immune activation in conditions such as IgA nephropathy and Sjogren’s syndrome [167,170,171]. TLR9 antagonism as a treatment for chronic liver disease has not yet been assessed in humans, but warrants attention due to accumulated evidence in animal models and in vitro studies.
(c) Toll-like Receptor 3. Another interesting but often overlooked target in liver inflammatory and fibrotic progression is TLR3. TLR3 activation is generally thought to have a protective and anti-inflammatory role in many models of liver disease. This role was clearly demonstrated in mice fed with a high-fat diet followed by binge drinking to induce liver injury [172]. TLR3 activation by polyinosinic-polycytidylic acid (polyI:C) attenuated liver fibrosis by increasing HSC and KC IL-10 expression, as well as reducing hepatic expression of TNF, IL-6 and CXCL2. TLR3 signaling is well defined in rodent natural killer (NK) cells, where activation of TLR3 results in a potent anti-fibrotic effect [173]. A study by Li et al. showed that hepatic NK cells can be synergistically activated by IL-18 and TLR3 ligand stimulation to induce HSCs apoptosis via TNF-related apoptosis-inducing ligand (TRAIL)-mediated degranulation [174]. On the contrary, exosome-mediated activation of TLR3 in HSCs has been shown to exacerbate liver fibrosis by enhancing IL-17A production by γδ T cells [175].
4.1.2. NLRP3
NLRP3 is an intracellular sensor that detects a broad range of microbial ligands, resulting in activation of the NLRP3 inflammasome. Hepatic NLRP3 inflammasome activation promotes caspase-1-dependent release of the pro-inflammatory cytokines IL-1β and IL-18 by KCs, as well as to gasdermin D-mediated pyroptotic cell death that has been demonstrated in patients with NASH and ASH. NLRP3 inflammasome hyper-activation has been linked to severe hepatic inflammation, fibrosis and hepatocyte pyroptosis in mice [176]. In addition, NLRP3 KO mice, who were fed on a high fat and caloric diet to establish NASH have shown enhanced hepatic MCP-1 expression and extensive M1 macrophage infiltration [177]. PAMP-responsive NLRP3 inflammasome inhibition with either MCC950 or a potent vitamin D receptor agonist, Calcipotriol, can alleviate fibrosis and partially reversed liver scarring in the murine models of NASH and experimental liver cholestasis respectively [178,179]. In humans, other investigational NLRP3 inhibitors such as NT-0167 and IFM-2427, which have demonstrated reduction in IL-1β and IL-18 in preclinical trials, are still being evaluated in a phase 1 clinical trial for treatment of numerous chronic inflammatory and fibrotic diseases [180,181].
4.2. Targeting Liver Macrophages
Hepatic macrophages hold a central position in maintaining homeostasis in the liver as well as in the pathogenesis of disease. Evidence suggests that KCs release CCL2 (C-C motif chemokine ligand 2) in response to acute insult to recruit pro-inflammatory and profibrogenic monocytes. KCs also possess a fibrogenic role during chronic liver disease by activating HSCs via secretion of TGFβ, platelet-derived growth factor (PDGF) and connective tissue growth factor (CTGF/CCN2). A profibrogenic niche is established in the liver via additional production of pro-inflammatory cytokines (TNF, IL-1β, IL-6) and chemokines (CCL1, CCL2 and CCL5) that exacerbate chronic inflammation and fibrosis progression. Activated HSCs undergo a phenotypic switch from a quiescent into proliferative, myofibroblast-like cells, exhibiting upregulated collagen synthesis, increased proliferation, migration and a relative resistance to apoptosis [182]. On the other hand, KCs also promote resolution of fibrosis and degradation of extracellular matrix via induction of metalloproteinases (MMP-9, -12 and -13) [35]. Given this role as first-line responders to liver injury and their dual functions in liver disease, hepatic macrophages are, in principle, intriguing therapeutic targets (Figure 4). Notwithstanding, macrophages exert a broad range of functions in the liver and consist of heterogeneous cellular subsets, thus rendering the development of macrophage-based interventional strategies targeting hepatic fibrosis a challenging task.
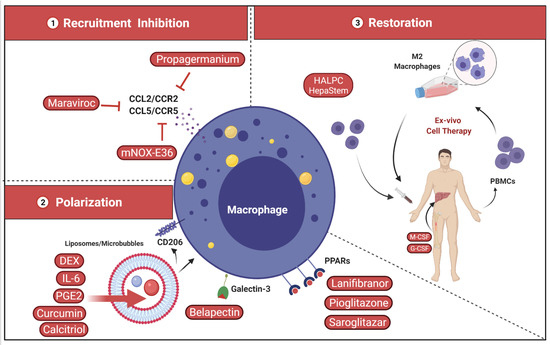
Figure 4.
Potential targets of hepatic macrophages to treat liver fibrosis. 1—Inhibition of inflammatory monocyte/macrophage recruitment through the inhibition of CCL2 (MCP-1) by RNA-based silencing (e.g., RNA-aptamer mNOX-E36), small molecule or monoclonal antibody (mAb). 2—Shaping and polarization of hepatic macrophage via biological engineering using nanoparticles or targeting drugs (e.g., dexamethasone [DEX] liposomes/microbubbles) from the pro-inflammatory to regenerative phenotype. 3—Restoration of hepatic macrophage count and function using IL-4 and CX3CL1 [183], macrophage colony stimulating factor (M-CSF) inhibitors or autologous macrophage-based cell therapy. CCL5/CCR5, C-C Chemokine ligand 5/C-C chemokine receptor type 5; CD206, Mannose receptor; HALPC, human allogeneic liver-derived progenitor cells; HepaStem® is an allogenic stem cell therapy product using human adult liver-derived progenitor cells; M2, macrophage type 2; G-CSF, granulocyte colony-stimulating factor; PPARs, peroxisome proliferator-activated receptors.
4.2.1. Inhibition of Inflammatory Monocyte Recruitment
The infiltration of inflammatory monocytes into the liver is critically regulated by the chemoattractant properties of several chemokines. The chemokine CCL2 and its receptor CCR2 represent the critical trigger for acute monocyte infiltration into the liver in alcoholic liver disease, NAFLD/NASH, and viral hepatitis, among others, representing a key initial step in the fibrosis that ensues [184]. During the early phase of liver injury in CCl4 induced liver injury model, inflammatory Ly 6Chigh monocytes directly activate HSCs in a TGF-β-dependent manner, with CCL2 playing a central role in their recruitment [185]. Indeed, targeted removal of infiltrated monocytes during fibrosis development using CD11b-diphtheria toxin receptor transgenic mice, reduced fibrosis in the CCl4 liver injury mouse model. Importantly, the depletion of these macrophages during resolution phase did not hamper HSC activation and ECM deposition, supporting the role of infiltrating monocytes in fibrosis initiation [186].
Due to the key role played by CCL2 in fibrosis initiation, interventions targeting CCL2-mediated recruitment are an attractive strategy to limit fibrosis initiation and progression. The CCL2 neutralizing RNA-aptamer mNOX-E36 has proven effective in ameliorating steatosis development and fibrosis progression in CCl4 or MCD models by preventing inflammatory monocyte recruitment into the liver [187]. Blockade of the CCL2 receptor CCR2 with the selective inhibitor Propagermanium has also been shown to reduce disease activity in a mouse model of NASH by reducing macrovesicular steatosis and lobular inflammation at early stages of disease [188].
The dual CCR2/CCR5 inhibitor, Cenicriviroc (CVC), has also been shown to effectively hamper CCL2-mediated monocyte recruitment and to exert antifibrotic effects in mouse models of ALD and NASH [189,190,191]. Despite promising results and success in early trials for treatment of liver fibrosis in NASH patients where improvement in fibrosis by ≥1 stage was achieved after 1 year of CVC (NASH CRN system) [192,193], the phase 3 clinical trial (AURORA) was discontinued due to lack of efficacy. (Tobira Therapeutics, Inc., NCT03028740) [194].
Maraviroc (Selzentry/Celsentri) is a sole CCL5 inhibitor that has been shown to reduce hepatic fibrosis progression and improve disease in murine NAFLD/NASH models [195]. Currently, Maraviroc is being evaluated in Phase 4 open-label study in combination with antiretroviral therapy in people living with HIV-1 as a treatment for NAFLD [196] (Brighton and Sussex University Hospitals NHS Trust, 2017-004141-24).
4.2.2. Shape and Polarization of Hepatic Macrophage Function
A promising strategy for treatment of liver fibrosis is via modulation of the functional switch between pro-inflammatory and regenerative liver macrophages. This approach uses bioengineered nanoparticles or “polarizing” drugs tailored to induce selective and functional reprogramming of hepatic macrophages [197]. The advantage of this approach is the absence of off-target effects when compared with conventional systemic therapies. This is mediated by the innate scavenging activity of KCs that drives the uptake of most nanomaterials and drug delivery systems in the liver. The addition of mannose or other carbohydrate-functionalized polymers to the surface of nanoparticles can potentially further improve delivery of anti-inflammatory drugs to hepatic macrophages due to their high expression of mannose receptors such as CD206. Other drug delivery systems, such as liposomes and microbubbles can be loaded with anti-inflammatory treatments such as corticosteroids (dexamethasone (DEX)), IL-4, IL-10 and PGE2, and have additionally been proposed/utilized as potential macrophage-specific treatment approaches in murine models [198].
The delivery of DEX-loaded vehicles has demonstrated efficacy in attenuating liver fibrosis in a murine model of CCl4 liver injury. Intravenous injections of DEX liposomes resulted in a significant reduction in fibrosis, inhibition of T-cell accumulation in liver and functional skewing of liver macrophages toward an M2 phenotype [199]. Modifying the surface chemistry of nanoparticle drug carriers can also induce immunomodulatory effects on hepatic macrophages [200]. Liposome-encapsulated lipophilic curcumin or 1,25-dihydroxy-vitamin D3 (calcitriol), when injected intravenously into mice with diet-induced NASH, shifted the hepatic dendritic cell inflammatory profile towards a regulatory phenotype, reduced inflammation and suppressed immune activation and collagen deposition [201].
Galectin-3 (Gal-3) is a β-galactoside-binding lectin expressed primarily in macrophages that stimulates TGF-β production, myofibroblast activation and extracellular matrix production. The galectin-3 inhibitor GR-MD-02 (Belapectin) has been shown to reduce hepatic fibrosis and portal hypertension when used in murine models of cirrhosis [202,203]. Unfortunately, in a phase 2b trial of 162 patients with NASH, cirrhosis, and portal hypertension, Belapectin had no significant effect on HVPG and fibrosis but proved effective at reducing the risk of variceal developments in selected patients with NASH-induced cirrhosis (Galectin Therapeutics Inc., NCT02462967) [204].
A family of therapeutic targets that has gained attention over recent years is the peroxisome proliferator-activated receptors (PPARα, β/δ, and γ); a group of nuclear transcription factors differentially expressed among hepatic cell types including macrophages. Dysregulated PPARs during chronic hepatic injuries contribute to liver disease progression and major metabolic dysfunctions. Moreover, beneficial effects from activating one or multiple PPAR isoforms on reversing fibrosis, as well as phenotypic enhancement of different liver cell types, have been observed in preclinical studies. Thus, strategies that modulate PPAR activity have the potential to induce macrophage polarization. Activation of PPAR-γ in particular has emerged as a key regulator of hepatic macrophage polarization, stimulating an anti-inflammatory M2 phenotype [205].
In vitro administration of the pan-PPAR agonist Lanifibranor has been shown to significantly reduce portal pressure, improve intrahepatic vascular resistance and prevent ascites in the rodent model of TAA or common bile duct ligation induced cirrhosis. The underlying mechanisms of the hemodynamic ameliorations involve marked deactivation of HSCs and inhibition of inflammatory macrophages probably via PPARδ agonism [206,207,208]. In humans, Lanifibranor exerted positive metabolic and anti-fibrotic effects in adult patients with NASH [208]. In a short 24-week clinical trial (NATIVE study), a single dose of Lanifibranor when given daily significantly decreased liver inflammation and prevented worsening of fibrosis in 49% of participants compared with 27% in the placebo arm [209,210]. Currently, Lanifibranor is being investigated in a phase 3 study of adults with NASH with stage 2/3 liver fibrosis (Inventia Pharma NCT04849728). Similarly, Pioglitazone and Saroglitazar, selective PPAR-γ and PPAR-α agonists, have been shown to improve NASH [211]. Pioglitazone reduced NAS by at least 2 points without worsening of fibrosis in more than half of patients [212] (University of Florida, NCT00994682). Likewise, Saroglitazar significantly improved liver stiffness measurement on FibroScan in NAFLD patients with diabetic dyslipidemia [213] (Zydus therapeutics Inc., NCT03061721).
4.2.3. Restoration of Hepatic Macrophage Count and Function
Macrophage polarization is a continuum of overlapping functional states that involve a plethora of signals and corresponding dynamic gene expression programs. During fibrogenesis, inflammatory monocytes are recruited to the inflamed liver, forming profibrotic macrophages. These cells phagocytose cellular debris, activating the ERK signaling cascade and forming restorative macrophages that orchestrate fibrinolysis. In line with this observation, it is speculated that phagocytosis elicits significant effects on macrophage phenotype and function to promote restorative differentiation pathways [214,215]. Therefore, induced phagocytic behavior in vivo has potential as a translational strategy for the treatment of hepatic fibrosis.
Another therapeutic intervention consists of autologous macrophage-based cell therapy, which entails ex vivo culturing of peripheral blood mononuclear cells under selective conditions to induce antifibrotic or fibrinolytic subsets. Following differentiation, macrophages are intravenously infused back into patients to hypothetically improve fibrosis. A first-in-human, phase 1, single-arm, dose escalation clinical trial in nine patients with compensated liver cirrhosis showed that administration of macrophages was safe, with no clinically relevant adverse reactions recorded during the infusion or in the immediate post-infusion period. Several non-invasive measures of liver fibrosis were improved following macrophage infusion, including transient elastography, serum-enhanced liver fibrosis score and the collagen turnover markers PRO-C3 and C3M, highlighting the potential antifibrotic effect of autologous monocyte-derived macrophage infusion in cirrhosis [216] (ISRCTN 10368050).
Another potential strategy utilizes bone marrow-derived stem cell (BMSC) engraftment onto the damaged liver, by using cytokines such as macrophage colony-stimulating factor (M-CSF) or granulocyte colony-stimulating factor (G-CSF) in advanced liver cirrhosis [217]. Both cytokines are hematopoietic growth factors that affect monocytopoiesis and monocyte release from bone marrow. In a single-center randomized trial of 50 patients with decompensated cirrhosis, a combination of G-CSF and erythropoietin (Darbepoetin α) improved survival by 1 additional year as compared to the placebo group [218] (Institute of Liver and Biliary Sciences, India, NCT01384565).
Cell-based therapies such as infusion of bona fide allogeneic liver-derived progenitors is another attractive strategy to dampen the pro-inflammatory hepatic milieu, inhibit HSC activation and fibrogenesis. HepaStem® is advertised as a highly advanced cell therapy platform consisting of human liver-derived stem cells that are ethically obtained from healthy donors and expanded in a current good manufacturing practice (cGMP)-compliant environment [219]. These progenitors have been tested in a phase 2a study in NASH, and a phase 2a study in ACLF, the first ever study to use stem cells to treat such indications. The results show that human allogeneic liver-derived progenitor cells (HALPC) infusion reduce systemic inflammatory markers and decrease altered liver function in surviving ACLF patients. The 28-day and 3-month survival rates were 83% and 71%, and no patient had ACLF at 3 months [220] (Promethera Biosciences (NCT04229901), EudraCT 2016-001177-32).
5. Conclusions
In light of the changing nature of CLD epidemiology, there is an ever-growing demand for novel targets and specific treatments that reverse or cease fibrosis progression. Interactions between the gut microbiome, the immune system and the liver are increasingly implicated in CLD progression and severity. Moreover, the steady advances over the last decade in basic research exploring mechanisms of hepatic fibrosis coupled with thorough dissection of the intricate gut–liver relationship have spurred promising avenues for identification of strategies to prevent liver fibrosis. Several of these strategies have already fueled a robust therapeutic pipeline across a variety of novel targets. Many of these emerging therapeutics have been systematized to target the intestinal mucosa, intestinal microbiome, or hepatic immune response. Additionally, some of these emerging therapies have shown beneficial effects on portal pressure and extrahepatic benefits such as improvement of body weight, lipid profile, glycemic control and others. Strategies involving combination therapies with different antifibrotic agents or monotherapy with multi-target drugs may be more effective given the complex mechanisms and pathways involved in hepatic fibrosis. However, standardized, stringent and unbiased interventional trials are still required for successful translation of any strategy into real world clinical practice.
Author Contributions
Writing—original draft preparation, review and editing (E.K., S.R., G.A.); S.R. and G.A. have equally contributed. Figures were created with BioRender.com. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ainsworth bequest to Western Sydney University.
Conflicts of Interest
The authors declare no conflict of interest.
List of Abbreviations
| AASLD | American Association for the Study of Liver Diseases |
| ACLF | Acute-on-chronic liver failure |
| ALD | Alcoholic liver disease |
| ASH | Alcoholic steatohepatitis |
| BAs | Bile acids |
| BT | Bacterial translocation |
| CCL2/MCP-1 | Chemokine (C-C motif) ligand 2/ Monocyte chemoattractant protein-1 |
| CCL5 | Chemokine (C-C motif) ligand 5 |
| CCl4 | Carbon tetrachloride |
| CCR5 | C-C chemokine receptor type 5 |
| CLD | Chronic liver disease |
| CTGF/CCN2 | Connective tissue growth factor |
| CVC | Cenicriviroc |
| DAMPs | Damage-associated molecular patterns |
| DMR | Duodenal mucosal resurfacing |
| DEX | Dexamethasone |
| ECM | Extracellular matrix |
| ESAL | European Association for the Study of the Liver |
| FGF19 | Fibroblast growth factor 19 |
| FGF21 | Fibroblast growth factor 21 |
| FMT | Fecal microbiota transplantation |
| FXR | Farnesoid X receptor |
| Gal-3 | Galectin-3 |
| G-CSF | Granulocyte colony-stimulating factor |
| GGG | Glucagon/GIP/GLP-1 |
| GIP | Glucose-dependent insulinotropic peptide |
| GLP-1 | Glucagon like peptide-1 |
| GLP-1R | Glucagon-like peptide-1 receptor |
| HE | Hepatic encephalopathy |
| HMGB1 | High-mobility group box 1 |
| HSCs | Hepatic stellate cells |
| HVPG | Hepatic venous pressure gradient |
| IL-1β | Interleukin one beta |
| IL-6 | Interleukin six |
| ISAPP | International Scientific Association for Probiotics and Prebiotics |
| KCs | Kupffer cells |
| LPS | Lipopolysaccharide |
| LBP | Lipopolysaccharide binding protein |
| LSECs | Liver sinusoidal endothelial cells |
| mAb | Monoclonal antibody |
| MAFLD | Metabolic associated fatty liver disease |
| MAMPS | Microbial -associated molecular patterns |
| M-CSF | Macrophage-colony stimulating factor |
| MCD | Methionine Choline- deficient |
| MELD | Model for end-stage liver disease |
| mtDNA | Mitochondrial DNA |
| NAS | NAFLD activity score |
| NASH | Non-alcoholic steatohepatitis |
| NF-κB | Nuclear factor-kappa B |
| NK | Natural killer |
| NSBBs | Nonselective beta blockers |
| OCA | Obeticholic acid |
| PAMPS | Pathogen-associated molecular patterns |
| PDGF | Platelet-derived growth factor |
| PPARs | Peroxisome proliferator-activated receptors |
| PRRs | Pattern recognition receptors |
| PTH | Portal hypertension |
| RCT | Randomized control trial |
| RIG-1 | Retinoic acid-inducible gene I |
| ROS | Reactive oxygen species |
| SBP | Spontaneous bacterial peritonitis |
| SCFA(s) | Short-chain fatty acids |
| SSD | Soluble solid dispersion |
| STING | Stimulator of interferon genes |
| TAA | Thioacetamide |
| TGF-β | Transforming growth factor beta |
| TGR5 | Takeda G-protein-coupled receptor 5 |
| TLRs | Toll-like receptors |
| TMA | Trimethylamine |
| TMAO | Trimethylamine-N-oxide |
| TNF-α | Tumor necrosis factor alpha |
| Uro A | Urolithin A |
| VH | Variceal hemorrhage |
| ZO-1 | Zonula occludens-1 |
| α-SMA | Alpha Smooth muscle actin |
References
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Zhai, M.; Liu, Z.; Long, J.; Zhou, Q.; Yang, L.; Zhou, Q.; Liu, S.; Dai, Y. The incidence trends of liver cirrhosis caused by nonalcoholic steatohepatitis via the GBD study 2017. Sci. Rep. 2021, 11, 5195. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases From 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef]
- Adams, L.A.; Roberts, S.K.; Strasser, S.I.; Mahady, S.E.; Powell, E.; Estes, C.; Razavi, H.; George, J. Nonalcoholic fatty liver disease burden: Australia, 2019–2030. J. Gastroenterol. Hepatol. 2020, 35, 1628–1635. [Google Scholar] [CrossRef] [Green Version]
- Collaborators, G.B.D.C. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef] [Green Version]
- Australian Institute of Health and Welfare. Leading Cause of Premature Mortality in Australia Fact Sheet: Liver Disease, Cat. no. PHE 199; AIHW: Canberra, Australia, 2015. [Google Scholar]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Hernaez, R.; Kramer, J.R.; Liu, Y.; Tansel, A.; Natarajan, Y.; Hussain, K.B.; Gines, P.; Sola, E.; Moreau, R.; Gerbes, A.; et al. Prevalence and short-term mortality of acute-on-chronic liver failure: A national cohort study from the USA. J. Hepatol. 2019, 70, 639–647. [Google Scholar] [CrossRef] [PubMed]
- WHO Global Health Observatory data repository. Liver Cirrhosis (15+), Age-Standardized Death Rates by Country for 2016. Available online: https://apps.who.int/gho/data/view.main.53420 (accessed on 10 July 2021).
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Bosch, J.; Abraldes, J.G.; Berzigotti, A.; Garcia-Pagan, J.C. The clinical use of HVPG measurements in chronic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, C.; Groszmann, R.J.; Garcia-Tsao, G.; Bosch, J.; Grace, N.; Burroughs, A.; Planas, R.; Escorsell, A.; Garcia-Pagan, J.C.; Makuch, R.; et al. Hepatic venous pressure gradient predicts development of hepatocellular carcinoma independently of severity of cirrhosis. J. Hepatol. 2009, 50, 923–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, V.; Moreau, R.; Kamath, P.S.; Jalan, R.; Gines, P.; Nevens, F.; Fernandez, J.; To, U.; Garcia-Tsao, G.; Schnabl, B. Acute-on-chronic liver failure in cirrhosis. Nat. Rev. Dis. Primers 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, R.; Jalan, R.; Gines, P.; Pavesi, M.; Angeli, P.; Cordoba, J.; Durand, F.; Gustot, T.; Saliba, F.; Domenicali, M.; et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013, 144, 1426–1437. [Google Scholar] [CrossRef]
- Gustot, T.; Stadlbauer, V.; Laleman, W.; Alessandria, C.; Thursz, M. Transition to decompensation and acute-on-chronic liver failure: Role of predisposing factors and precipitating events. J. Hepatol. 2021, 75, S36–S48. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The microbiota in cirrhosis and its role in hepatic decompensation. J. Hepatol. 2021, 75, S67–S81. [Google Scholar] [CrossRef]
- Reiberger, T.; Ferlitsch, A.; Payer, B.A.; Mandorfer, M.; Heinisch, B.B.; Hayden, H.; Lammert, F.; Trauner, M.; Peck-Radosavljevic, M.; Vogelsang, H.; et al. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J. Hepatol. 2013, 58, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.G.; Mendoza, Y.P.; Bosch, J. Beta-blockers in cirrhosis: Evidence-based indications and limitations. JHEP Rep. 2020, 2, 100063. [Google Scholar] [CrossRef] [Green Version]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Rep. 2019, 1, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. Publisher Correction: The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 785. [Google Scholar] [CrossRef]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Baffy, G. Potential mechanisms linking gut microbiota and portal hypertension. Liver Int. 2019, 39, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, G.; Gustot, T.; Mookerjee, R.P.; Garcia-Pagan, J.C.; Fallon, M.B.; Shah, V.H.; Moreau, R.; Jalan, R. Inflammation and portal hypertension—the undiscovered country. J. Hepatol. 2014, 61, 155–163. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Grander, C.; Moschen, A.R. How does the microbiome affect liver disease? Clin. Liver Dis. 2016, 8, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Davis, B.C.; Bajaj, J.S. The Human Gut Microbiome in Liver Diseases. Semin. Liver Dis. 2017, 37, 128–140. [Google Scholar] [CrossRef]
- Cho, I.; Yamanishi, S.; Cox, L.; Methe, B.A.; Zavadil, J.; Li, K.; Gao, Z.; Mahana, D.; Raju, K.; Teitler, I.; et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 2012, 488, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Eksteen, B.; Curbishley, S.M. Immunology of the gut and liver: A love/hate relationship. Gut 2008, 57, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A. Metabolic liver disease—what’s in a name? Nat. Rev. Endocrinol. 2021, 17, 79–80. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.L.; Chen, H.; Wang, C.L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Goel, A.; Gupta, M.; Aggarwal, R. Gut microbiota and liver disease. J. Gastroenterol. Hepatol. 2014, 29, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebicka, J.; Reiberger, T.; Laleman, W. Gut-Liver Axis Links Portal Hypertension to Acute-on-Chronic Liver Failure. Visc. Med. 2018, 34, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Halilbasic, E.; Fuchs, C.; Traussnigg, S.; Trauner, M. Farnesoid X Receptor Agonists and Other Bile Acid Signaling Strategies for Treatment of Liver Disease. Dig. Dis. 2016, 34, 580–588. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Schwabl, P.; Reiberger, T.; Trauner, M. Liver Capsule: FXR agonists against liver disease. Hepatology 2016, 64, 1773. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Farre, R.; Trebicka, J.; Komuta, M.; Roskams, T.; Klein, S.; Elst, I.V.; Windmolders, P.; Vanuytsel, T.; Nevens, F.; et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 2014, 59, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kuruba, R.; Wilson, A.; Gao, X.; Zhang, Y.; Li, S. Inhibition of endothelin-1-mediated contraction of hepatic stellate cells by FXR ligand. PLoS ONE 2010, 5, e13955. [Google Scholar] [CrossRef] [Green Version]
- Ubeda, M.; Lario, M.; Munoz, L.; Borrero, M.J.; Rodriguez-Serrano, M.; Sanchez-Diaz, A.M.; Del Campo, R.; Lledo, L.; Pastor, O.; Garcia-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Farre, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am. J. Pathol. 2015, 185, 409–419. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Mookerjee, R.; Rosselli, M.; Pieri, G.; Beecher-Jones, T.; Hooshmand-Rad, R.; Chouhan, M.; Mehta, G.; Jalan, R.; Shapiro, D. Effects of the FXR agonist obeticholic acid on hepatic venous pressure gradient (HVPG) in alcoholic cirrhosis: A proof of concept phase 2a study. J. Hepatol. 2014, 60, S7–S8. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Pares, A.; Mason, A.; et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, P.; Wei, G.; Huang, P.; Li, W.; Qi, X.; Lin, Y.; Vaid, K.A.; Wang, J.; Zhang, S.; Li, Y.; et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 2020, 40, 1655–1669. [Google Scholar] [CrossRef]
- ENYO PHARMA. Vonafexor (EYP001) in NASH. 2019. Available online: http://www.enyopharma.com/pipeline/vonafexor-in-nash/ (accessed on 15 July 2021).
- Schwabl, P.; Hambruch, E.; Seeland, B.A.; Hayden, H.; Wagner, M.; Garnys, L.; Strobel, B.; Schubert, T.L.; Riedl, F.; Mitteregger, D.; et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J. Hepatol. 2017, 66, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef]
- Schwabl, P.; Hambruch, E.; Budas, G.R.; Supper, P.; Burnet, M.; Liles, J.T.; Birkel, M.; Brusilovskaya, K.; Konigshofer, P.; Peck-Radosavljevic, M.; et al. The Non-Steroidal FXR Agonist Cilofexor Improves Portal Hypertension and Reduces Hepatic Fibrosis in a Rat NASH Model. Biomedicines 2021, 9, 60. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Y.; Liu, Y.; Wang, W.; Tian, X.; Chen, S.; Lu, Y.; Du, J.; Cai, W. A nonbile acid farnesoid X receptor agonist tropifexor potently inhibits cholestatic liver injury and fibrosis by modulating the gut-liver axis. Liver Int. 2021. [Google Scholar] [CrossRef]
- Lucas, K.J.; Lopez, P.; Lawitz, E.; Sheikh, A.; Aizenberg, D.; Hsia, S.; Boon Bee, G.G.; Vierling, J.; Frias, J.; White, J. Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig. Liver Dis. 2021, 52, e38. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated With the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Renga, B.; Cipriani, S.; Carino, A.; Simonetti, M.; Zampella, A.; Fiorucci, S. Reversal of Endothelial Dysfunction by GPBAR1 Agonism in Portal Hypertension Involves a AKT/FOXOA1 Dependent Regulation of H2S Generation and Endothelin-1. PLoS ONE 2015, 10, e0141082. [Google Scholar] [CrossRef] [Green Version]
- Klindt, C.; Reich, M.; Hellwig, B.; Stindt, J.; Rahnenfuhrer, J.; Hengstler, J.G.; Kohrer, K.; Schoonjans, K.; Haussinger, D.; Keitel, V. The G Protein-Coupled Bile Acid Receptor TGR5 (Gpbar1) Modulates Endothelin-1 Signaling in Liver. Cells 2019, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Macnaughtan, J.; Ranchal, I.; Soeda, J.; Sawhney, R.; Oben, J.; Davies, N.; Mookerjee, R.; Marchesi, J.; Cox, J.; Jalan, R. O091: Oral therapy with non-absorbable carbons of controlled porosity (YAQ-001) selectively modulates stool microbiome and its function and this is associated with restoration of immune function and inflammasome activation. J. Hepatol. 2015, 62, S240. [Google Scholar] [CrossRef]
- Haidry, R.J.; van Baar, A.C.; Galvao Neto, M.P.; Rajagopalan, H.; Caplan, J.; Levin, P.S.; Bergman, J.J.; Rodriguez, L.; Deviere, J.; Thompson, C.C. Duodenal mucosal resurfacing: Proof-of-concept, procedural development, and initial implementation in the clinical setting. Gastrointest. Endosc. 2019, 90, 673–681. [Google Scholar] [CrossRef]
- Van Baar, A.C.G.; Holleman, F.; Crenier, L.; Haidry, R.; Magee, C.; Hopkins, D.; Rodriguez Grunert, L.; Galvao Neto, M.; Vignolo, P.; Hayee, B.; et al. Endoscopic duodenal mucosal resurfacing for the treatment of type 2 diabetes mellitus: One year results from the first international, open-label, prospective, multicentre study. Gut 2020, 69, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology 2002, 122, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribble, F.M.; Reimann, F. Metabolic Messengers: Glucagon-like peptide 1. Nat. Metab. 2021, 3, 142–148. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A.; Clausen, W.H.; Elbrond, B.; Gough, S.C.; Tomlinson, J.W.; Newsome, P.N. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: Individual patient data meta-analysis of the LEAD program. Aliment. Pharmacol. Ther. 2013, 37, 234–242. [Google Scholar] [CrossRef]
- Nahra, R.; Wang, T.; Gadde, K.M.; Oscarsson, J.; Stumvoll, M.; Jermutus, L.; Hirshberg, B.; Ambery, P. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults With Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care 2021. [Google Scholar] [CrossRef]
- Choi, J.; Kim, J.K.; Lee, S.M.; Kwon, H.; Lee, J.; Bae, S.; Kim, D.; Choi, I.Y. 1830-P:Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in CDHFD-Induced NASH and Fibrosis Mice. Am. Diabetes Assoc. Diabetes 2020, 69 (Suppl. 1). [Google Scholar] [CrossRef]
- Abdelmalek, M.; Choi, J.; Kim, Y.; Seo, K.; Hompesch, M.; Baek, S. LBP03-HM15211, a novel GLP-1/GIP/Glucagon triple-receptor co-agonist significantly reduces liver fat and body weight in obese subjects with non-alcoholic fatty liver disease: A Phase 1b/2a, multi-center, randomized, placebo-controlled trial. J. Hepatol. 2020, 73, S124. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; Investigators, N.N. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Choudhary, N.S.; Singh, M.K.; Wasir, J.S.; Kaur, P.; Gill, H.K.; Bano, T.; Farooqui, K.J.; et al. Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: Randomised controlled trial (D-LIFT trial). Diabetologia 2020, 63, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.R.; Johnston, I.M.; Nolan, J.D.; Vassie, C.; Pruzanski, M.E.; Shapiro, D.A. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment. Pharmacol. Ther. 2015, 41, 54–64. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled, phase 2a trial. Nat. Med. 2021, 27, 1262–1271. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2019, 30, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clement, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci. Rep. 2016, 6, 34055. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de la Hera, A.; Gonzalez, M.; Moya, J.L.; Calleja, J.L.; Monserrat, J.; Ruiz-del-Arbol, L.; Alvarez-Mon, M. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology 2003, 37, 208–217. [Google Scholar] [CrossRef]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taura, P.; Fuster, J.; Garcia-Valdecasas, J.C.; Lacy, A.; Suarez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar] [CrossRef]
- Wiest, R.; Garcia-Tsao, G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005, 41, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Saxinger, L.; Ma, M.; Prado, V.; Fernandez, J.; Kumar, D.; Gonzalez-Abraldes, J.; Keough, A.; Bastiampillai, R.; Carbonneau, M.; et al. Bacterial infections in acute variceal hemorrhage despite antibiotics-a multicenter study of predictors and clinical impact. United Eur. Gastroenterol. J. 2017, 5, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadamrad, S.; McCoy, K.D.; Geuking, M.B.; Sagesser, H.; Kirundi, J.; Macpherson, A.J.; De Gottardi, A. Attenuated portal hypertension in germ-free mice: Function of bacterial flora on the development of mesenteric lymphatic and blood vessels. Hepatology 2015, 61, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calanni, F.; Renzulli, C.; Barbanti, M.; Viscomi, G.C. Rifaximin: Beyond the traditional antibiotic activity. J. Antibiot. 2014, 67, 667–670. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. Electronic address and L. European Association for the Study of the, L. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460. [Google Scholar] [CrossRef] [Green Version]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Takaya, H.; Saikawa, S.; Furukawa, M.; Sato, S.; Kawaratani, H.; Kitade, M.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Rifaximin ameliorates hepatic encephalopathy and endotoxemia without affecting the gut microbiome diversity. World J. Gastroenterol. 2017, 23, 8355–8366. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heimanson, Z.; Israel, R.; Sanyal, A. SAT-014-Efficacy of rifaximin soluble solid dispersion in patients with early decompensated cirrhosis and a Conn score of 0: A post hoc analysis of a randomized, double-blind, placebo-controlled trial. J. Hepatol. 2019, 70, e631. [Google Scholar] [CrossRef]
- Zhu, Q.; Zou, L.; Jagavelu, K.; Simonetto, D.A.; Huebert, R.C.; Jiang, Z.D.; DuPont, H.L.; Shah, V.H. Intestinal decontamination inhibits TLR4 dependent fibronectin-mediated cross-talk between stellate cells and endothelial cells in liver fibrosis in mice. J. Hepatol. 2012, 56, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachogiannakos, J.; Saveriadis, A.S.; Viazis, N.; Theodoropoulos, I.; Foudoulis, K.; Manolakopoulos, S.; Raptis, S.; Karamanolis, D.G. Intestinal decontamination improves liver haemodynamics in patients with alcohol-related decompensated cirrhosis. Aliment. Pharmacol. Ther. 2009, 29, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Kimer, N.; Pedersen, J.S.; Busk, T.M.; Gluud, L.L.; Hobolth, L.; Krag, A.; Moller, S.; Bendtsen, F.; Copenhagen Rifaximin Study, G. Rifaximin has no effect on hemodynamics in decompensated cirrhosis: A randomized, double-blind, placebo-controlled trial. Hepatology 2017, 65, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.L.; Kim, M.Y.; Jang, Y.O.; Baik, S.K.; Kwon, S.O. Rifaximin and Propranolol Combination Therapy Is More Effective than Propranolol Monotherapy for the Reduction of Portal Pressure: An Open Randomized Controlled Pilot Study. Gut Liver 2017, 11, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, Y.; Kawaratani, H.; Kaya, D.; Tsuji, Y.; Ozutsumi, T.; Furukawa, M.; Kitagawa, K.; Sato, S.; Nishimura, N.; Sawada, Y.; et al. Effective Combination Therapy of Angiotensin-II Receptor Blocker and Rifaximin for Hepatic Fibrosis in Rat Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5589. [Google Scholar] [CrossRef] [PubMed]
- Hennenberg, M.; Trebicka, J.; Buecher, D.; Heller, J.; Sauerbruch, T. Lack of effect of norfloxacin on hyperdynamic circulation in bile duct-ligated rats despite reduction of endothelial nitric oxide synthase function: Result of unchanged vascular Rho-kinase? Liver Int. 2009, 29, 933–941. [Google Scholar] [CrossRef]
- Kemp, W.; Colman, J.; Thompson, K.; Madan, A.; Vincent, M.; Chin-Dusting, J.; Kompa, A.; Krum, H.; Roberts, S. Norfloxacin treatment for clinically significant portal hypertension: Results of a randomised double-blind placebo-controlled crossover trial. Liver Int. 2009, 29, 427–433. [Google Scholar] [CrossRef]
- Moreau, R.; Elkrief, L.; Bureau, C.; Perarnau, J.M.; Thevenot, T.; Saliba, F.; Louvet, A.; Nahon, P.; Lannes, A.; Anty, R.; et al. Effects of Long-term Norfloxacin Therapy in Patients With Advanced Cirrhosis. Gastroenterology 2018, 155, 1816–1827. [Google Scholar] [CrossRef] [Green Version]
- Rasaratnam, B.; Kaye, D.; Jennings, G.; Dudley, F.; Chin-Dusting, J. The effect of selective intestinal decontamination on the hyperdynamic circulatory state in cirrhosis. A randomized trial. Ann. Intern. Med. 2003, 139, 186–193. [Google Scholar] [CrossRef]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhiman, R.K.; Rana, B.; Agrawal, S.; Garg, A.; Chopra, M.; Thumburu, K.K.; Khattri, A.; Malhotra, S.; Duseja, A.; Chawla, Y.K. Probiotic VSL#3 reduces liver disease severity and hospitalization in patients with cirrhosis: A randomized, controlled trial. Gastroenterology 2014, 147, 1327–1337 e1323. [Google Scholar] [CrossRef] [PubMed]
- Duseja, A.; Acharya, S.K.; Mehta, M.; Chhabra, S.; Rana, S.; Das, A.; Dattagupta, S.; Dhiman, R.K.; Chawla, Y.K. High potency multistrain probiotic improves liver histology in non-alcoholic fatty liver disease (NAFLD): A randomised, double-blind, proof of concept study. BMJ Open Gastroenterol. 2019, 6, e000315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincon, D.; Vaquero, J.; Hernando, A.; Galindo, E.; Ripoll, C.; Puerto, M.; Salcedo, M.; Frances, R.; Matilla, A.; Catalina, M.V.; et al. Oral probiotic VSL#3 attenuates the circulatory disturbances of patients with cirrhosis and ascites. Liver Int. 2014, 34, 1504–1512. [Google Scholar] [CrossRef]
- Jayakumar, S.; Carbonneau, M.; Hotte, N.; Befus, A.D.; St Laurent, C.; Owen, R.; McCarthy, M.; Madsen, K.; Bailey, R.J.; Ma, M.; et al. VSL#3 (R) probiotic therapy does not reduce portal pressures in patients with decompensated cirrhosis. Liver Int. 2013, 33, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Moncrief, K.; Madsen, K.; Arrieta, M.C.; Owen, R.J.; Bain, V.G.; Wong, W.W.; Ma, M.M. Effects of probiotic therapy on portal pressure in patients with cirrhosis: A pilot study. Liver Int. 2009, 29, 1110–1115. [Google Scholar] [CrossRef]
- Gupta, N.; Kumar, A.; Sharma, P.; Garg, V.; Sharma, B.C.; Sarin, S.K. Effects of the adjunctive probiotic VSL#3 on portal haemodynamics in patients with cirrhosis and large varices: A randomized trial. Liver Int. 2013, 33, 1148–1157. [Google Scholar] [CrossRef]
- Kurtz, C.B.; Millet, Y.A.; Puurunen, M.K.; Perreault, M.; Charbonneau, M.R.; Isabella, V.M.; Kotula, J.W.; Antipov, E.; Dagon, Y.; Denney, W.S.; et al. An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Rose, C.F.; Amodio, P.; Bajaj, J.S.; Dhiman, R.K.; Montagnese, S.; Taylor-Robinson, S.D.; Vilstrup, H.; Jalan, R. Hepatic encephalopathy: Novel insights into classification, pathophysiology and therapy. J. Hepatol. 2020, 73, 1526–1547. [Google Scholar] [CrossRef]
- Sharma, B.C.; Sharma, P.; Agrawal, A.; Sarin, S.K. Secondary prophylaxis of hepatic encephalopathy: An open-label randomized controlled trial of lactulose versus placebo. Gastroenterology 2009, 137, 885–891. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients With Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065. [Google Scholar] [CrossRef]
- Garcia-Lezana, T.; Raurell, I.; Bravo, M.; Torres-Arauz, M.; Salcedo, M.T.; Santiago, A.; Schoenenberger, A.; Manichanh, C.; Genesca, J.; Martell, M.; et al. Restoration of a healthy intestinal microbiota normalizes portal hypertension in a rat model of nonalcoholic steatohepatitis. Hepatology 2018, 67, 1485–1498. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Gavis, E.A.; Fagan, A.; Wade, J.B.; Thacker, L.R.; Fuchs, M.; Patel, S.; Davis, B.; Meador, J.; Puri, P.; et al. A Randomized Clinical Trial of Fecal Microbiota Transplant for Alcohol Use Disorder. Hepatology 2021, 73, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B.; Kunze, W.A.; Clerc, N. Nutrient tasting and signaling mechanisms in the gut. II. The intestine as a sensory organ: Neural, endocrine, and immune responses. Am. J. Physiol. 1999, 277, G922–G928. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeiro, M.H.; Ramirez, M.J.; Milagro, F.I.; Martinez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [Green Version]
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2015, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, H.; Li, B.; Lee, C.; Alganabi, M.; Zheng, S.; Pierro, A. Beneficial effects of butyrate in intestinal injury. J. Pediatr. Surg. 2020, 55, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Juanola, O.; Ferrusquia-Acosta, J.; Garcia-Villalba, R.; Zapater, P.; Magaz, M.; Marin, A.; Olivas, P.; Baiges, A.; Bellot, P.; Turon, F.; et al. Circulating levels of butyrate are inversely related to portal hypertension, endotoxemia, and systemic inflammation in patients with cirrhosis. FASEB J. 2019, 33, 11595–11605. [Google Scholar] [CrossRef] [PubMed]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Sanyal, A. Abstract: Icosabutate, A Novel Structurally Engineered Fatty Acid, Significantly Reduces Relevant Markers of NASH and Fibrosis in 16 Weeks: Interim Analysis Results of the ICONA Trial. Available online: https://easl.eu/press-release/treatment-advances-for-non-alcoholic-fatty-liver-disease-nafld-announced-at-ilc-2021/ (accessed on 15 July 2021).
- Singh, R.; Chandrashekharappa, S.; Bodduluri, S.R.; Baby, B.V.; Hegde, B.; Kotla, N.G.; Hiwale, A.A.; Saiyed, T.; Patel, P.; Vijay-Kumar, M.; et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun. 2019, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Jala, V.R.; Singh, R.; Chandrashekharappa, S.; Joshi-Barve, S.; McClain, C.; Bodduluri, B.; Vemula, P.K. Gut microbial metabolites as therapeutics to treat of alcoholic liver disease. J. Immunol. 2020, 204, 83.17. [Google Scholar]
- Schon, H.T.; Bartneck, M.; Borkham-Kamphorst, E.; Nattermann, J.; Lammers, T.; Tacke, F.; Weiskirchen, R. Pharmacological Intervention in Hepatic Stellate Cell Activation and Hepatic Fibrosis. Front. Pharmacol. 2016, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Le, T.H.; Shahidipour, H.; Read, S.A.; Ahlenstiel, G. The Role of Gut-Derived Microbial Antigens on Liver Fibrosis Initiation and Progression. Cells 2019, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Zeromski, J.; Kierepa, A.; Brzezicha, B.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Pattern Recognition Receptors: Significance of Expression in the Liver. Arch. Immunol. Ther. Exp. 2020, 68, 29. [Google Scholar] [CrossRef]
- Engelmann, C.; Sheikh, M.; Sharma, S.; Kondo, T.; Loeffler-Wirth, H.; Zheng, Y.B.; Novelli, S.; Hall, A.; Kerbert, A.J.C.; Macnaughtan, J.; et al. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. J. Hepatol. 2020, 73, 102–112. [Google Scholar] [CrossRef]
- Liaunardy-Jopeace, A.; Gay, N.J. Molecular and cellular regulation of toll-like receptor-4 activity induced by lipopolysaccharide ligands. Front. Immunol. 2014, 5, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Liu, M.; Sehrawat, T.S.; Shah, V.H. Regulation and functional roles of chemokines in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Aoyama, T.; Paik, Y.H.; Seki, E. Toll-like receptor signaling and liver fibrosis. Gastroenterol Res. Pract. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Feng, J.; Du, J.; Zhuo, Z.; Yang, S.; Zhang, W.; Wang, W.; Zhang, S.; Iwakura, Y.; Meng, G.; et al. Macrophage-derived IL-1alpha promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell Mol. Immunol. 2018, 15, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazawa, T.; Tsujimoto, T.; Kawaratani, H.; Fukui, H. Therapeutic approach to regulate innate immune response by Toll-like receptor 4 antagonist E5564 in rats with D-galactosamine-induced acute severe liver injury. J. Gastroenterol. Hepatol. 2009, 24, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, T.; Tsujimoto, T.; Kawaratani, H.; Fukui, H. Salvage effect of E5564, Toll-like receptor 4 antagonist on d-galactosamine and lipopolysaccharide-induced acute liver failure in rats. J. Gastroenterol. Hepatol. 2010, 25, 1009–1012. [Google Scholar] [CrossRef]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Monnet, E.; Lapeyre, G.; Poelgeest, E.V.; Jacqmin, P.; Graaf, K.; Reijers, J.; Moerland, M.; Burggraaf, J.; Min, C. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin. Pharmacol. Ther. 2017, 101, 200–208. [Google Scholar] [CrossRef]
- Bennett, R.G.; Simpson, R.L.; Hamel, F.G. Serelaxin increases the antifibrotic action of rosiglitazone in a model of hepatic fibrosis. World J. Gastroenterol. 2017, 23, 3999–4006. [Google Scholar] [CrossRef]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef]
- Vergis, N.; Atkinson, S.R.; Knapp, S.; Maurice, J.; Allison, M.; Austin, A.; Forrest, E.H.; Masson, S.; McCune, A.; Patch, D.; et al. In Patients With Severe Alcoholic Hepatitis, Prednisolone Increases Susceptibility to Infection and Infection-Related Mortality, and Is Associated With High Circulating Levels of Bacterial DNA. Gastroenterology 2017, 152, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, S.; Marcos, M.; Gattu, A.; Catalano, D.; Szabo, G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS ONE 2014, 9, e96864. [Google Scholar] [CrossRef]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Gabele, E.; Muhlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Scholmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Farooq, A.; Malik, A.; Trawick, B.N.; Berberich, D.W.; McClurg, J.P.; Galen, K.P.; Mehal, W. A novel small-molecule enantiomeric analogue of traditional (-)-morphinans has specific TLR9 antagonist properties and reduces sterile inflammation-induced organ damage. J. Immunol. 2013, 190, 4297–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaker, M.E.; Trawick, B.N.; Mehal, W.Z. The novel TLR9 antagonist COV08-0064 protects from ischemia/reperfusion injury in non-steatotic and steatotic mice livers. Biochem. Pharmacol. 2016, 112, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Fukui, R.; Motoi, Y.; Shibata, T.; Saitoh, S.I.; Sato, R.; Miyake, K. The protective effect of the anti-Toll-like receptor 9 antibody against acute cytokine storm caused by immunostimulatory DNA. Sci. Rep. 2017, 7, 44042. [Google Scholar] [CrossRef]
- Gilboa-Geffen, A.; Wolf, Y.; Hanin, G.; Melamed-Book, N.; Pick, M.; Bennett, E.R.; Greenberg, D.S.; Lester, S.; Rischmueller, M.; Soreq, H. Activation of the alternative NFkappaB pathway improves disease symptoms in a model of Sjogren’s syndrome. PLoS ONE 2011, 6, e28727. [Google Scholar] [CrossRef]
- Kiripolsky, J.; Kramer, J.M. Current and Emerging Evidence for Toll-Like Receptor Activation in Sjogren’s Syndrome. J. Immunol. Res. 2018, 2018, 1246818. [Google Scholar] [CrossRef]
- Byun, J.S.; Suh, Y.G.; Yi, H.S.; Lee, Y.S.; Jeong, W.I. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in mice. J. Hepatol. 2013, 58, 342–349. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Song, H.; Li, H.; Cui, A.; Liu, Y.; Su, L.; Crispe, I.N.; Tu, Z. Activated NK cells kill hepatic stellate cells via p38/PI3K signaling in a TRAIL-involved degranulation manner. J. Leukoc. Biol. 2019, 105, 695–704. [Google Scholar] [CrossRef]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.S.; Lee, Y.S.; Park, S.H.; Jang, M.J.; Jo, E.; Kim, S.C.; Han, Y.M.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.-Y.; Liu, C.; Li, Z.-R.; Niu, C.; Wu, J. NLRP3 deficiency did not attenuate NASH development under high fat calorie diet plus high fructose and glucose in drinking water. Lab. Investig. 2021, 101, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Qu, J.; Yuan, Z.; Pan, R.; Li, K. Calcipotriol Inhibits NLRP3 Signal Through YAP1 Activation to Alleviate Cholestatic Liver Injury and Fibrosis. Front. Pharmacol. 2020, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- NodThera is Unlocking the Significant Therapeutic Potential of NLRP3 Inflammasome Activation Inhibitors Through Our Novel Drug Discovery Platform. Available online: https://www.nodthera.com/approach-progress/ (accessed on 27 August 2021).
- Advancing Our Pipeline. Available online: https://www.ifmthera.com/pipeline/ (accessed on 27 August 2021).
- Xu, R.; Zhang, Z.; Wang, F.S. Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell Mol. Immunol. 2012, 9, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Zimmermann, H.W.; Roderburg, C.; Gassler, N.; Wasmuth, H.E.; Luedde, T.; Trautwein, C.; Tacke, F. The fractalkine receptor CX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology 2010, 52, 1769–1782. [Google Scholar] [CrossRef]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Mockel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. [Google Scholar] [CrossRef]
- Mulder, P.; van den Hoek, A.M.; Kleemann, R. The CCR2 Inhibitor Propagermanium Attenuates Diet-Induced Insulin Resistance, Adipose Tissue Inflammation and Non-Alcoholic Steatohepatitis. PLoS ONE 2017, 12, e0169740. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambade, A.; Lowe, P.; Kodys, K.; Catalano, D.; Gyongyosi, B.; Cho, Y.; Iracheta-Vellve, A.; Adejumo, A.; Saha, B.; Calenda, C.; et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology 2019, 69, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.J.; Fuchs, B.C.; Masia, R.; Holmes, J.A.; Salloum, S.; Sojoodi, M.; Ferreira, D.S.; Rutledge, S.M.; Caravan, P.; Alatrakchi, N.; et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol. Commun. 2018, 2, 529–545. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [Green Version]
- NIH, U.S. National Library of Medicine Clinicaltrials.gov. AURORA: Phase 3 Study for the Efficacy and Safety of CVC for the Treatment of Liver Fibrosis in Adults With NASH, ClinicalTrials.gov Identifier, NCT03028740. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03028740 (accessed on 10 July 2021).
- Perez-Martinez, L.; Perez-Matute, P.; Aguilera-Lizarraga, J.; Rubio-Mediavilla, S.; Narro, J.; Recio, E.; Ochoa-Callejero, L.; Oteo, J.A.; Blanco, J.R. Maraviroc, a CCR5 antagonist, ameliorates the development of hepatic steatosis in a mouse model of non-alcoholic fatty liver disease (NAFLD). J. Antimicrob. Chemother. 2014, 69, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, D.; Gilleece, Y.; Verma, S.; Abramowicz, I.; Bremner, S.; Perry, N. Protocol for a phase IV, open-label feasibility study investigating non-invasive markers of hepatic fibrosis in people living with HIV-1 and non-alcoholic fatty liver disease randomised to receiving optimised background therapy (OBT) plus maraviroc or OBT alone. BMJ Open 2020, 10, e035596. [Google Scholar] [CrossRef]
- Colino, C.I.; Lanao, J.M.; Gutierrez-Millan, C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 2020, 11, 218. [Google Scholar] [CrossRef] [Green Version]
- Ergen, C.; Heymann, F.; Al Rawashdeh, W.; Gremse, F.; Bartneck, M.; Panzer, U.; Pola, R.; Pechar, M.; Storm, G.; Mohr, N.; et al. Targeting distinct myeloid cell populations in vivo using polymers, liposomes and microbubbles. Biomaterials 2017, 114, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Scheyda, K.M.; Warzecha, K.T.; Rizzo, L.Y.; Hittatiya, K.; Luedde, T.; Storm, G.; Trautwein, C.; Lammers, T.; Tacke, F. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 2015, 37, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Bygd, H.C.; Ma, L.; Bratlie, K.M. Physicochemical properties of liposomal modifiers that shift macrophage phenotype. Mater. Sci. Eng. C. Mater. Biol. Appl. 2017, 79, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Maradana, M.R.; Yekollu, S.K.; Zeng, B.; Ellis, J.; Clouston, A.; Miller, G.; Talekar, M.; Bhuyan, Z.A.; Mahadevaiah, S.; Powell, E.E.; et al. Immunomodulatory liposomes targeting liver macrophages arrest progression of nonalcoholic steatohepatitis. Metabolism 2018, 78, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Iacobini, C.; Menini, S.; Ricci, C.; Blasetti Fantauzzi, C.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Traber, P.G.; Chou, H.; Zomer, E.; Hong, F.; Klyosov, A.; Fiel, M.I.; Friedman, S.L. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 2013, 8, e75361. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [Green Version]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andres-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernandez-Iglesias, A.; Broqua, P.; Junien, J.L.; Wettstein, G.; Bosch, J.; et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Marra, F.; Efsen, E.; Romanelli, R.G.; Caligiuri, A.; Pastacaldi, S.; Batignani, G.; Bonacchi, A.; Caporale, R.; Laffi, G.; Pinzani, M.; et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology 2000, 119, 466–478. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrene, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Francque, S.; Bedossa, P.; Ratziu, V.; Anstee, Q.; Bugianesi, E.; Sanyal, A.; Loomba, R.; Harrison, S.A.; Balabanska, R.I.; Mateva, L.; et al. The panPPAR agonist lanifibranor induces both resolution of NASH and regression of fibrosis after 24 weeks of treatment in non-cirrhotic nash: Results of the NATIVE phase 2b trial. Hepatology 2020, 72, 9A. [Google Scholar]
- Sven, M.F.; Pierre, B.; Manal, F.A.; Quentin, M.A.; Elisabetta, B.; Vlad, R.; Philippe, H.M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar] [CrossRef]
- Goyal, O.; Nohria, S.; Goyal, P.; Kaur, J.; Sharma, S.; Sood, A.; Chhina, R.S. Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: A prospective, observational, real world study. Sci. Rep. 2020, 10, 21117. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-alpha/gamma Agonist, for Treatment of Nonalcoholic Fatty Liver Disease: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, C.; Dupuis, T.; Jouvion, G.; Eberl, G.; Disson, O.; Lecuit, M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 2015, 42, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [Green Version]
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat. Med. 2019, 25, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Stutchfield, B.M.; Antoine, D.J.; Mackinnon, A.C.; Gow, D.J.; Bain, C.C.; Hawley, C.A.; Hughes, M.J.; Francis, B.; Wojtacha, D.; Man, T.Y.; et al. CSF1 Restores Innate Immunity After Liver Injury in Mice and Serum Levels Indicate Outcomes of Patients With Acute Liver Failure. Gastroenterology 2015, 149, 1896–1909. [Google Scholar] [CrossRef] [Green Version]
- Kedarisetty, C.K.; Anand, L.; Bhardwaj, A.; Bhadoria, A.S.; Kumar, G.; Vyas, A.K.; David, P.; Trehanpati, N.; Rastogi, A.; Bihari, C.; et al. Combination of granulocyte colony-stimulating factor and erythropoietin improves outcomes of patients with decompensated cirrhosis. Gastroenterology 2015, 148, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Promethera Biosciences. HepaStem: Toward an Alternative to Liver Transplantation. Available online: https://www.nature.com/articles/d43747-020-00724-x (accessed on 10 July 2021).
- Nevens, F.; Gustot, T.; Laterre, P.F.; Lasser, L.L.; Haralampiev, L.E.; Vargas, V.; Lyubomirova, D.; Albillos, A.; Najimi, M.; Michel, S.; et al. A phase II study of human allogeneic liver-derived progenitor cell therapy for acute-on-chronic liver failure and acute decompensation. JHEP Rep. 2021, 3, 100291. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).