The Space of Disse: The Liver Hub in Health and Disease

 ,
,  ,
,  , ,
, , 
Abstract
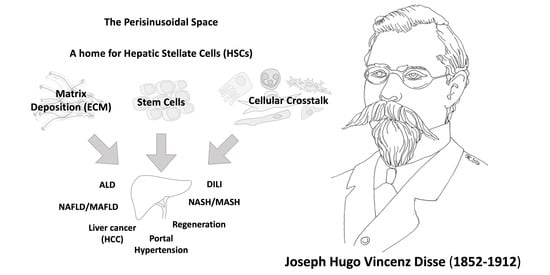
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
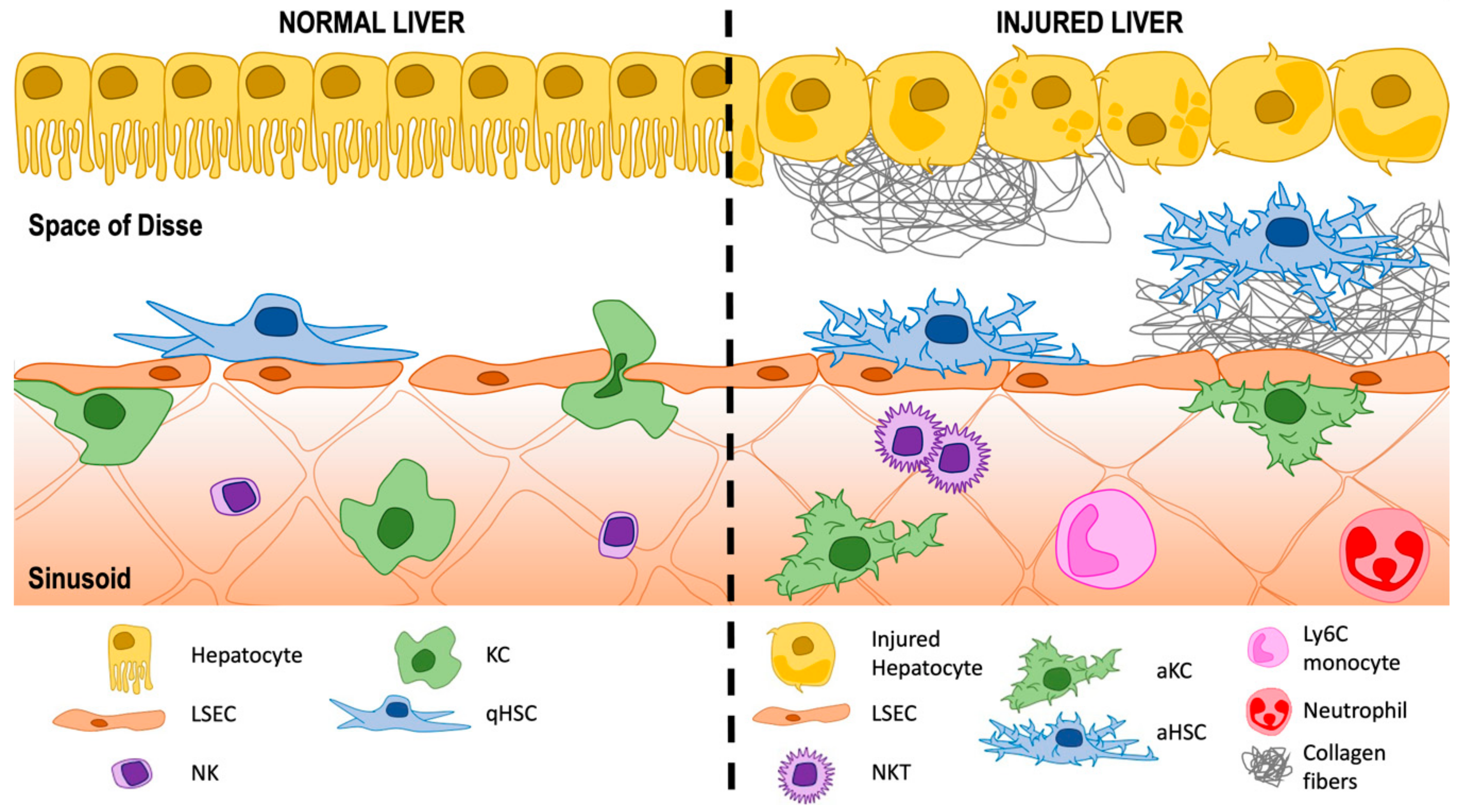
2. The Hepatic Sinusoids
3. A Historic Perspective of the Space of Disse: Home for Hepatic Stellate Cells (HSCs)
3.1. Activation of HSCs
3.1.1. Intracellular Pathways Involved in HSC Activation
3.1.2. Extracellular Interactions Involved in HSC Activation
HSCs and Liver Sinusoidal Endothelial Cells (LSECs)
HSCs and Macrophages—Both Resident KCs and Infiltrating Cells
HSCs and T Lymphocytes
HSCs and Other Cell-Types
3.1.3. Crosstalk Cells-Stimulus Involved in HSC Activation
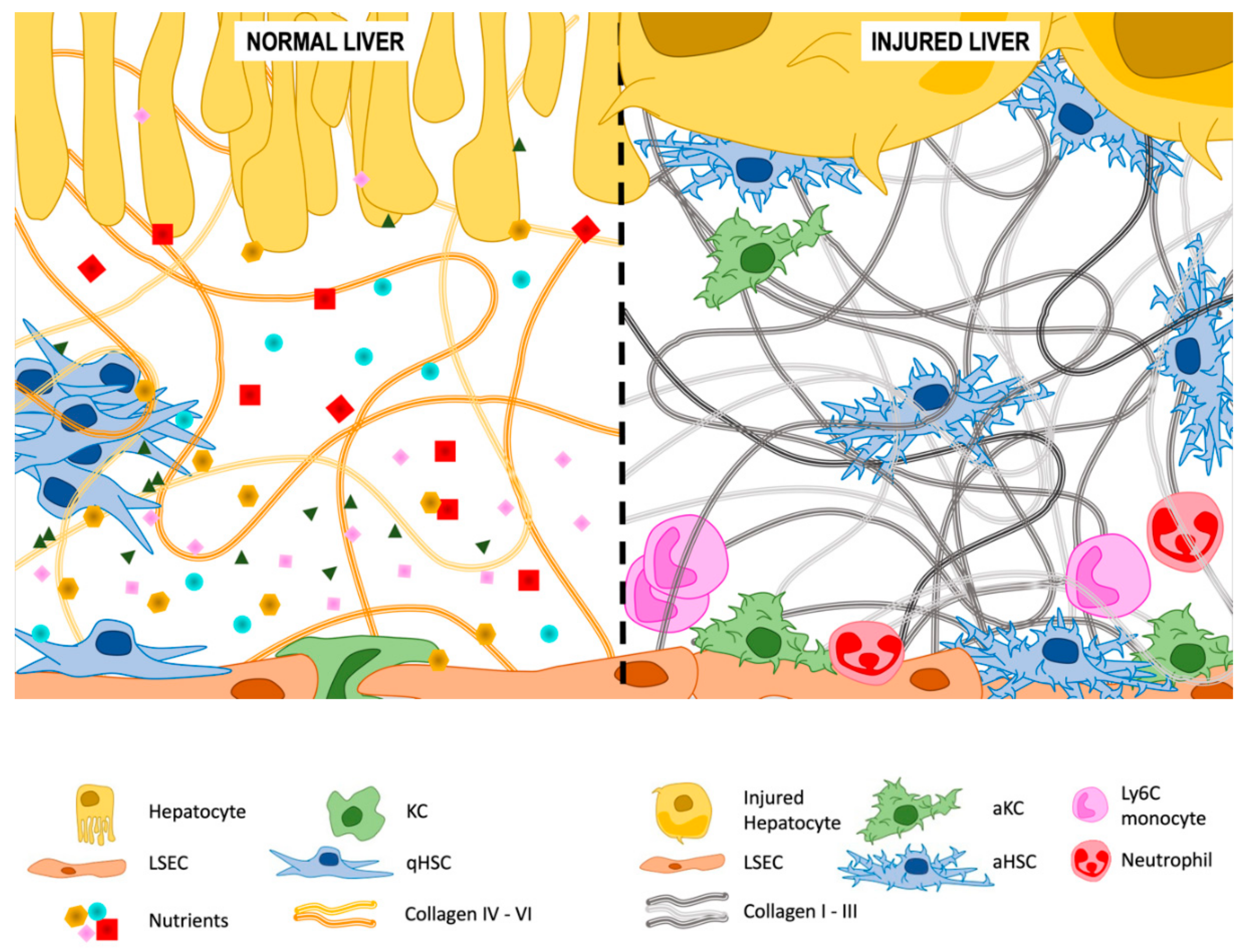
3.2. Extracellular Matrix (ECM) Deposition
3.2.1. Type IV and VI Collagen
3.2.2. Type I and III Collagen
3.2.3. Other Components of the ECM
3.3. Microvilli
3.4. HSCs as Stem Cells
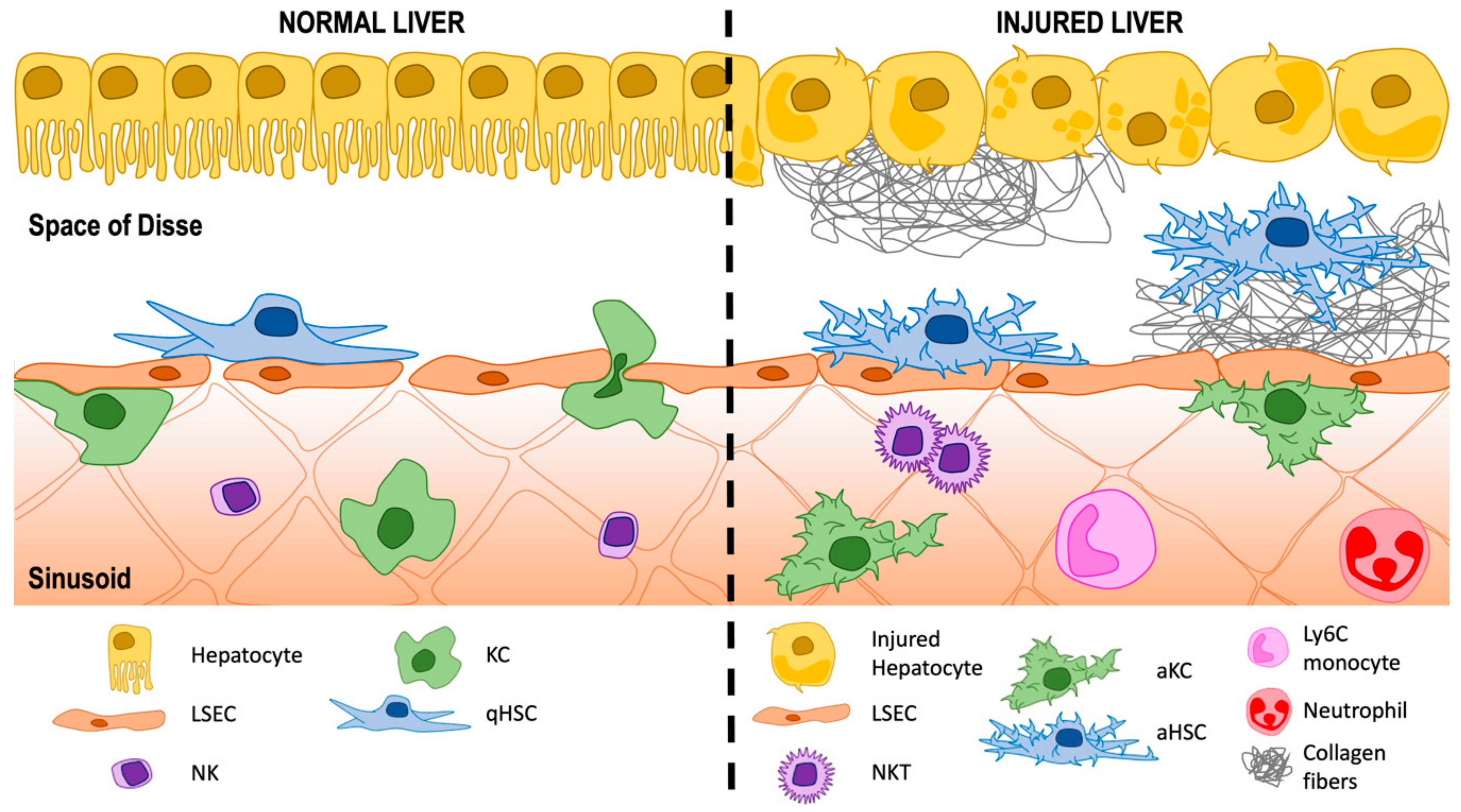
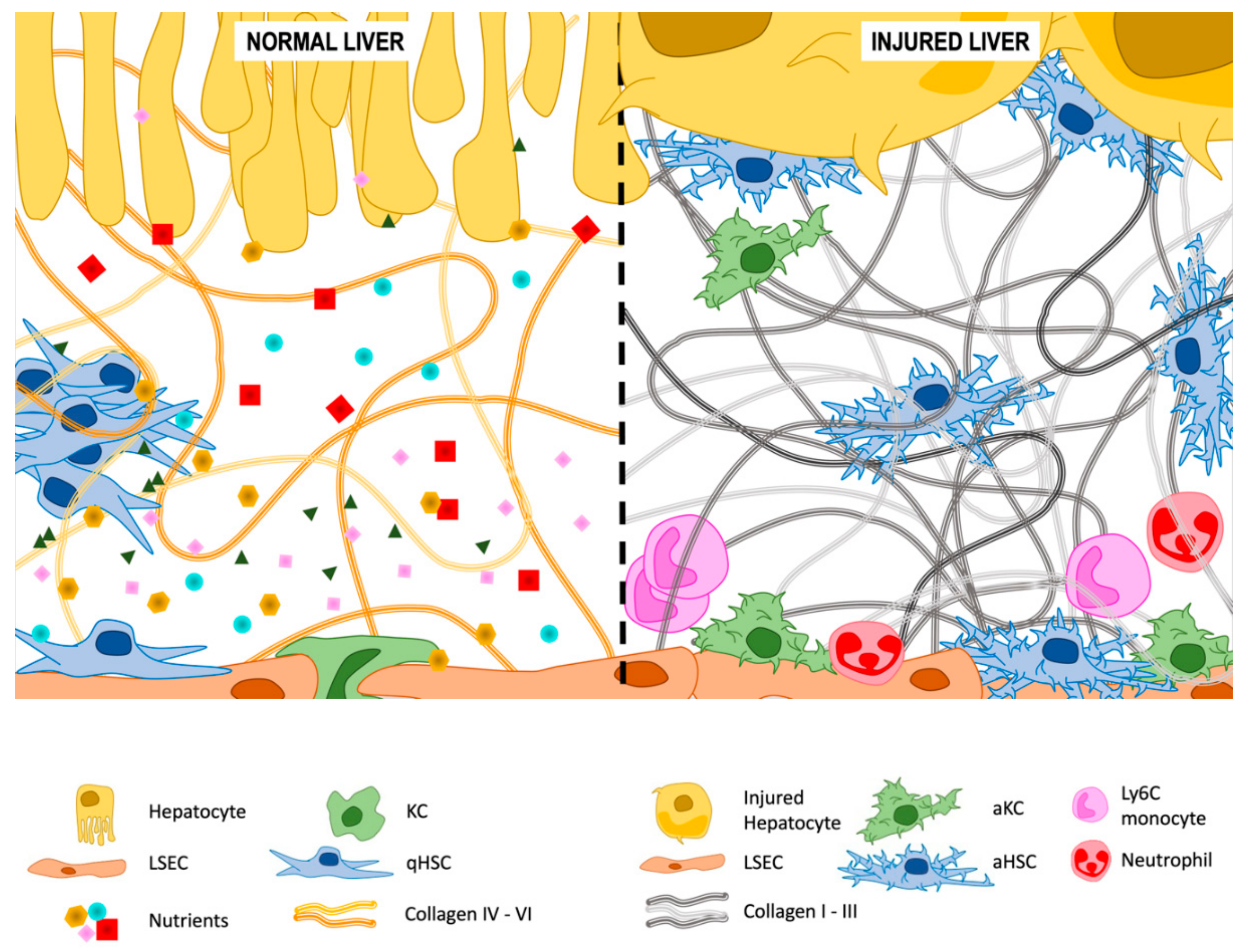
4. Involvement of the Space of Disse in Liver Disease
4.1. Hepatotoxicity
4.1.1. Drug-Induced Liver Injury (DILI)
4.1.2. Alcoholic Liver Disease (ALD)
4.1.3. NAFLD/MAFLD and NASH/MASH
4.1.4. Portal Hypertension
4.1.5. Chronic Cholestatic Liver Diseases
4.2. Liver Regeneration
4.3. Progression from Fibrosis to Cirrhosis
4.4. Hepatocellular Carcinoma (HCC)
4.5. Hemochromatosis
4.6. Other Diseases Related with the Space of Disse
5. Therapeutic Intervention in the Space of Disse
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arráez-Aybar, L.A.A.; Arias, J.; Mérida-Velasco, J.R. Disse and his Space. Indian J. Anat. 2018, 7, 4. [Google Scholar]
- Brunt, E.M.; Gouw, A.S.H.; Hubscher, S.G.; Tiniakos, D.G.; Bedossa, P.; Burt, A.D.; Callea, F.; Clouston, A.D.; Dienes, H.P.; Goodman, Z.D.; et al. Pathology of the liver sinusoids. Histopathology 2014, 64, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Wisse, E.; Luo, D.; Vermijlen, D.; Kanellopoulou, C.; De Zanger, R.; Braet, F. On the Function of Pit Cells, the Liver-Specific Natural Killer Cells. Semin. Liver Dis. 1997, 17, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Aydin, M.M.; Akcali, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Covas, D.T.; Panepucci, R.A.; Fontes, A.M.; Orellana, W.A.S., Jr.; Freitas, M.C.C.; Neder, L.; Santos, A.R.D.; Peres, L.C.; Jamur, M.C.; Zago, M.A.; et al. Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Exp. Hematol. 2008, 36, 642–654. [Google Scholar] [CrossRef]
- Chinnadurai, R.; Sands, J.; Rajan, D.; Liu, X.; Arafat, D.; Das, R.; Anania, F.A.; Gibson, G.; Kisseleva, T.; Galipeau, J. Molecular Genetic and Immune Functional Responses Distinguish Bone Marrow Mesenchymal Stromal Cells from Hepatic Stellate Cells. Stem Cells 2019, 37, 1075–1082. [Google Scholar] [CrossRef]
- Gandhi, C.R. Hepatic stellate cell activation and pro-fibrogenic signals. J. Hepatol. 2017, 67, 1104–1105. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Wang, X.; Zou, Y.; Zhong, Y. Key role of liver sinusoidal endothelial cells in liver fibrosis. Biosci. Trends 2017, 11, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Smedsrød, B. Clearance function of scavenger endothelial cells. Comp. Hepatol. 2004, 3, S22. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeve, L.D.; Maretti-Mira, A.C. Liver Sinusoidal Endothelial Cell: An Update. Semin. Liver Dis. 2017, 37, 377–387. [Google Scholar] [PubMed]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrød, B. The scavenger endothelial cell: A new player in homeostasis and immunity. Am. J. Physiol. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedsrød, B.; Pertoft, H.; Eggertsen, G.; Sundström, C. Functional and morphological characterization of cultures of Kupffer cells and liver endothelial cells prepared by means of density separation in Percoll, and selective substrate adherence. Cell Tissue Res. 1985, 241, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Laakso, T.; Sjöholm, I. Biodegradable Microspheres X: Some Properties of Polyacryl Starch Microparticles Prepared from Acrylic Acid-Esterified Starch. J. Pharm. Sci. 1987, 76, 935–939. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 Signaling in Inflammatory, Kupffer Cells, and Hepatic Stellate Cells Exacerbates Liver Fibrosis in Mice. Gastroenterology 2012, 143, 765–776.e3. [Google Scholar] [CrossRef] [Green Version]
- Maricic, I.; Sheng, H.; Marrero, I.; Seki, E.; Kisseleva, T.; Chaturvedi, S.; Molle, N.; Mathews, S.A.; Gao, B.; Kumar, V. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology 2015, 61, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, S.; Li, M. TGF-β Control of Adaptive Immune Tolerance: A Break from Treg Cells. BioEssays 2018, 40, e1800063. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S. Natural killer and natural killer T cells in liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Fasbender, F.; Widera, A.; Henjstler, J.G.; Watz, C. Natural Killer Cells and Liver Fibrosis. Front. Immunol. 2016, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Disse, J. Ueber die Lymphbahnen der Säugethierleber. Arch. Mikrosk. Anat. 1890, 36, 203–224. [Google Scholar] [CrossRef]
- Gillavry, M. Zur Anatomie der Leber; Wiener Sitzungsber: Vienna, Austria, 1864; p. 50. [Google Scholar]
- Hering, E. Von der Leber; Stricker’s Handbuch der Lehre von den Geweben: Leipzig, Germany, 1868; p. 429. [Google Scholar]
- Henle, J. Eingeweidelehre, 1875.
- Krause, W. Allgemeine Anatomie, 1876.
- Von Kupffer, C. Ueber die Sternzellen der Leber. Arch. Mikrosk. Anat. 1876, 12, 353–358. [Google Scholar] [CrossRef]
- Von Kupffer, C. Ueber die sogennanten Sternzellen der S–ugethierle-ber. Arch. Mikrosk. Anat. 1899, 54, 254–288. [Google Scholar] [CrossRef] [Green Version]
- Rothe, P. Ueber die Sternzellen der Leber; Münchener Diss: Munich, Germany, 1882. [Google Scholar]
- Nicolescu, P.; Rouiller, C. Relations between the endothelial cells of the liver sinusoids and the Kupffer cells. Electron microscopic study. Z. Zellforsch. Mikrosk. Anat. 1967, 76, 313–338. [Google Scholar] [CrossRef]
- Wake, K.; Motomatsu, K.; Senoo, H.; Masuda, A.; Adachi, E. Improved Kupffer’s gold chloride method for demonstrating the stellate cells storing retinol (vitamin A) in the liver and extrahepatic organs of vertebrates. Stain Technol. 1986, 61, 193–200. [Google Scholar] [CrossRef]
- Wake, K. Liver perivascular cells revealed by gold- and silver-impregnation methods and electron microscopy. In Biopathology of the Liver; Springer Nature: Cham, Switzerland, 1988; pp. 23–36. [Google Scholar]
- Wisse, E. An electron microscopic study of the fenestrated endothelial lining of rat liver sinusoids. J. Ultrastruct. Res. 1970, 31, 125–150. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Mehal, W.Z.; Iredale, J.; Friedman, S.L. Scraping fibrosis: Expressway to the core of fibrosis. Nat. Med. 2011, 17, 552–553. [Google Scholar] [CrossRef]
- Yang, L.; Kwon, J.; Popov, Y.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular Endothelial Growth Factor Promotes Fibrosis Resolution and Repair in Mice. Gastroenterology 2014, 146, 1339–1350.e1. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.; Yamasaki, G.; Johnson, R.J.; Friedman, S.L. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Investig. 1994, 94, 1563–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzani, M. PDGF and signal transduction in hepatic stellate cells. Front. Biosci. 2002, 7, 1720–1726. [Google Scholar] [CrossRef]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.-C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hosida, Y.; et al. Beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantari-Mimoun, C.; Castells, M.; Klose, R.; Meinecke, A.-K.; Lemberger, U.J.; Rautou, P.-E.; Pinot-Roussel, H.; Badoual, C.; Schrödter, K.; Österreicher, C.H.; et al. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology 2014, 61, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Brigstock, D.R. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 2012, 17, 2495–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.V.; Dooley, S. TGF-beta/Smad signaling in the injured liver. Z Gastroenterol. 2006, 44, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.E.; McDonnell, M.A.; Law, B.K.; Moses, H.L. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J. Biol. Chem. 1999, 274, 37413–37420. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, H.; Ninomiya-Tsuji, J.; Masuyama, N.; Nishita, M.; Fujisawa, J.; Shibuya, H.; Matsumoto, K.; Nishida, E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem. 1999, 274, 27161–27167. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-J.; Tao, H.; Li, J. Hedgehog signaling pathway as key player in liver fibrosis: New insights and perspectives. Expert Opin. Ther. Targets 2014, 18, 1011–1021. [Google Scholar] [CrossRef]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereora, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philips, G.M.; Chan, I.S.; Swiderska, M.; Schroder, V.T.; Guy, C.; Karaca, G.F.; Moylan, C.; Venkatraman, T.; Feuerlein, S.; Syn, W.-K.; et al. Hedgehog Signaling Antagonist Promotes Regression of Both Liver Fibrosis and Hepatocellular Carcinoma in a Murine Model of Primary Liver Cancer. PLoS ONE 2011, 6, e23943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Dong, Y.; Kumar, V.; Almawash, S.; Mahato, R.I. The use of micelles to deliver potential hedgehog pathway inhibitor for the treatment of liver fibrosis. Theranostics 2019, 9, 7537–7555. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Yan, M.; Wang, C.; Meng, X.; Xiang, Z.; Qiu, Y.; Han, X. Microcystin-leucine-arginine induces liver fibrosis by activating the Hedgehog pathway in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2020, 533, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Ju, H.-L.; Cho, K.J.; Kim, D.Y.; Han, K.-H.; Eun, J.W.; Nam, S.W.; Ribback, S.; Dombrowski, F.; et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J. Hepatol. 2016, 64, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy Releases Lipid That Promotes Fibrogenesis by Activated Hepatic Stellate Cells in Mice and in Human Tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.S.; Hasegawa, D.; Goossens, N.; Tsuchida, T.; Athwal, V.; Sun, X.; Robinson, C.L.; Bhattacharya, D.; Chou, H.-I.; Zhang, D.Y.; et al. The XBP1 Arm of the Unfolded Protein Response Induces Fibrogenic Activity in Hepatic Stellate Cells Through Autophagy. Sci. Rep. 2016, 6, 39342. [Google Scholar] [CrossRef]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic Reticulum Stress in Hepatic Stellate Cells Promotes Liver Fibrosis via PERK-Mediated Degradation of HNRNPA1 and Up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193.e8. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Venugopal, S.K.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.H.; Devaraj, S.; Shah, V.H.; Gershwin, M.E.; et al. Reduced Nicotinamide Adenine Dinucleotide Phosphate Oxidase 2 Plays a Key Role in Stellate Cell Activation and Liver Fibrogenesis In Vivo. Gastroenterology 2010, 139, 1375–1384.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A High-Cholesterol Diet Exacerbates Liver Fibrosis in Mice via Accumulation of Free Cholesterol in Hepatic Stellate Cells. Gastroenterology 2012, 142, 152–164.e10. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Tamura, S.; Kiso, S.; Matsumoto, H.; Saji, Y.; Yoshida, Y.; Fukui, K.; Maeda, N.; Nishizawa, H.; Nagaretani, H.; et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 2003, 125, 1796–1807. [Google Scholar] [CrossRef] [PubMed]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: Role of short-form leptin receptors and osteopontin. Am. J. Physiol. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombes, J.D.; Choi, S.S.; Swiderska-Syn, M.; Manka, P.P.; Reid, D.T.; Palma, E.; Briones-Orta, M.A.; Xie, G.; Younis, R.; Kitamura, N.; et al. Osteopontin is a proximal effector of leptin-mediated non-alcoholic steatohepatitis (NASH) fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.-S.; Lee, Y.-S.; Byun, J.-S.; Seo, W.; Jeong, J.-M.; Park, O.; Duester, G.; Haseba, T.; Kim, S.C.; Park, K.G.; et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer cells in mice. Hepatology 2014, 60, 1044–1053. [Google Scholar] [CrossRef] [Green Version]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef]
- Chiu, Y.-S.; Wei, C.-C.; Lin, Y.-J.; Hsu, Y.-H.; Chang, C.-P. IL-20 and IL-20R1 antibodies protect against liver fibrosis. Hepatology 2014, 60, 1003–1014. [Google Scholar] [CrossRef]
- Jiao, J.; Ooka, K.; Fey, H.; Fiel, M.I.; Rahmman, A.H.; Kojima, K.; Hoshida, Y.; Chen, X.; De Paula, T.; Vetter, D.; et al. Interleukin-15 receptor α on hepatic stellate cells regulates hepatic fibrogenesis in mice. J. Hepatol. 2016, 65, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Kiziltas, S. Toll-like receptors in pathophysiology of liver diseases. World J. Hepatol. 2016, 8, 1354–1369. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.-S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Zhou, C.; York, S.R.; Chen, J.Y.; Pondick, J.V.; Motola, D.L.; Chung, R.T.; Mullen, A.C. Long noncoding RNAs expressed in human hepatic stellate cells form networks with extracellular matrix proteins. Genome Med. 2016, 8, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kifayathullah, L.; Arunachalam, J.P.; Bodda, C.; Agbemenyah, H.; Laccone, F.; Mannan, A. MeCP2270 Mutant Protein Is Expressed in Astrocytes as well as in Neurons and Localizes in the Nucleus. Cytogenet. Genome Res. 2010, 129, 290–297. [Google Scholar] [CrossRef]
- Kweon, S.M.; Chi, F.; Higashiyama, R.; Lai, K.; Tsukamoto, H. Wnt Pathway Stabilizes MeCP2 Protein to Repress PPAR-gamma in Activation of Hepatic Stellate Cells. PLoS ONE 2016, 11, e0156111. [Google Scholar] [CrossRef]
- Tian, W.; Fan, Z.; Li, J.; Hao, C.; Li, M.; Xu, H.; Wu, X.; Zhou, B.; Zhang, L.; Fang, M.; et al. Myocardin-related transcription factor A (MRTF-A) plays an essential role in hepatic stellate cell activation by epigenetically modulating TGF-beta signaling. Int. J. Biochem. Cell. Biol. 2016, 71, 35–43. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Deleve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef]
- Bonder, C.S.; Norman, M.U.; Swain, M.G.; Zbytnuik, L.D.; Yamanouchi, J.; Santamaria, P.; Ajuebor, M.; Salmi, M.; Jalkanen, S.; Kubes, P. Rules of Recruitment for Th1 and Th2 Lymphocytes in Inflamed Liver: A Role for Alpha-4 Integrin and Vascular Adhesion Protein-1. Immunity 2005, 23, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munder, M.; Eichmann, K.; Modolell, M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: Competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J. Immunol. 1998, 160, 5347–5354. [Google Scholar] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating Monocyte-Derived Macrophages and Resident Kupffer Cells Display Different Ontogeny and Functions in Acute Liver Injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.-H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-Like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef]
- Langhans, B.; Krämer, B.; Louis, M.; Nischalke, H.D.; Hüneburg, R.; Staratschek-Jox, A.; Odenthal, M.; Manekeller, S.; Schepke, M.; Kalff, J.; et al. Intrahepatic IL-8 producing Foxp3+CD4+ regulatory T cells and fibrogenesis in chronic hepatitis C. J. Hepatol. 2013, 59, 229–235. [Google Scholar] [CrossRef]
- Radaeva, S.; Wang, L.; Radaev, S.; Jeong, W.-I.; Park, O.; Gao, B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am. J. Physiol. Liver Physiol. 2007, 293, G809–G816. [Google Scholar] [CrossRef]
- Mantovani, S.; Mele, D.; Oliviero, B.; Barbarini, G.; Varchetta, S.; Mondelli, M.U. NKp30 isoforms in patients with chronic hepatitis C virus infection. Immunology 2015, 146, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Gur, C.; Doron, S.; Kfir-Erenfeld, S.; Horwitz, E.; Abu-Tair, L.; Safadi, R.; Mandelboim, O. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut 2012, 61, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Muhanna, N.; Abu Tair, L.; Doron, S.; Amer, J.; Azzeh, M.; Mahamid, M.; Friedman, S.; Safadi, R. Amelioration of hepatic fibrosis by NK cell activation. Gut 2010, 60, 90–98. [Google Scholar] [CrossRef]
- Stegmann, K.A.; Björkström, N.K.; Veber, H.; Ciesek, S.; Riese, P.; Wiegand, J.; Hadem, J.; Suneetha, P.V.; Jaroszewicz, J.; Wang, C.; et al. Interferon-α–Induced TRAIL on Natural Killer Cells Is Associated with Control of Hepatitis C Virus Infection. Gastroenterology 2010, 138, 1885–1897.e10. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, F.; Olinga, P.; Poelstra, K.; Beljaars, L. Targeted Therapies in Liver Fibrosis: Combining the Best Parts of Platelet-Derived Growth Factor BB and Interferon Gamma. Front. Med. 2015, 2, 72. [Google Scholar] [CrossRef] [Green Version]
- Glässner, A.; Eisenhardt, M.; Krämer, B.; Körner, C.; Coenen, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Investig. 2012, 92, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Jiao, J.; Sastre, D.; Fiel, M.I.; Lee, U.E.; Ghiassi-Nejad, Z.; Ginhoux, F.; Vivier, E.; Friedman, S.L.; Merad, M.; Aloman, C. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology 2011, 55, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.W.; Montuori, N. FPRs: Linking innate immune system and fibrosis. Oncotarget 2015, 6, 18736–18737. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guixé-Muntet, S.; Ortega-Ribera, M.; Wang, C.; Selicean, S.; Andreu, I.; Kechagia, J.Z.; Fondevila, C.; Roca-Cusachs, P.; Dufour, J.-F.; Bosch, J.; et al. Nuclear deformation mediates liver cell mechanosensing in cirrhosis. JHEP Rep. 2020, 2, 100145. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Sand, J.M.B.; Gudmann, N.S.; Karsdal, M.A. Biochemistry of Collagens, Laminins and Elastin; Academic Press: London, UK, 2019. [Google Scholar]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-beta as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Beingker, N.M. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Murase, K.; Kato, J.; Kobune, M.; Sato, T.; Kawano, Y.; Takimoto, R.; Takada, K.; Miyanishi, K.; Matsunaga, T.; et al. Resolution of liver cirrhosis using vitamin A–coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 2008, 26, 431–442. [Google Scholar] [CrossRef]
- Han, Y.-P. Matrix metalloproteinases, the pros and cons, in liver fibrosis. J. Gastroenterol. Hepatol. 2006, 21, S88–S91. [Google Scholar] [CrossRef]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Hernandez, A.; Amenta, P.S. The extracellular matrix in hepatic regeneration. FASEB J. 1995, 9, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W. Lipoproteins and the liver sieve: The role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, atherosclerosis, and cirrhosis. Hepatology 1995, 21, 863–874. [Google Scholar] [PubMed]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, J.-E.; Brégerie, O.; Robert, A.; Debey, P.; Brechot, C.; Desdouets, C. Liver Cell Polyploidization: A Pivotal Role for Binuclear Hepatocytes. J. Biol. Chem. 2003, 278, 19095–19101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margall-Ducos, G.; Celton-Morizur, S.; Couton, D.; Brégerie, O.; Desdouets, C. Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J. Cell Sci. 2007, 120, 3633–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Iwakiri, Y. The Hepatic Lymphatic Vascular System: Structure, Function, Markers, and Lymphangiogenesis. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 733–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, O.; Ohtani, Y. Lymph Circulation in the Liver. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2008, 291, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Author Correction: Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2019, 25, 701. [Google Scholar] [CrossRef]
- Häussinger, D.; Kordes, C. Space of Disse: A stem cell niche in the liver. Biol. Chem. 2019, 401, 81–95. [Google Scholar] [CrossRef]
- Rohn, F.; Kordes, C.; Castoldi, M.; Götze, S.; Poschmann, G.; Stühler, K.; Herebian, D.; Benk, A.S.; Geiger, F.; Zhang, T.; et al. Laminin-521 promotes quiescence in isolated stellate cells from rat liver. Biomaterials 2018, 180, 36–51. [Google Scholar] [CrossRef]
- GBD Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 2287–2323. [Google Scholar]
- Mokdad, A.A.; Lopez, A.D.; Shahraz, S.; Lozano, R.; Stanaway, J.; Murray, C.; Naghavi, M. Liver cirrhosis mortality in 187 countries between 1980 and 2010: A systematic analysis. BMC Med. 2014, 12, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracia-Sancho, J.; Marrone, G.; Fernández-Iglesias, A. Hepatic microcirculation and mechanisms of portal hypertension. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; Bossche, B.V.D.; De Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, R.J.; Chalasani, N.; Björnsson, E.S.; Suzuki, A.; Kullak-Ublick, G.A.; Watkins, P.B.; Devarbhavi, H.; Merz, M.; Lucena, M.I.; Kaplowitz, N.; et al. Drug-induced liver injury. Nat. Rev. Dis. Prim. 2019, 5, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Global Status Report on Alcohol and Health. 2018. Available online: https://apps.who.int/iris/bitstream/handle/10665/274603/9789241565639-eng.pdf?ua=1 (accessed on 28 January 2021).
- Sussman, N.L.; Lucey, M.R. Alcohol and Alcoholic Liver Disease. Clin. Liver Dis. 2019, 23, xiii–xiv. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Tiribelli, C. Is it time to change NAFLD and NASH nomenclature? Lancet Gastroenterol. Hepatol. 2017, 2, 547–548. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Law, K.; Brunt, E.M. Nonalcoholic fatty liver disease. Clin. Liver Dis. 2010, 14, 591–604. [Google Scholar] [CrossRef]
- Pinzani, M. Pathophysiology of Non-Alcoholic Steatohepatitis and Basis for Treatment. Dig. Dis. 2011, 29, 243–248. [Google Scholar] [CrossRef]
- Gunarathne, L.S.; Rajapaksha, H.; Shackel, N.; Angus, P.W.; Herath, C.B. Cirrhotic portal hypertension: From pathophysiology to novel therapeutics. World J. Gastroenterol. 2020, 26, 6111–6140. [Google Scholar] [CrossRef] [PubMed]
- Angeli, P.; Fernandez-Varo, G.; Libera, V.D.; Fasolato, S.; Galioto, A.; Arroyo, V.; Sticca, A.; Guarda, S.; Gatta, A.; Jimenez, W. The role of nitric oxide in the pathogenesis of systemic and splanchnic vasodilation in cirrhotic rats before and after the onset of ascites. Liver Int. 2005, 25, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Berzigotti, A.; Bosch, J. Pharmacologic Management of Portal Hypertension. Clin. Liver Dis. 2014, 18, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Colle, I.O.; De Vriese, A.S.; Van Vlierberghe, H.R.; Lameire, N.H.; De Vos, M.M. Vascular hyporesponsiveness in the mesenteric artery of anaesthetized rats with cirrhosis and portal hypertension: An in-vivo study. Eur. J. Gastroenterol. Hepatol. 2004, 16, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M. Molecular pathophysiology of portal hypertension. Hepatology 2015, 61, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pras, E.; Gallego, J.; Coch, L.; Mejias, M.; Fernandez-Miranda, G.; Pardal, R.; Bosch, J.; Mendez, R.; Fernandez, M. Role and therapeutic potential of vascular stem/progenitor cells in pathological neovascularisation during chronic portal hypertension. Gut 2016, 66, 1306–1320. [Google Scholar] [CrossRef]
- Deliwala, S.; Sundus, S.; Haykal, T.; Elbedawi, M.M.; Bachuwa, G. Small Duct Primary Sclerosing Cholangitis: An Underdiagnosed Cause of Chronic Liver Disease and Cirrhosis. Cureus 2020, 12, e7298. [Google Scholar] [CrossRef] [Green Version]
- Georgiev, P.; Jochum, W.; Heinrich, S.; Jang, J.H.; Nocito, A.; Dahm, F.; Clavien, P.-A. Characterization of time-related changes after experimental bile duct ligation. BJS 2008, 95, 646–656. [Google Scholar] [CrossRef]
- Kopp, J.L.; Grompe, M.; Sander, M. Stem cells versus plasticity in liver and pancreas regeneration. Nat. Cell Biol. 2016, 18, 238–245. [Google Scholar] [CrossRef]
- Valizadeh, A.; Majidinia, M.; Samadi-Kafil, H.; Yousefi, M.; Yousefi, B. The roles of signaling pathways in liver repair and regeneration. J. Cell. Physiol. 2019, 234, 14966–14974. [Google Scholar] [CrossRef]
- Balabaud, C.; Bioulac-Sage, P.; Desmoulière, A. The role of hepatic stellate cells in liver regeneration. J. Hepatol. 2004, 40, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, I.A.; Field, J.; Farrell, G.C. Leptin-specific mechanisms for impaired liver regeneration in ob/ob mice after toxic injury. Gastroenterology 2003, 124, 1451–1464. [Google Scholar] [CrossRef]
- Collin de l’Hortet, A.; Zerrad-Saadi, A.; Prip-Buus, C.; Fauveau, V.; Helmy, N.; Ziol, M.; Vons, C.; Billot, K.; Baud, V.; Gilgenkrantz, H.; et al. GH administration rescues fatty liver regeneration impairment by restoring GH/EGFR pathway deficiency. Endocrinology 2014, 155, 2545–2554. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.T.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.K.; Yim, H.J. Reversal of liver cirrhosis: Current evidence and expectations. Korean J. Intern. Med. 2017, 32, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Brissot, P. Haemochromatosis. Nat. Rev. Dis. Primers. 2018, 4, 18016. [Google Scholar] [CrossRef]
- Porter, J.L.; Rawla, P. Hemochromatosis; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Weiss, B.M.S. Onco-Nephrology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 99–106. [Google Scholar]
- Fan, C.Q.; Crawford, J.M. Sinusoidal Obstruction Syndrome (Hepatic Veno-Occlusive Disease). J. Clin. Exp. Hepatol. 2014, 4, 332–346. [Google Scholar] [CrossRef] [Green Version]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, D.; De Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharmacol. 2017, 174, 3107–3117. [Google Scholar] [CrossRef] [Green Version]
- Hammad, S.; Cavalcanti, E.; Werle, J.; Caruso, M.L.; Dropmann, A.; Ignazzi, A.; Ebert, M.P.; Dooley, S.; Giannelli, G. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch. Toxicol. 2018, 92, 2297–2309. [Google Scholar] [CrossRef]
- Zhang, H.; Ju, B.; Zhang, X.; Zhu, Y.; Nie, Y.; Xu, Y.; Lei, Q. Magnolol Attenuates Concanavalin A-induced Hepatic Fibrosis, Inhibits CD4(+) T Helper 17 (Th17) Cell Differentiation and Suppresses Hepatic Stellate Cell Activation: Blockade of Smad3/Smad4 Signalling. Basic Clin. Pharmacol. Toxicol. 2017, 120, 560–570. [Google Scholar] [CrossRef]
- Ahn, J.; Son, M.K.; Jung, K.H.; Kim, K.; Kim, G.J.; Lee, S.-H.; Hong, S.-S.; Park, S.G. Aminoacyl-tRNA synthetase interacting multi-functional protein 1 attenuates liver fibrosis by inhibiting TGFbeta signaling. Int. J. Oncol. 2016, 48, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Ganai, A.A.; Husain, M. Genistein attenuates D-GalN induced liver fibrosis/chronic liver damage in rats by blocking the TGF-beta/Smad signaling pathways. Chem. Biol. Interact. 2017, 261, 80–85. [Google Scholar] [CrossRef]
- Liu, J.; Kong, D.; Qiu, J.; Xie, Y.; Lu, Z.; Zhou, C.; Liu, X.; Zhang, R.; Wang, Y. Praziquantel ameliorates CCl4 -induced liver fibrosis in mice by inhibiting TGF-beta/Smad signalling via up-regulating Smad7 in hepatic stellate cells. Br. J. Pharmacol. 2019, 176, 4666–4680. [Google Scholar] [CrossRef]
- Zhao, X.; Li, R.; Liu, Y.; Zhang, X.; Zhang, M.; Zeng, Z.; Wu, L.; Gao, X.; Lan, T.; Wang, Y. Polydatin protects against carbon tetrachloride-induced liver fibrosis in mice. Arch. Biochem. Biophys. 2017, 629, 1–7. [Google Scholar] [CrossRef]
- Lin, L.; Gong, H.; Li, R.; Huang, J.; Cai, M.; Lan, T.; Huang, W.; Guo, Y.; Zhou, Z.; An, Y.; et al. Nanodrug with ROS and pH Dual-Sensitivity Ameliorates Liver Fibrosis via Multicellular Regulation. Adv. Sci. 2020, 7, 1903138. [Google Scholar] [CrossRef]
- Harrison, S.A.; Goodman, Z.; Jabbar, A.; Vemulapalli, R.; Younes, Z.H.; Freilich, B.; Sheikh, M.Y.; Schattenberg, J.M.; Kayali, Z.; Zivony, A.; et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 2020, 72, 816–827. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Bosch, J.; Kayali, Z.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.; Satapathy, S.K.; Ghabril, M.; Shiffman, M.L.; Younes, Z.H.; et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 2020, 72, 885–895. [Google Scholar] [CrossRef]
- Schuster-Gaul, S.; Geisler, L.J.; McGeough, M.D.; Johnson, C.D.; Zagorska, A.; Li, L.; Wree, A.; Barry, V.; Mikaelian, I.; Jih, L.J.; et al. ASK1 inhibition reduces cell death and hepatic fibrosis in an Nlrp3 mutant liver injury model. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Qu, J.; Yuan, Z.; Wang, G.; Wang, X.; Li, K.-W. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharmacol. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Iimuro, Y.; Nishio, T.; Morimoto, T.; Nitta, T.; Stefanovic, B.; Choi, S.K.; Brenner, D.A.; Yamaoka, Y. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology 2003, 124, 445–458. [Google Scholar] [CrossRef]
- Du, C.; Jiang, M.; Wei, X.; Qin, J.; Xu, H.; Wang, Y.; Zhang, Y.; Zhou, D.; Xue, H.; Zheng, S.; et al. Transplantation of human matrix metalloproteinase-1 gene-modified bone marrow-derived mesenchymal stem cell attenuates CCL4-induced liver fibrosis in rats. Int. J. Mol. Med. 2018, 41, 3175–3184. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wang, P.; Cong, M.; Zhang, D.; Liu, L.; Li, H.; Zhai, Q.; Li, Z.; Jia, J.; You, H. Matrix metalloproteinase-1 induction by diethyldithiocarbamate is regulated via Akt and ERK/miR222/ETS-1 pathways in hepatic stellate cells. Biosci. Rep. 2016, 36, e00371. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bañuelos, J.; Siller-Lopez, F.; Miranda, A.; Aguilar, L.K.; Aguilar-Cordova, E.; Armendariz-Borunda, J. Cirrhotic rat livers with extensive fibrosis can be safely transduced with clinical-grade adenoviral vectors. Evidence of cirrhosis reversion. Gene Ther. 2002, 9, 127–134. [Google Scholar] [CrossRef]
- Liu, J.; Cai, X.; Li, J.; Li, L.; Tang, J.; Feng, X.; Chen, J.; Liang, Y.; Jin, R.; Xie, A.; et al. Adenoviral delivery of truncated MMP-8 fused with the hepatocyte growth factor mutant 1K1 ameliorates liver cirrhosis and promotes hepatocyte proliferation. Drug Des. Dev. Ther. 2015, 9, 5655–5667. [Google Scholar] [CrossRef] [Green Version]
- Roderfeld, M.; Weiskirchen, R.; Wagner, S.; Berres, M.; Henkel, C.; Grötzinger, J.; Gressner, A.M.; Matern, S.; Roeb, E. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. FASEB J. 2006, 20, 444–454. [Google Scholar] [CrossRef]
- Lu, C.; Zou, Y.; Liu, Y.; Niu, Y. Rosmarinic acid counteracts activation of hepatic stellate cells via inhibiting the ROS-dependent MMP-2 activity: Involvement of Nrf2 antioxidant system. Toxicol. Appl. Pharmacol. 2017, 318, 69–78. [Google Scholar] [CrossRef]
- Guixé-Muntet, S.; Zhu, C.-P.; Xie, W.-F.; Gracia-Sancho, J. Novel therapeutics for portal hypertension and fibrosis in chronic liver disease. Pharmacol. Ther. 2020, 215, 107626. [Google Scholar] [CrossRef]
- Atef, M.M.; Hafez, Y.M.; Alshenawy, H.A.; Emam, M.N. Ameliorative effects of autophagy inducer, simvastatin on alcohol-induced liver disease in a rat model. J. Cell. Biochem. 2019, 120, 7679–7688. [Google Scholar] [CrossRef]
- Bosch, J.; Gracia-Sancho, J.; Abraldes, J.G. Cirrhosis as new indication for statins. Gut 2020, 69, 953–962. [Google Scholar] [CrossRef]
- Ghoreshi, Z.-A.-S.; Kabirifar, R.; Khodarahmi, A.; Karimollah, A.; Moradi, A. The preventive effect of atorvastatin on liver fibrosis in the bile duct ligation rats via antioxidant activity and down-regulation of Rac1 and NOX1. Iran. J. Basic Med. Sci. 2020, 23, 30–35. [Google Scholar]
- Kamal, S.; Khan, M.A.; Seth, A.; Cholankeril, G.; Gupta, D.; Singh, U.; Kamal, F.; Howden, C.W.; Stave, C.; Nair, S.; et al. Beneficial Effects of Statins on the Rates of Hepatic Fibrosis, Hepatic Decompensation, and Mortality in Chronic Liver Disease: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2017, 112, 1495–1505. [Google Scholar] [CrossRef]
- Kaplan, D.E. The Use of Statins in Patients with Cirrhosis. Gastroenterol. Hepatol. 2018, 14, 485–487. [Google Scholar]
- Liu, Z.; Zhang, X.; Xiao, Q.; Ye, S.; Lai, C.-H.; Luo, J.; Huang, X.; Wang, W.; Zeng, C.; Zhong, Z.; et al. Pretreatment Donors after Circulatory Death with Simvastatin Alleviates Liver Ischemia Reperfusion Injury through a KLF2-Dependent Mechanism in Rat. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Motawi, T.M.; Atta, H.M.; Sadik, N.A.E.-H.; Azzam, M. The Therapeutic Effects of Bone Marrow-Derived Mesenchymal Stem Cells and Simvastatin in a Rat Model of Liver Fibrosis. Cell Biophys. 2013, 68, 111–125. [Google Scholar] [CrossRef]
- Nozari, E.; Moradi, A.; Samadi, M. Effect of Atorvastatin, Curcumin, and Quercetin on miR-21 and miR-122 and their correlation with TGFbeta1 expression in experimental liver fibrosis. Life Sci. 2020, 259, 118293. [Google Scholar] [CrossRef]
- Yim, C.-S.; Jeong, Y.-S.; Lee, S.-Y.; Pyeon, W.; Ryu, H.-M.; Lee, J.-H.; Lee, K.-R.; Maeng, H.-J.; Chung, S.-J. Specific Inhibition of the Distribution of Lobeglitazone to the Liver by Atorvastatin in Rats: Evidence for a Rat Organic Anion Transporting Polypeptide 1B2–Mediated Interaction in Hepatic Transport. Drug Metab. Dispos. 2017, 45, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Yu, M.; Zhang, H.; Xie, D.; Nie, W.; Shi, K. Suppression of miR-143-3p contributes to the anti-fibrosis effect of atorvastatin on myocardial tissues via the modulation of Smad2 activity. Exp. Mol. Pathol. 2020, 112, 104346. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Bosch, J.; García-Pagán, J.C. Enhanced vasoconstrictor prostanoid production by sinusoidal endothelial cells increases portal perfusion pressure in cirrhotic rat livers. J. Hepatol. 2007, 47, 220–227. [Google Scholar] [CrossRef]
- Kruger, A.; Fuchs, B.C.; Masia, R.; Holmes, J.A.; Salloum, S.; Sojoodi, M.; Ferreira, D.D.S.; Rutledge, S.M.; Caravan, P.; Alatrakchi, N.; et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol. Commun. 2018, 2, 529–545. [Google Scholar] [CrossRef]
- Liang, F.; Giordano, C.; Shang, D.; Li, Q.; Petrof, B.J. The dual CCR2/CCR5 chemokine receptor antagonist Cenicriviroc reduces macrophage infiltration and disease severity in Duchenne muscular dystrophy (Dmdmdx-4Cv) mice. PLoS ONE 2018, 13, e0194421. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Park, H.; Bae, E.J. Efficacy of evogliptin and cenicriviroc against nonalcoholic steatohepatitis in mice: A comparative study. Korean J. Physiol. Pharmacol. 2019, 23, 459–466. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz-García, C.; Fernández-Iglesias, A.; Gracia-Sancho, J.; Arráez-Aybar, L.A.; Nevzorova, Y.A.; Cubero, F.J. The Space of Disse: The Liver Hub in Health and Disease. Livers 2021, 1, 3-26. https://doi.org/10.3390/livers1010002
Sanz-García C, Fernández-Iglesias A, Gracia-Sancho J, Arráez-Aybar LA, Nevzorova YA, Cubero FJ. The Space of Disse: The Liver Hub in Health and Disease. Livers. 2021; 1(1):3-26. https://doi.org/10.3390/livers1010002
Chicago/Turabian StyleSanz-García, Carlos, Anabel Fernández-Iglesias, Jordi Gracia-Sancho, Luis Alfonso Arráez-Aybar, Yulia A. Nevzorova, and Francisco Javier Cubero. 2021. "The Space of Disse: The Liver Hub in Health and Disease" Livers 1, no. 1: 3-26. https://doi.org/10.3390/livers1010002
APA StyleSanz-García, C., Fernández-Iglesias, A., Gracia-Sancho, J., Arráez-Aybar, L. A., Nevzorova, Y. A., & Cubero, F. J. (2021). The Space of Disse: The Liver Hub in Health and Disease. Livers, 1(1), 3-26. https://doi.org/10.3390/livers1010002