
Modification of Epoxidized Soybean Oil for the Preparation of Amorphous, Nonretrogradable, and Hydrophobic Starch Films


 , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Soybean Oil Cinnamates
2.3. Preparation of Starch-Based Film
2.4. Characterization of ESO Derivatives (Soybean Oil Cinnamates)
2.5. Characterization of Starch-Based Films
3. Results and Discussion
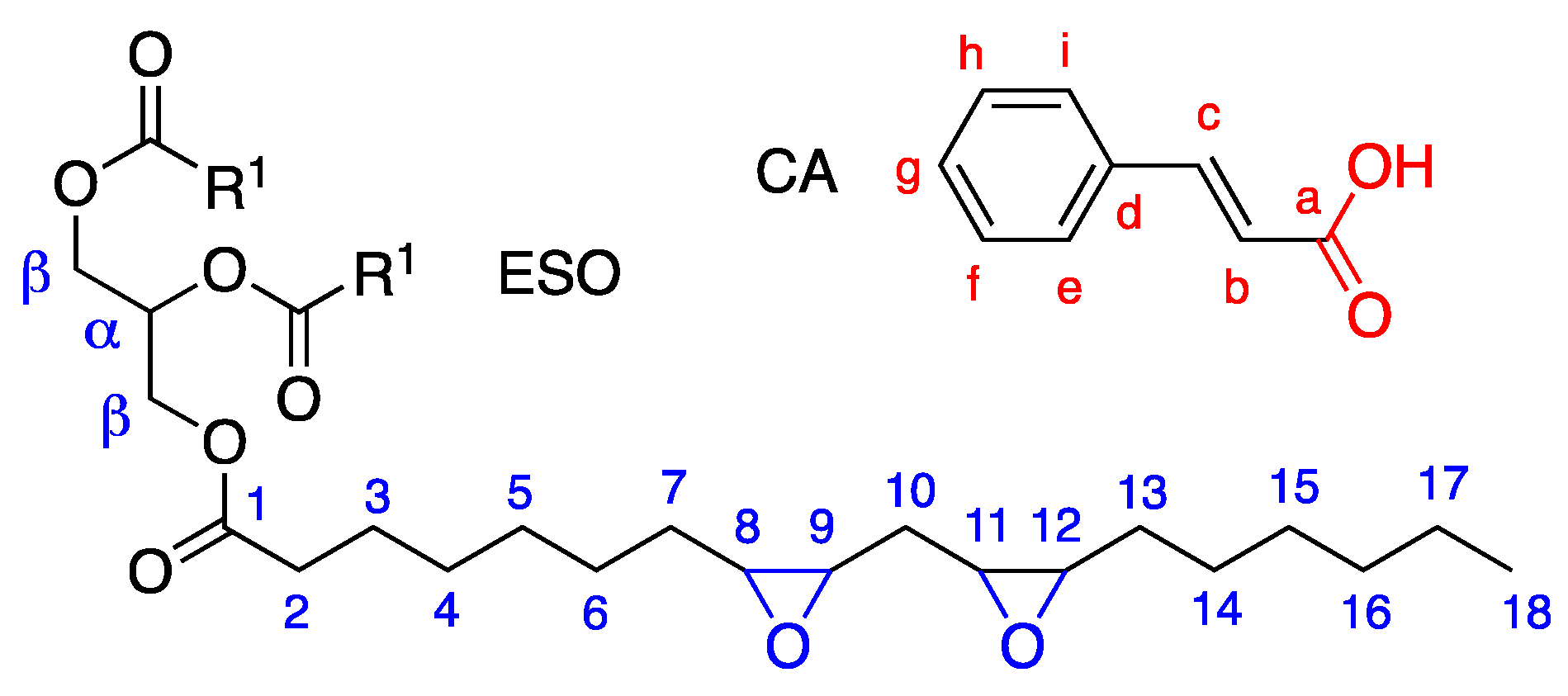
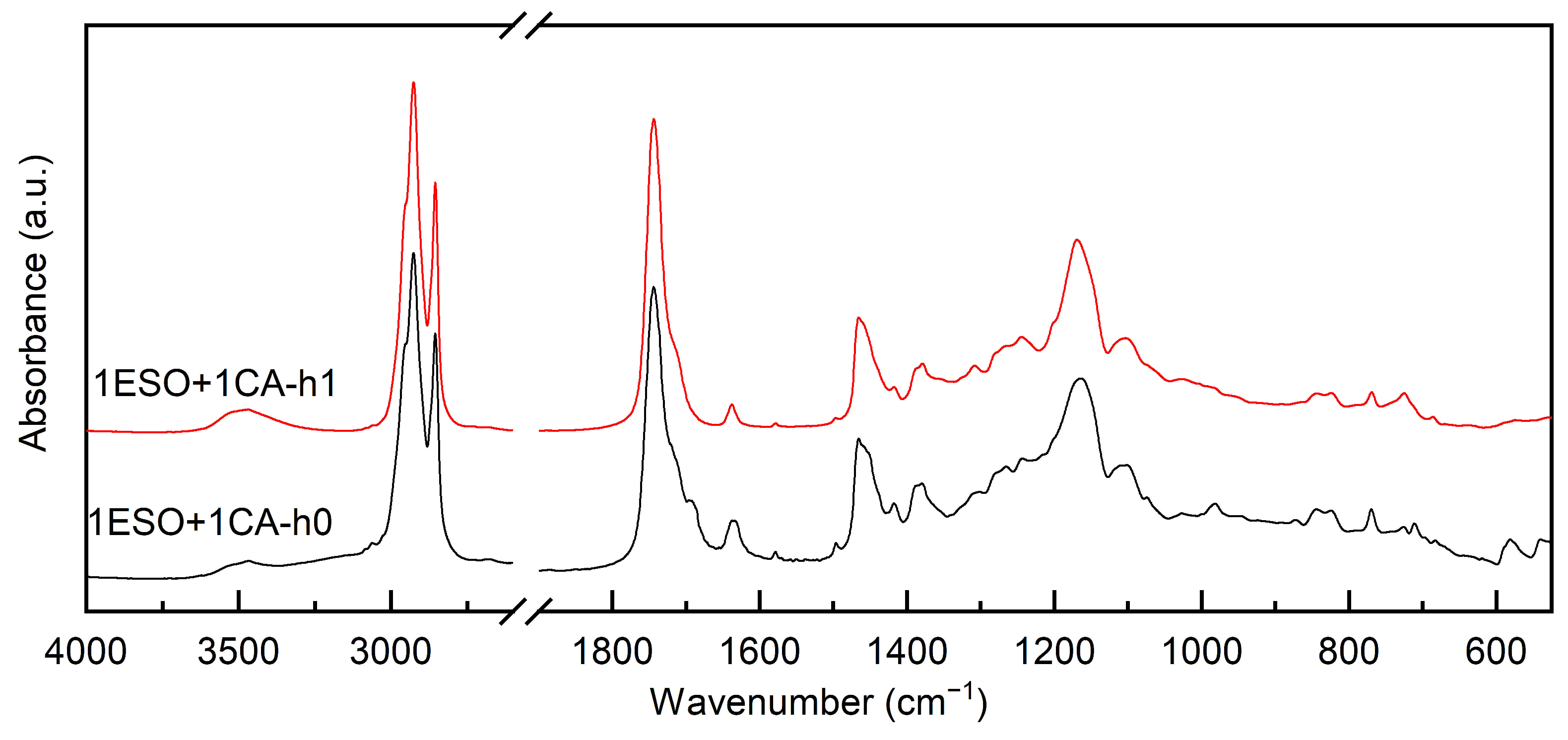
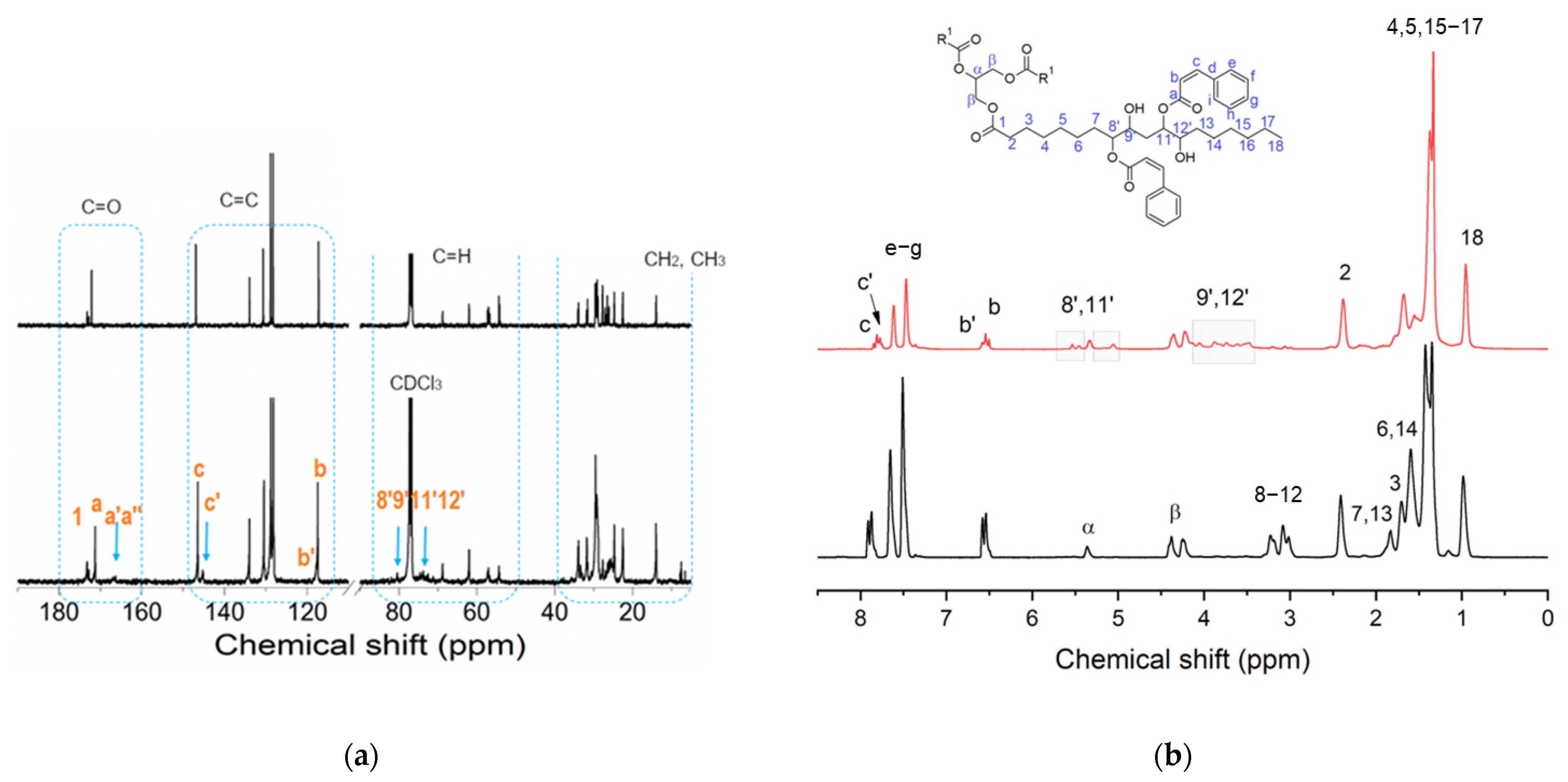
3.1. Modification of the Soybean-Epoxidized Oil by Addition of Cinnamic Acid: Effects of Thermal Cycles and Stoichiometry
3.2. ESO Cinnamates as Additives for Starch-Based Film Production
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ESO | epoxidized soybean oil |
| CA | cinnamic acid |
| MW | molar mass |
| NMR | nuclear magnetic resonance |
| FTIR | Fourier transform infrared |
| XRD | X-ray diffraction |
References
- Ratnayake, W.S.; Jackson, D.S. Chapter 5 Starch Gelatinization. In Advances in Food and Nutrition Research; Academic Press: Cambridge, MA, USA, 2008; Volume 55, pp. 221–268. [Google Scholar]
- Xie, F.; Liu, P.; Yu, L. Processing of Plasticized Starch-Based Materials. In Starch Polymers; Elsevier: Amsterdam, The Netherlands, 2014; pp. 257–289. ISBN 978-0-444-53730-0. [Google Scholar]
- Li, C. Unraveling the Complexities of Starch Retrogradation: Insights from Kinetics, Molecular Interactions, and Influences of Food Ingredients. Food Rev. Int. 2024, 40, 3159–3182. [Google Scholar] [CrossRef]
- Adewale, P.; Yancheshmeh, M.S.; Lam, E. Starch Modification for Non-Food, Industrial Applications: Market Intelligence and Critical Review. Carbohydr. Polym. 2022, 291, 119590. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.J.F. Chapter 15—Starch: Major Sources, Properties and Applications as Thermoplastic Materials. In Monomers, Polymers and Composites from Renewable Resources; Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 321–342. ISBN 978-0-08-045316-3. [Google Scholar]
- Haq, F.; Yu, H.; Wang, L.; Teng, L.; Haroon, M.; Khan, R.U.; Mehmood, S.; Bilal-Ul-Amin; Ullah, R.S.; Khan, A.; et al. Advances in Chemical Modifications of Starches and Their Applications. Carbohydr. Res. 2019, 476, 12–35. [Google Scholar] [CrossRef]
- Otache, M.A.; Duru, R.U.; Achugasim, O.; Abayeh, O.J. Advances in the Modification of Starch via Esterification for Enhanced Properties. J. Polym. Environ. 2021, 29, 1365–1379. [Google Scholar] [CrossRef]
- Petroni, S.; Fernanda Orsini, S.; Bugnotti, D.; Callone, E.; Dirè, S.; Zoia, L.; Bongiovanni, R.; Vacche, S.D.; Vitale, A.; Raimondo, L.; et al. Photocrosslinkable Starch Cinnamyl Ethers as Bioinspired Bio-Based Polymers. J. Mater. Chem. B 2025, 13, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, L.; Tao, S.; Huang, Z.; Qi, H. Freely Moldable Modified Starch as a Sustainable and Recyclable Plastic. Biomacromolecules 2021, 22, 2676–2683. [Google Scholar] [CrossRef]
- Nguyen, M.T.P.; Escribà-Gelonch, M.; Hessel, V.; Coad, B.R. A Review of the Current and Future Prospects for Producing Bioplastic Films Made from Starch and Chitosan. ACS Sustain. Chem. Eng. 2024, 12, 1750–1768. [Google Scholar] [CrossRef]
- Koh, J.J.; Zhang, X.; He, C. Fully Biodegradable Poly(Lactic Acid)/Starch Blends: A Review of Toughening Strategies. Int. J. Biol. Macromol. 2018, 109, 99–113. [Google Scholar] [CrossRef]
- Muñoz-Gimena, P.F.; Oliver-Cuenca, V.; Peponi, L.; López, D. A Review on Reinforcements and Additives in Starch-Based Composites for Food Packaging. Polymers 2023, 15, 2972. [Google Scholar] [CrossRef]
- Singh, G.P.; Bangar, S.P.; Yang, T.; Trif, M.; Kumar, V.; Kumar, D. Effect on the Properties of Edible Starch-Based Films by the Incorporation of Additives: A Review. Polymers 2022, 14, 1987. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, P.; Li, Y.; Li, J.; Li, X.; Yang, J.; Ji, M.; Li, F.; Zhang, C. Recent Advances and Future Challenges of the Starch-Based Bio-Composites for Engineering Applications. Carbohydr. Polym. 2023, 307, 120627. [Google Scholar] [CrossRef]
- Bugnotti, D.; Dalle Vacche, S.; Esposito, L.H.; Callone, E.; Orsini, S.F.; Ceccato, R.; D’Arienzo, M.; Bongiovanni, R.; Dirè, S.; Vitale, A. Structure of Starch–Sepiolite Bio-Nanocomposites: Effect of Processing and Matrix–Filler Interactions. Polymers 2023, 15, 1207. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Cheng, H.; He, K.; McClements, D.J.; Jin, Z.; Xu, Z.; Meng, M.; Peng, X.; Chen, L. Recent Progress in the Hydrophobic Modification of Starch-Based Films. Food Hydrocoll. 2024, 151, 109860. [Google Scholar] [CrossRef]
- Terrié, C.; Mahieu, A.; Lequart, V.; Martin, P.; Leblanc, N.; Joly, N. Towards the Hydrophobization of Thermoplastic Starch Using Fatty Acid Starch Ester as Additive. Molecules 2022, 27, 6739. [Google Scholar] [CrossRef] [PubMed]
- Belhassen, R.; Vilaseca, F.; Mutjé, P.; Boufi, S. Thermoplasticized Starch Modified by Reactive Blending with Epoxidized Soybean Oil. Ind. Crops Prod. 2014, 53, 261–267. [Google Scholar] [CrossRef]
- Meng, L.; Li, S.; Yang, W.; Simons, R.; Yu, L.; Liu, H.; Chen, L. Improvement of Interfacial Interaction between Hydrophilic Starch Film and Hydrophobic Biodegradable Coating. ACS Sustain. Chem. Eng. 2019, 7, 9506–9514. [Google Scholar] [CrossRef]
- Li, C.; Ju, B.; Zhang, S. Fully Bio-Based Hydroxy Ester Vitrimer Synthesized by Crosslinking Epoxidized Soybean Oil with Doubly Esterified Starch. Carbohydr. Polym. 2023, 302, 120442. [Google Scholar] [CrossRef]
- Qian, Z.; Liu, S.; Du, G.; Wang, S.; Shen, Y.; Zhou, X.; Jiang, S.; Niu, H.; Duan, Z.; Li, T. Versatile Epoxidized Soybean Oil-Based Resin with Excellent Adhesion and Film-Forming Property. ACS Sustain. Chem. Eng. 2023, 11, 5315–5324. [Google Scholar] [CrossRef]
- Moser, B.R.; Cermak, S.C.; Doll, K.M.; Kenar, J.A.; Sharma, B.K. A Review of Fatty Epoxide Ring Opening Reactions: Chemistry, Recent Advances, and Applications. J. Am. Oil Chem. Soc. 2022, 99, 801–842. [Google Scholar] [CrossRef]
- Yang, J.; Dong, X.; Wang, J.; Ching, Y.C.; Liu, J.; Li, C.; Baikeli, Y.; Li, Z.; Mohammed Al-Hada, N.; Xu, S. Synthesis and Properties of Bioplastics from Corn Starch and Citric Acid-Epoxidized Soybean Oil Oligomers. J. Mater. Res. Technol. 2022, 20, 373–380. [Google Scholar] [CrossRef]
- Yang, J.; Xu, S.; Ching, Y.C.; Wang, R.; Al-Hada, N.M.; Li, C.; Lv, D.; Zhang, Z. Facile Synthesis of Cassava Starch-Based Bioplastics Modified by Citric Acid-Epoxidized Soybean Oil Oligomers. J. Appl. Polym. Sci. 2023, 140, e54334. [Google Scholar] [CrossRef]
- Qi, M.; Xu, Y.-J.; Rao, W.-H.; Luo, X.; Chen, L.; Wang, Y.-Z. Epoxidized Soybean Oil Cured with Tannic Acid for Fully Bio-Based Epoxy Resin. RSC Adv. 2018, 8, 26948–26958. [Google Scholar] [CrossRef]
- Annuur, R.M.; Triana, D.; Ernawati, T.; Murai, Y.; Aswad, M.; Hashimoto, M.; Tachrim, Z.P. A Review of Cinnamic Acid’s Skeleton Modification: Features for Antibacterial-Agent-Guided Derivatives. Molecules 2024, 29, 3929. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Chen, Y.F.; Zhong, Z.; Lu, F.; Du, Y.; Liu, L.; Huang, Y. Preparation of the Flexible Soybean Oil-Based Material via [2 + 2] Cycloaddition Photo-Polymerization. J. Appl. Polym. Sci. 2021, 138, 49925. [Google Scholar] [CrossRef]
- D’Arienzo, M.; Diré, S.; Masneri, V.; Rovera, D.; Di Credico, B.; Callone, E.; Mascotto, S.; Pegoretti, A.; Ziarelli, F.; Scotti, R. Tailoring the Dielectric and Mechanical Properties of Polybutadiene Nanocomposites by Using Designed Ladder-like Polysilsesquioxanes. ACS Appl. Nano Mater. 2018, 1, 3817–3828. [Google Scholar] [CrossRef]
- Tran, T.-N.; Mauro, C.D.; Graillot, A.; Mija, A. Chemical Reactivity and the Influence of Initiators on the Epoxidized Vegetable Oil/Dicarboxylic Acid System. Macromolecules 2020, 53, 2526–2538. [Google Scholar] [CrossRef]
- Blank, W.J.; He, Z.A.; Picci, M. Catalysis of the Epoxy-Carboxyl Reaction. J. Coat. Technol. 2002, 74, 33–41. [Google Scholar] [CrossRef]
- Jang, K.-S.; Eom, Y.-S.; Choi, K.-S.; Bae, H.-C. Crosslinkable Deoxidizing Hybrid Adhesive of Epoxy–Diacid for Electrical Interconnections in Semiconductor Packaging. Polym. Int. 2018, 67, 1241–1247. [Google Scholar] [CrossRef]
- Supanchaiyamat, N.; Shuttleworth, P.S.; Hunt, A.J.; Clark, J.H.; Matharu, A.S. Thermosetting Resin Based on Epoxidised Linseed Oil and Bio-Derived Crosslinker. Green Chem. 2012, 14, 1759–1765. [Google Scholar] [CrossRef]
- Schuchardt, U.; Sercheli, R.; Vargas, R.M. Transesterification of Vegetable Oils: A Review. J. Braz. Chem. Soc. 1998, 9, 199–210. [Google Scholar] [CrossRef]
- Ahn, B.-J.K.; Kraft, S.; Sun, X.S. Solvent-Free Acid-Catalyzed Ring-Opening of Epoxidized Oleochemicals Using Stearates/Stearic Acid, and Its Applications. J. Agric. Food Chem. 2012, 60, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Warren, F.J.; Gidley, M.J.; Flanagan, B.M. Infrared Spectroscopy as a Tool to Characterise Starch Ordered Structure—A Joint FTIR–ATR, NMR, XRD and DSC Study. Carbohydr. Polym. 2016, 139, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Šoltýs, A.; Hronský, V.; Šmídová, N.; Olčák, D.; Ivanič, F.; Chodák, I. Solid-State 1H and 13C NMR of Corn Starch Plasticized with Glycerol and Urea. Eur. Polym. J. 2019, 117, 19–27. [Google Scholar] [CrossRef]
- Smits, A.L.M.; Ruhnau, F.C.; Vliegenthart, J.F.G.; van Soest, J.J.G. Ageing of Starch Based Systems as Observed with FT-IR and Solid State NMR Spectroscopy. Starch-Stärke 1998, 50, 478–483. [Google Scholar] [CrossRef]
- Voelkel, R. High-Resolution Solid-State 13C-NMR Spectroscopy of Polymers [New Analytical Methods (37)]. Angew. Chem. Int. Ed. Engl. 1988, 27, 1468–1483. [Google Scholar] [CrossRef]
- Dirè, S.; Callone, E.; Ceccato, R.; Parrino, F.; Di Credico, B.; Mostoni, S.; Scotti, R.; D’Arienzo, M. Structural Effects of TiO2 Nanoparticles in Photocurable Ladder-like Polysilsesquioxane Nanocomposites. J. Sol-Gel Sci. Technol. 2023, 114, 1–13. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | g of ESO per 1 g of CA | Epoxy:Acid Equivalents | Process Conditions |
|---|---|---|---|
| Stoichiometric conditions | |||
| 1ESO+1CA-h0 | 1.62 | 1:1 | Mixing for 24 h at room T |
| 1ESO+1CA-h1 | 1.62 | 1:1 | Heating for 1 h at 90 °C |
| 1ESO+1CA-h2 | 1.62 | 1:1 | Heating for 6 h at 90 °C |
| 1ESO+1CA-h3 | 1.62 | 1:1 | Heating for 1 h at 90 °C + 2 h at 180 °C |
| 1ESO+1CA-h4 | 1.62 | 1:1 | Heating for 1 h at 90 °C + 3 h 120 °C + 2 h at 180 °C |
| Non-stoichiometric conditions | |||
| 2ESO+1CA-h3 | 3.25 | 2:1 | Heating for 1 h 90 °C + 2 h at 180 °C |
| 3ESO+1CA-h3 | 4.87 | 3:1 | Heating for 1 h 90 °C + 2 h at 180 °C |
| 1ESO+3CA-h3 | 0.54 | 1:3 | Heating for 1 h 90 °C + 2 h at 180 °C |
| Film Label | Composition |
|---|---|
| Y | Yuca starch |
| YG | Yuca starch + glycerol (40% w/w) |
| Y20ESO-CA | Yuca starch + 1ESO+1CA-h3 (20% w/w) |
| Y40ESO-CA | Yuca starch + 1ESO+1CA-h3 (40% w/w) |
| Sample | Residual Nepoxy (±0.02) a | Epoxide Conversion b | Ester Formation c |
|---|---|---|---|
| 1ESO+1CA-h0 | 4.60 | 0% | |
| 1ESO+1CA-h1 | n.a. | 42% d | 19% e |
| 1ESO+1CA-h2 | 1.20 | 73% | 24% |
| 1ESO+1CA-h3 | 0 | 100% | 42% |
| 1ESO+1CA-h4 | 0 | 100% | 27% |
| Sample | Residual Nepoxy a | Epoxide Conversion by 13C-NMR b | Epoxide Conversion by FTIR c | CA Conversion by FTIR d | Ester Formation by 13C-NMR e |
|---|---|---|---|---|---|
| 1ESO+1CA-h3 | 0 | 100% | 99% | 49% | 42% |
| 2ESO+1CA-h3 | 0.28 | 94% | 96% | 47% | 69% |
| 3ESO+1CA-h3 | 2.09 | 54% | 74% | 53% | 53% |
| 1ESO+3CA-h3 | 0 | 100% | 99% | 25% | 14% |
| Sample | T1ρ(H) (ms) |
|---|---|
| Y | 4.6 ± 0.1 |
| YG | 2.2 ± 0.1 |
| Y20ESO-CINNII | 4.5 ± 0.1 |
| Y40ESO-CINNII | 4.3 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalle Vacche, S.; Esposito, L.H.; Bugnotti, D.; Callone, E.; Orsini, S.F.; D’Arienzo, M.; Cipolla, L.; Petroni, S.; Vitale, A.; Bongiovanni, R.; et al. Modification of Epoxidized Soybean Oil for the Preparation of Amorphous, Nonretrogradable, and Hydrophobic Starch Films. Polysaccharides 2025, 6, 40. https://doi.org/10.3390/polysaccharides6020040
Dalle Vacche S, Esposito LH, Bugnotti D, Callone E, Orsini SF, D’Arienzo M, Cipolla L, Petroni S, Vitale A, Bongiovanni R, et al. Modification of Epoxidized Soybean Oil for the Preparation of Amorphous, Nonretrogradable, and Hydrophobic Starch Films. Polysaccharides. 2025; 6(2):40. https://doi.org/10.3390/polysaccharides6020040
Chicago/Turabian StyleDalle Vacche, Sara, Leandro Hernan Esposito, Daniele Bugnotti, Emanuela Callone, Sara Fernanda Orsini, Massimiliano D’Arienzo, Laura Cipolla, Simona Petroni, Alessandra Vitale, Roberta Bongiovanni, and et al. 2025. "Modification of Epoxidized Soybean Oil for the Preparation of Amorphous, Nonretrogradable, and Hydrophobic Starch Films" Polysaccharides 6, no. 2: 40. https://doi.org/10.3390/polysaccharides6020040
APA StyleDalle Vacche, S., Esposito, L. H., Bugnotti, D., Callone, E., Orsini, S. F., D’Arienzo, M., Cipolla, L., Petroni, S., Vitale, A., Bongiovanni, R., & Dirè, S. (2025). Modification of Epoxidized Soybean Oil for the Preparation of Amorphous, Nonretrogradable, and Hydrophobic Starch Films. Polysaccharides, 6(2), 40. https://doi.org/10.3390/polysaccharides6020040