
The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming?

 , and
, and 
{kind=link}
{kind=link}
Abstract
1. Introduction
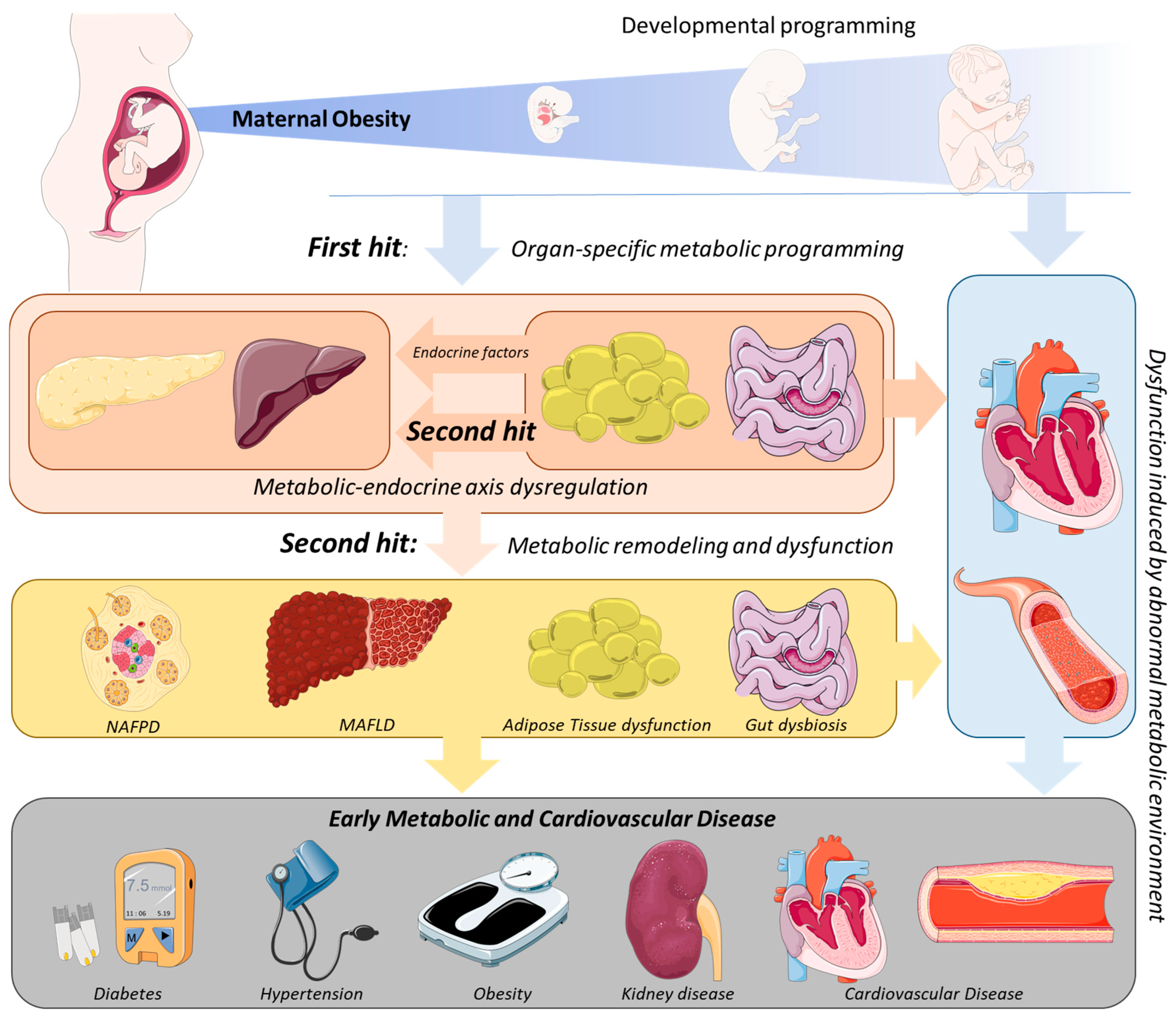
2. Mechanistic Links between MO and the Development of Metabolic Disease in the Offspring
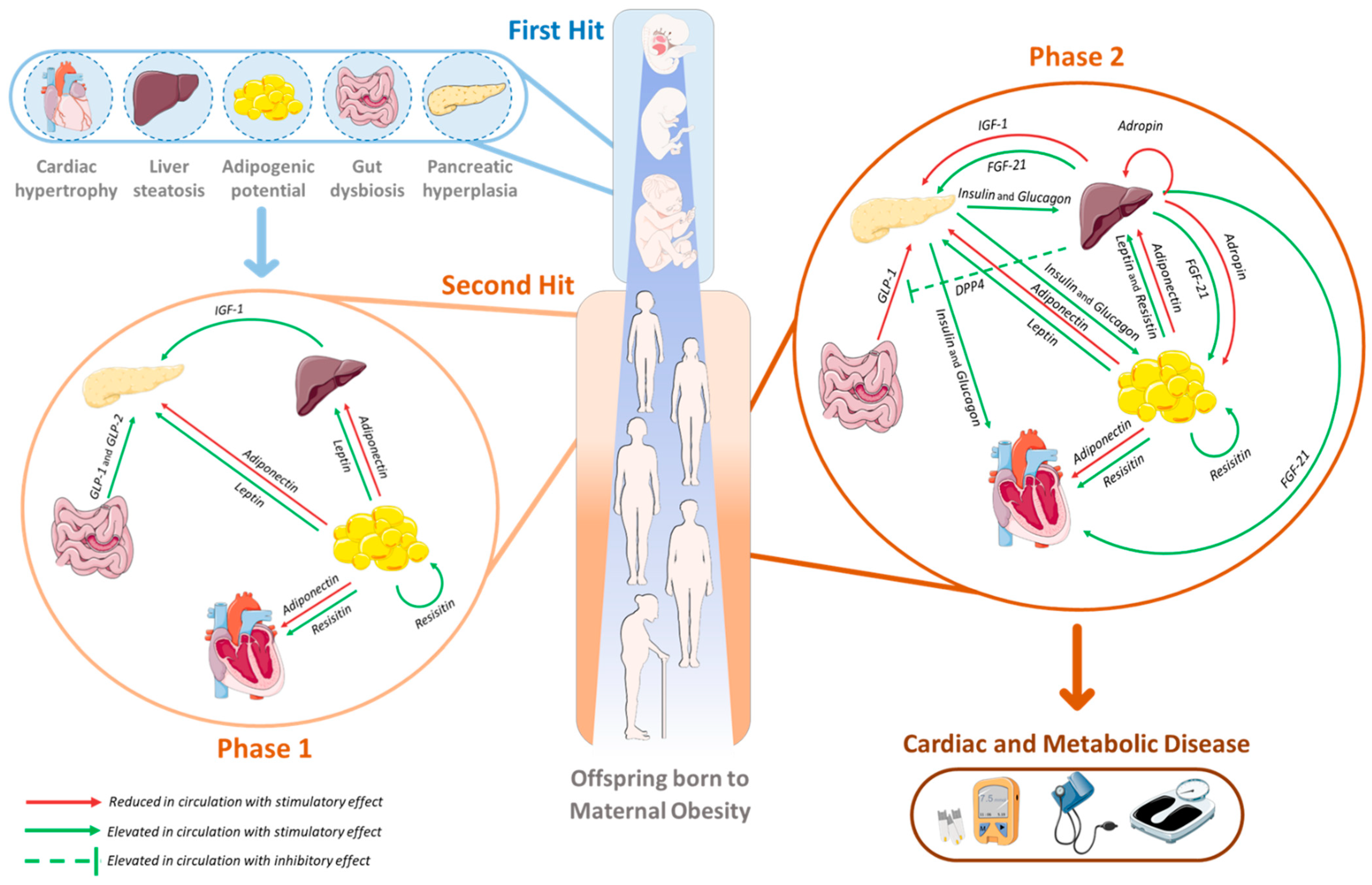
3. The Second Hit of Developmental Programming by MO: The Endocrine–Metabolic Axis
3.1. Adipose Tissue: An Endocrine Organ Involved in MO-Offspring’s Programming
3.2. Impact of MO Programming of Endocrine–Metabolic Axis Dysregulation in Offspring’s Hepatic Disease Development
3.3. The Contribution of the Gut–Pancreas Axis to the Endocrine–Metabolic Imbalance in MO Offspring
3.4. Relation of MO Programmed Metabolic Dysfunction with Cardiovascular Disease Development in the Offspring
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- González-Plaza, E.; Bellart, J.; Martínez-Verdú, M.; Arranz, A.; Luján-Barroso, L.; Seguranyes, G. Prevalencia de sobrepeso y obesidad preconcepcional en mujeres gestantes, y relación con los resultados maternos y perinatales. Enfermería Clínica 2021, 32, S23–S30. [Google Scholar] [CrossRef] [PubMed]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, R.; Felix, J.; Duijts, L.; Jaddoe, V.W. Childhood consequences of maternal obesity and excessive weight gain during pregnancy. Acta Obstet. Gynecol. Scand. 2014, 93, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356, j1. [Google Scholar] [CrossRef]
- Mouzon, S.H.-D.; Lassance, L. Endocrine and metabolic adaptations to pregnancy; impact of obesity. Horm. Mol. Biol. Clin. Investig. 2015, 24, 65–72. [Google Scholar] [CrossRef]
- Kriebs, J.M. Obesity in pregnancy—Addressing risks to improve outcomes. J. Perinat. Neonatal Nurs. 2014, 28, 32–40. [Google Scholar] [CrossRef]
- Bodnar, L.M.; Catov, J.M.; Klebanoff, M.A.; Ness, R.B.; Roberts, J.M. Prepregnancy Body Mass Index and the Occurrence of Severe Hypertensive Disorders of Pregnancy. Epidemiology 2007, 18, 234–239. [Google Scholar] [CrossRef]
- Torloni, M.R.; Betrán, A.P.; Horta, B.L.; Nakamura, M.U.; Atallah, A.N.; Moron, A.F.; Valente, O. Prepregnancy BMI and the risk of gestational diabetes: A systematic review of the literature with meta-analysis: Diagnostic in Obesity and Complications. Obes. Rev. 2009, 10, 194–203. [Google Scholar] [CrossRef]
- Callaghan, W.M.; Chu, S.Y.; Kim, Y.S.; Schmid, C.H.; Lau, J.; England, J.L.; Dietz, M.P. Maternal obesity and risk of gestational. Diabetes Care 2007, 30, 2070–2076. [Google Scholar]
- Solomon, C.G. A prospective study of pregravid determinants of gestational diabetes mellitus. J. Am. Med. Assoc. 1997, 278, 1078. [Google Scholar] [CrossRef]
- Chu, S.Y.; Kim, S.Y.; Schmid, C.H.; Dietz, P.M.; Callaghan, W.M.; Lau, J.; Curtis, K.M. Maternal obesity and risk of cesarean delivery: A meta-analysis. Obes. Rev. 2007, 8, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Nijland, M.; Ford, S.P.; Nathanielsz, P. Prenatal origins of adult disease. Curr. Opin. Obstet. Gynecol. 2008, 20, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2016, 5, 53–64. [Google Scholar] [CrossRef]
- Howell, K.; Powell, T.L. Effects of maternal obesity on placental function and fetal development. Reproduction 2017, 153, R97–R108. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Arah, O.A.; Liew, Z.; Cnattingius, S.; Olsen, J.; Sorensen, H.T.; Qin, G.; Li, J. Maternal diabetes during pregnancy and early onset of cardiovascular disease in offspring: Population based cohort study with 40 years of follow-up. BMJ 2019, 367, l6398. [Google Scholar] [CrossRef]
- Grilo, L.F.; Tocantins, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafome, P.; Pereira, S.P. Metabolic Disease Programming: From Mitochondria to Epigenetics, Glucocorticoid Signalling and Beyond. Eur. J. Clin. Investig. 2021, 51, e13625. [Google Scholar] [CrossRef]
- Yu, Z.; Han, S.; Zhu, J.; Sun, X.; Ji, C.; Guo, X. Pre-Pregnancy Body Mass Index in Relation to Infant Birth Weight and Offspring Overweight/Obesity: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e61627. [Google Scholar] [CrossRef]
- Gaillard, R.; Steegers, E.A.P.; Franco, O.; Hofman, A.; Jaddoe, V.W.V. Maternal weight gain in different periods of pregnancy and childhood cardio-metabolic outcomes. The Generation R Study. Int. J. Obes. 2014, 39, 677–685. [Google Scholar] [CrossRef]
- Mamun, A.A.; O’Callaghan, M.; Callaway, L.; Williams, G.; Najman, J.; Lawlor, D.A. Associations of gestational weight gain with offspring body mass index and blood pressure at 21 years of age: Evidence from a birth cohort study. Circulation 2009, 119, 1720–1727. [Google Scholar] [CrossRef]
- Hochner, H.; Friedlander, Y.; Calderon-Margalit, R.; Meiner, V.; Sagy, Y.; Avgil-Tsadok, M.; Burger, A.; Savitsky, B.; Siscovick, D.S.; Manor, O. Associations of maternal prepregnancy body mass index and gestational weight gain with adult offspring cardiometabolic risk factors: The Jerusalem Perinatal Family Follow-up Study. Circulation 2012, 125, 1381–1389. [Google Scholar] [CrossRef]
- Eriksson, J.; Sandboge, S.; Salonen, M.; Kajantie, E.; Osmond, C. Maternal weight in pregnancy and offspring body composition in late adulthood: Findings from the Helsinki Birth Cohort Study (HBCS). Ann. Med. 2015, 47, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Andres, A.; Hull, H.R.; Shankar, K.; Casey, P.H.; Cleves, M.A.; Badger, T.M. Longitudinal body composition of children born to mothers with normal weight, overweight, and obesity. Obesity 2015, 23, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Bronson, S.C.; Seshiah, V. Transgenerational transmission of non-communicable diseases: How to break the vicious cycle? Cureus 2021, 13, 18754. [Google Scholar] [CrossRef] [PubMed]
- Menting, M.D.; Mintjens, S.; van de Beek, C.; Frick, C.J.; Ozanne, S.E.; Limpens, J.; Roseboom, T.J.; Hooijmans, C.R.; van Deutekom, A.W.; Painter, R.C. Maternal obesity in pregnancy impacts offspring cardiometabolic health: Systematic review and meta-analysis of animal studies. Obes. Rev. 2019, 20, 675–685. [Google Scholar] [CrossRef]
- Nunes, F.; Frantz, E.D.C.; Lannes, W.R.; Menezes, M.C.D.S.; Mandarim-De-Lacerda, C.; Souza-Mello, V. Pregestational maternal obesity impairs endocrine pancreas in male F1 and F2 progeny. Nutrition 2015, 31, 380–387. [Google Scholar] [CrossRef]
- Turdi, S.; Ge, W.; Hu, N.; Bradley, K.M.; Wang, X.; Ren, J. Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. J. Mol. Cell. Cardiol. 2012, 55, 117–129. [Google Scholar] [CrossRef]
- Rivera, H.M.; Kievit, P.; Kirigiti, M.A.; Bauman, L.A.; Baquero, K.; Blundell, P.; Dean, T.A.; Valleau, J.C.; Takahashi, D.L.; Frazee, T.; et al. Maternal high-fat diet and obesity impact palatable food intake and dopamine signaling in nonhuman primate offspring. Obesity 2015, 23, 2157–2164. [Google Scholar] [CrossRef]
- Rajia, S.; Chen, H.; Morris, M.J. Maternal overnutrition impacts offspring adiposity and brain appetite markers-modulation by postweaning diet. J. Neuroendocr. 2010, 22, 905–914. [Google Scholar] [CrossRef]
- Long, N.M.; George, L.A.; Uthlaut, A.B.; Smith, D.T.; Nijland, M.; Nathanielsz, P.; Ford, S.P. Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring1. J. Anim. Sci. 2010, 88, 3546–3553. [Google Scholar] [CrossRef]
- Chen, H.; Simar, D.; Morris, M.J. Maternal obesity impairs brain glucose metabolism and neural response to hyperglycemia in male rat offspring. J. Neurochem. 2013, 129, 297–303. [Google Scholar] [CrossRef]
- McCurdy, C.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Investig. 2009, 119, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Hirschmugl, B.; Perazzolo, S.; Sengers, B.G.; Lewis, R.M.; Gruber, M.; Desoye, G.; Wadsack, C. Placental mobilization of free fatty acids contributes to altered materno-fetal transfer in obesity. Int. J. Obes. 2021, 45, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Hay, W.W. Placental-fetal glucose exchange and fetal glucose metabolism. Trans. Am. Clin. Clim. Assoc. 2006, 117, 321–340. [Google Scholar]
- Martin-Gronert, M.S.; Fernandez-Twinn, D.S.; Poston, L.; Ozanne, S.E. Altered hepatic insulin signalling in male offspring of obese mice. J. Dev. Orig. Heal. Dis. 2010, 1, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Tocantins, C.; Diniz, M.S.; Grilo, L.F.; Pereira, S.P. The birth of cardiac disease: Mechanisms linking gestational diabetes mellitus and early onset of cardiovascular disease in offspring. WIREs Mech. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.D.; Sabey, K.H.; Knutson, A.J.; Gandy, T.C.T.; Louwagie, E.J.; Lauterboeck, L.; Mdaki, K.S.; Baack, M.L. Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. Int. J. Mol. Sci. 2019, 20, 3090. [Google Scholar] [CrossRef]
- Mdaki, K.S.; Larsen, T.D.; Wachal, A.L.; Schimelpfenig, M.D.; Weaver, L.J.; Dooyema, S.D.R.; Louwagie, E.J.; Baack, M.L. Maternal high-fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am. J. Physiol. Circ. Physiol. 2016, 310, H681–H692. [Google Scholar] [CrossRef]
- Yokomizo, H.; Inoguchi, T.; Sonoda, N.; Sakaki, Y.; Maeda, Y.; Inoue, T.; Hirata, E.; Takei, R.; Ikeda, N.; Fujii, M.; et al. Maternal high-fat diet induces insulin resistance and deterioration of pancreatic β-cell function in adult offspring with sex differences in mice. Am. J. Physiol. Metab. 2014, 306, E1163–E1175. [Google Scholar] [CrossRef]
- Nicholas, L.M.; Rattanatray, L.; MacLaughlin, S.M.; Ozanne, S.E.; Kleemann, D.O.; Walker, S.K.; Morrison, J.L.; Zhang, S.; Muhlhäusler, B.S.; Martin-Gronert, M.S.; et al. Differential effects of maternal obesity and weight loss in the periconceptional period on the epigenetic regulation of hepatic insulin-signaling pathways in the offspring. FASEB J. 2013, 27, 3786–3796. [Google Scholar] [CrossRef]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.S.; Alfaradhi, M.Z.; Martin-Gronert, M.S.; Duque-Guimaraes, D.E.; Piekarz, A.; Ferland-McCollough, D.; Bushell, M.; Ozanne, S.E. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol. Metab. 2014, 3, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Borengasser, S.J.; Zhong, Y.; Kang, P.; Lindsey, F.; Ronis, M.J.J.; Badger, T.M.; Gomez-Acevedo, H.; Shankar, K. Maternal Obesity Enhances White Adipose Tissue Differentiation and Alters Genome-Scale DNA Methylation in Male Rat Offspring. Endocrinology 2013, 154, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Hiramatsu, Y. Effects of a High-Fat Diet Exposure in UTERO on the Metabolic Syndrome-Like Phenomenon in Mouse Offspring through Epigenetic Changes in Adipocytokine Gene Expression. Endocrinology 2012, 153, 2823–2830. [Google Scholar] [CrossRef]
- Zambrano, E.; Ibáñez, C.; Martínez-Samayoa, P.M.; Lomas, C.; Durand-Carbajal, M.; González, G.L.R. Maternal Obesity: Lifelong Metabolic Outcomes for Offspring from Poor Developmental Trajectories During the Perinatal Period. Arch. Med. Res. 2016, 47, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Ross, M.G. Maternal-infant nutrition and development programming of offspring appetite and obesity. Nutr. Rev. 2020, 78, 25–31. [Google Scholar] [CrossRef]
- Morris, M.J.; Chen, H. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int. J. Obes. 2008, 33, 115–122. [Google Scholar] [CrossRef]
- Shrestha, N.; Ezechukwu, H.C.; Holland, O.J.; Hryciw, D.H. Developmental programming of peripheral diseases in offspring exposed to maternal obesity during pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 2020, 319, R507–R516. [Google Scholar] [CrossRef]
- Torell, F.; Bennett, K.; Cereghini, S.; Rännar, S.; Lundstedt-Enkel, K.; Moritz, T.; Haumaitre, C.; Trygg, J.; Lundstedt, T. Multi-Organ Contribution to the Metabolic Plasma Profile Using Hierarchical Modelling. PLoS ONE 2015, 10, e0129260. [Google Scholar] [CrossRef]
- Ito, M.; Adachi-Akahane, S. Inter-organ Communication in the Regulation of Lipid Metabolism: Focusing on the Network Between the Liver, Intestine, and Heart. J. Pharmacol. Sci. 2013, 123, 312–317. [Google Scholar] [CrossRef]
- Jacome-Sosa, M.; Parks, E.J.; Bruno, R.S.; Tasali, E.; Lewis, G.F.; O Schneeman, B.; Rains, T.M. Postprandial Metabolism of Macronutrients and Cardiometabolic Risk: Recent Developments, Emerging Concepts, and Future Directions12. Adv. Nutr. Int. Rev. J. 2016, 7, 364–374. [Google Scholar] [CrossRef]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M. The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef] [PubMed]
- Nogues, P.; Santos, E.D.; Jammes, H.; Berveiller, P.; Arnould, L.; Vialard, F.; Dieudonné, M. Maternal obesity influences expression and DNA methylation of the adiponectin and leptin systems in human third-trimester placenta. Clin. Epigen. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kelstrup, L.; Clausen, T.D.; Mathiesen, E.R.; Hansen, T.; Holst, J.J.; Damm, P. Incretin and Glucagon Levels in Adult Offspring Exposed to Maternal Diabetes in Pregnancy. J. Clin. Endocrinol. Metab. 2015, 100, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Kim, J.-S.; Jo, M.-J.; Cho, E.; Ahn, S.-Y.; Kwon, Y.-J.; Ko, G.-J. The Roles and Associated Mechanisms of Adipokines in Development of Metabolic Syndrome. Molecules 2022, 27, 334. [Google Scholar] [CrossRef]
- Lustig, R.H.; Collier, D.; Kassotis, C.; Roepke, T.A.; Kim, M.J.; Blanc, E.; Barouki, R.; Bansal, A.; Cave, M.C.; Chatterjee, S.; et al. Obesity I: Overview and molecular and biochemical mechanisms. Biochem. Pharmacol. 2022, 199, 115012. [Google Scholar] [CrossRef]
- Janochova, K.; Haluzik, M.; Buzga, M. Visceral fat and insulin resistance—What we know? Biomed. Pap. 2019, 163, 19–27. [Google Scholar] [CrossRef]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Sayın, O.; Tokgöz, Y.; Arslan, N. Investigation of adropin and leptin levels in pediatric obesity-related nonalcoholic fatty liver disease. J. Pediatr. Endocrinol. Metab. 2014, 27, 479–484. [Google Scholar] [CrossRef]
- Walker, C.D.; Naef, L.; d’Asti, E.; Long, H.; Xu, Z.; Moreau, A.; Azeddine, B. Perinatal maternal fat intake affects metabolism and hippocampal function in the offspring: A potential role for leptin. Ann. N. Y. Acad. Sci. 2008, 1144, 189–202. [Google Scholar] [CrossRef]
- Mingrone, G.; Manco, M.; Mora, M.E.V.; Guidone, C.; Iaconelli, A.; Gniuli, D.; Leccesi, L.; Chiellini, C.; Ghirlanda, G. Influence of Maternal Obesity on Insulin Sensitivity and Secretion in Offspring. Diabetes Care 2008, 31, 1872–1876. [Google Scholar] [CrossRef]
- Kelly, A.C.; Powell, T.L.; Jansson, T. Placental function in maternal obesity. Clin. Sci. 2020, 134, 961–984. [Google Scholar] [CrossRef] [PubMed]
- Vernini, J.M.; Moreli, J.B.; Costa, R.A.A.; Negrato, C.A.; Rudge, M.V.C.; Calderon, I.M.P. Maternal adipokines and insulin as biomarkers of pregnancies complicated by overweight and obesity. Diabetol. Metab. Syndr. 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, A.; Yamauchi, T.; Ito, Y.; Hada, Y.; Maki, T.; Takekawa, S.; Kamon, J.; Kobayashi, M.; Suzuki, R.; Hara, K.; et al. Insulin/Foxo1 pathway regulates expression levels of adiponectin receptors and adiponectin sensitivity. J. Biol. Chem. 2004, 279, 30817–30822. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Ospina, A.; Castaño-Moreno, E.; Munoz-Munoz, E.; Krause, B.J.; Uauy, R.; Casanello, P.; Castro-Rodriguez, J.A. Maternal obesity is associated with higher cord blood adipokines in offspring most notably in females. J. Pediatr. Gastroenterol. Nutr. 2021, 73, 264–270. [Google Scholar] [PubMed]
- Dosch, N.C.; Guslits, E.F.; Weber, M.B.; Murray, S.E.; Ha, B.; Coe, C.L.; Auger, A.P.; Kling, P.J. Maternal Obesity Affects Inflammatory and Iron Indices in Umbilical Cord Blood. Physiol. Behav. 2016, 172, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Machinal-Quélin, F.; Dieudonné, M.N.; Leneveu, M.C.; Pecquery, R.; Giudicelli, Y. Proadipogenic effect of leptin on rat preadipocytes in vitro: Activation of MAPK and STAT3 signaling pathways. Am. J. Physiol. Physiol. 2002, 282, C853–C863. [Google Scholar] [CrossRef]
- Palhinha, L.; Liechocki, S.; Hottz, E.D.; Pereira, J.A.D.S.; De Almeida, C.J.; Vieira, P.; Bozza, P.T.; Maya-Monteiro, C.M. Leptin Induces Proadipogenic and Proinflammatory Signaling in Adipocytes. Front. Endocrinol. 2019, 10, 841. [Google Scholar] [CrossRef]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef]
- Herrera, E.; Lasunción, M.A.; Huerta, L.; Martín-Hidalgo, A. Plasma leptin levels in rat mother and offspring during pregnancy and lactation. Neonatology 2000, 78, 315–320. [Google Scholar] [CrossRef]
- Paul, H.A.; Bomhof, M.R.; Vogel, H.J.; Reimer, R.A. Diet-induced changes in maternal gut microbiota and metabolomic profiles influence programming of offspring obesity risk in rats. Sci. Rep. 2016, 6, 20683. [Google Scholar] [CrossRef]
- Shankar, K.; Kang, P.; Harrell, A.; Zhong, Y.; Marecki, J.C.; Ronis, M.J.J.; Badger, T.M. Maternal Overweight Programs Insulin and Adiponectin Signaling in the Offspring. Endocrinology 2010, 151, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-J.; Lee, D.S.; Kim, W.K.; Han, B.S.; Lee, S.C.; Bae, K.-H. Metabolic Adaptation in Obesity and Type II Diabetes: Myokines, Adipokines and Hepatokines. Int. J. Mol. Sci. 2016, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Wang, J.; Whiteman, E.L.; Birnbaum, M.J.; Lazar, M.A. Activation of SOCS-3 by Resistin. Mol. Cell. Biol. 2005, 25, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Visfatin induces inflammation and insulin resistance via the NF-κB and STAT3 signaling pathways in hepatocytes. J. Diabetes Res. 2019, 2019, 4021623. [Google Scholar] [CrossRef] [PubMed]
- Simões, N.F.; Domingos, A.L.G.; Oliveira, F.L.P.D.; Caldas, I.S.; Guedes, M.R.; Fajardo, V.C.; Freitas, S.N.D. Resistin and visfatin concentrations are related to central obesity and inflammation in Brazilian children. Nutrire 2018, 43, 7. [Google Scholar] [CrossRef]
- Nourbakhsh, M.; Nourbakhsh, M.; Gholinejad, Z.; Razzaghy-Azar, M. Visfatin in obese children and adolescents and its association with insulin resistance and metabolic syndrome. Scand. J. Clin. Lab. Investig. 2015, 75, 183–188. [Google Scholar] [CrossRef]
- Tie, H.-T.; Xia, Y.-Y.; Zeng, Y.-S.; Zhang, Y.; Dai, C.-L.; Guo, J.J.; Zhao, Y. Risk of childhood overweight or obesity associated with excessive weight gain during pregnancy: A meta-analysis. Arch. Gynecol. Obstet. 2013, 289, 247–257. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.S.; Hjort, L.; Novakovic, B.; Ozanne, S.E.; Saffery, R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia 2019, 62, 1789–1801. [Google Scholar] [CrossRef]
- Perng, W.; Gillman, M.W.; Mantzoros, C.S.; Oken, E. A prospective study of maternal prenatal weight and offspring cardiometabolic health in mid-childhood. Ann. Epidemiol. 2014, 24, 793. [Google Scholar] [CrossRef]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Yki–Järvinen, H. Increased Liver Fat, Impaired Insulin Clearance, and Hepatic and Adipose Tissue Insulin Resistance in Type 2 Diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef]
- Shou, J.; Chen, P.-J.; Xiao, W.-H. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol. Metab. Syndr. 2020, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, M.; Mitchell, S.J.; Wu, L.E.; White, M.; Cordwell, S.; Mach, J.; Solon-Biet, S.; Boyer, D.; Nines, D.; Das, A.; et al. Ultrastructure of the liver microcirculation influences hepatic and systemic insulin activity and provides a mechanism for age-related insulin resistance. Aging Cell 2016, 15, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, J.-W.; Targher, G. Links between metabolic syndrome and metabolic dysfunction-associated fatty liver disease. Trends Endocrinol. Metab. 2021, 32, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Heal. 2019, 16, 3415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Niu, Y.; Gu, H.; Lu, S.; Zhang, W.; Li, X.; Yang, Z.; Qin, L.; Su, Q. Low serum adiponectin is a predictor of progressing to nonalcoholic fatty liver disease. J. Clin. Lab. Anal. 2018, 33, e22709. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Bril, F.; Barb, D.; Portillo-Sanchez, P.; Biernacki, D.; Lomonaco, R.; Suman, A.; Weber, M.H.; Budd, J.T.; Lupi, M.E.; Cusi, K. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology 2016, 65, 1132–1144. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef]
- Long, W.; Hui Ju, Z.; Fan, Z.; Jing, W.; Qiong, L. The effect of recombinant adeno-associated virus-adiponectin (rAAV2/1-Acrp30) on glycolipid dysmetabolism and liver morphology in diabetic rats. Gen. Comp. Endocrinol. 2014, 206, 1–7. [Google Scholar] [CrossRef]
- Jung, T.W.; Youn, B.-S.; Choi, H.Y.; Lee, S.Y.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Kim, B.-H.; Baik, S.H.; Choi, K.M. Salsalate and adiponectin ameliorate hepatic steatosis by inhibition of the hepatokine fetuin-A. Biochem. Pharmacol. 2013, 86, 960–969. [Google Scholar] [CrossRef]
- Buckley, A.J.; Keserü, B.; Briody, J.; Thompson, M.; Ozanne, S.; Thompson, C.H. Altered body composition and metabolism in the male offspring of high fat–fed rats. Metabolism 2005, 54, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Marques, V.; Afonso, M.B.; Bierig, N.; Duarte-Ramos, F.; Santos-Laso, A.; Jimenez-Agüero, R.; Eizaguirre, E.; Bujanda, L.; Pareja, M.J.; Luís, R.; et al. Adiponectin, Leptin, and IGF-1 Are Useful Diagnostic and Stratification Biomarkers of NAFLD. Front. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, S.; Cui, C.-J.; Zhang, Y.; Yang, S.-H.; Li, J.-J. Leptin decreases the expression of low-density lipoprotein receptor via PCSK9 pathway: Linking dyslipidemia with obesity. J. Transl. Med. 2016, 14, 276. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.G.; Trevaskis, J.L.; Lam, D.D.; Sutton, G.M.; Koza, R.A.; Chouljenko, V.N.; Kousoulas, K.G.; Rogers, P.M.; Kesterson, R.A.; Thearle, M.; et al. Identification of Adropin as a Secreted Factor Linking Dietary Macronutrient Intake with Energy Homeostasis and Lipid Metabolism. Cell Metab. 2008, 8, 468–481. [Google Scholar] [CrossRef]
- Gao, S.; McMillan, R.P.; Zhu, Q.; Lopaschuk, G.D.; Hulver, M.W.; Butler, A.A. Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol. Metab. 2015, 4, 310–324. [Google Scholar] [CrossRef]
- Stevens, J.R.; Kearney, M.L.; St-Onge, M.P.; Stanhope, K.L.; Havel, P.J.; Kanaley, J.A.; Thyfault, J.P.; Weiss, E.P.; Butler, A.A. Inverse association between carbohydrate consumption and plasma adropin concentrations in humans. Obesity 2016, 24, 1731–1740. [Google Scholar] [CrossRef]
- Butler, A.A.; Tam, C.S.; Stanhope, K.L.; Wolfe, B.M.; Ali, M.R.; O’Keeffe, M.; St-Onge, M.-P.; Ravussin, E.; Havel, P.J. Low Circulating Adropin Concentrations with Obesity and Aging Correlate with Risk Factors for Metabolic Disease and Increase after Gastric Bypass Surgery in Humans. J. Clin. Endocrinol. Metab. 2012, 97, 3783–3791. [Google Scholar] [CrossRef]
- Aydin, S.; Kuloglu, T.; Aydin, S. Copeptin, adropin and irisin concentrations in breast milk and plasma of healthy women and those with gestational diabetes mellitus. Peptides 2013, 47, 66–70. [Google Scholar] [CrossRef]
- Dugas, C.; Perron, J.; Kearney, M.; Mercier, R.; Tchernof, A.; Marc, I.; Weisnagel, S.J.; Robitaille, J. Postnatal Prevention of Childhood Obesity in Offspring Prenatally Exposed to Gestational Diabetes mellitus: Where Are We Now. Obes. Facts 2017, 10, 396–406. [Google Scholar] [CrossRef]
- Wang, W.-J.; Wang, S.; Yang, M.-N.; Dong, Y.; He, H.; Fang, F.; Huang, R.; Yu, X.-G.; Zhang, G.-H.; Zhao, X.; et al. Fetuin-A in Infants Born Small- or Large-for-Gestational-Age. Front. Endocrinol. 2020, 11, 567955. [Google Scholar] [CrossRef]
- Peter, A.; Kovarova, M.; Staiger, H.; Machann, J.; Schick, F.; Königsrainer, A.; Königsrainer, I.; Schleicher, E.; Fritsche, A.; Häring, H.-U.; et al. The hepatokines fetuin-A and fetuin-B are upregulated in the state of hepatic steatosis and may differently impact on glucose homeostasis in humans. Am. J. Physiol. Metab. 2018, 314, E266–E273. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-J.; Zhang, L.; Zheng, T.; Zhang, G.-H.; Du, K.; Yang, M.-N.; He, H.; Wang, S.; Wang, W.; Zhang, J.; et al. Fetuin-A and fetal growth in gestational diabetes mellitus. BMJ Open Diabetes Res. Care 2020, 8, e000864. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fang, Q.; Gao, F.; Fan, J.; Zhou, J.; Wang, X.; Zhang, H.; Pan, X.; Bao, Y.; Xiang, K.; et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J. Hepatol. 2010, 53, 934–940. [Google Scholar] [CrossRef]
- Kampmann, F.B.; Thuesen, A.C.B.; Hjort, L.; A Bjerregaard, A.; E Chavarro, J.; Frystyk, J.; Bjerre, M.; Tetens, I.; Olsen, S.; Vaag, A.A.; et al. Increased leptin, decreased adiponectin and FGF21 concentrations in adolescent offspring of women with gestational diabetes. Eur. J. Endocrinol. 2019, 181, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Moeckli, B.; Delaune, V.; Prados, J.; Tihy, M.; Peloso, A.; Oldani, G.; Delmi, T.; Slits, F.; Gex, Q.; Rubbia-Brandt, L.; et al. Impact of Maternal Obesity on Liver Disease in the Offspring: A Comprehensive Transcriptomic Analysis and Confirmation of Results in a Murine Model. Biomedicines 2022, 10, 294. [Google Scholar] [CrossRef]
- Kandzija, N.; Zhang, W.; Motta-Mejia, C.; Mhlomi, V.; McGowan-Downey, J.; James, T.; Cerdeira, A.S.; Tannetta, D.; Sargent, I.; Redman, C.W.; et al. Placental extracellular vesicles express active dipeptidyl peptidase IV; levels are increased in gestational diabetes mellitus. J. Extracell. Vesicles 2019, 8, 1617000. [Google Scholar] [CrossRef] [PubMed]
- Montaniel, K.R.C.; Bucher, M.; Maloyan, A. The role of inflammation and dipeptidyl peptidase IV in the developmental programming of obesity and insulin resistance. Circulation 2018, 138, A15395. [Google Scholar]
- Baumeier, C.; Schlüter, L.; Saussenthaler, S.; Laeger, T.; Rödiger, M.; Alaze, S.A.; Fritsche, L.; Häring, H.-U.; Stefan, N.; Fritsche, A.; et al. Elevated hepatic DPP4 activity promotes insulin resistance and non-alcoholic fatty liver disease. Mol. Metab. 2017, 6, 1254–1263. [Google Scholar] [CrossRef]
- Zhou, L.; Xiao, X. The role of gut microbiota in the effects of maternal obesity during pregnancy on offspring metabolism. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Gérard, P. Gut microbiota and obesity. Cell. Mol. Life Sci. 2016, 73, 147–162. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host Remodeling of the Gut Microbiome and Metabolic Changes during Pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, E.; Marín, M.L.; Martín, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernández, L.; Rodríguez, J.M. Is meconium from healthy newborns actually sterile? Res. Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.M.; Panigrahi, P. Is a foetus developing in a sterile environment? Lett. Appl. Microbiol. 2014, 59, 572–579. [Google Scholar] [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef]
- Wankhade, U.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Fernandez-Kim, S.-O.; Townsend, R.L.; Kruger, C.; Carmouche, R.; Newman, S.; Salbaum, J.M.; Berthoud, H.-R. Maternal obese-type gut microbiota differentially impact cognition, anxiety and compulsive behavior in male and female offspring in mice. PLoS ONE 2017, 12, e0175577. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Karlsson, F.; Mardinoglu, A.; Nookaew, I.; Bordel, S.; Nielsen, J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 2013, 3, srep02532. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.M.; Antony, K.M.; Ma, J.; Prince, A.L.; Showalter, L.; Moller, M.; Aagaard, K.M. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 2016, 8, 77. [Google Scholar] [CrossRef]
- Clarke, S.; Murphy, E.F.; O’Sullivan, O.; Ross, R.; O’Toole, P.; Shanahan, F.; Cotter, P.D. Targeting the Microbiota to Address Diet-Induced Obesity: A Time Dependent Challenge. PLoS ONE 2013, 8, e65790. [Google Scholar] [CrossRef]
- Soderborg, T.K.; Clark, S.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Patrício, B.; Lioci, G.; Macedo, M.P.; Gastaldelli, A. Gut-Pancreas-Liver Axis as a Target for Treatment of NAFLD/NASH. Int. J. Mol. Sci. 2020, 21, 5820. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Millán, E.; Guillén, C. Multi-Organ Crosstalk with Endocrine Pancreas: A Focus on How Gut Microbiota Shapes Pancreatic Beta-Cells. Biomolecules 2022, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Atanes, P.; Ashik, T.; Persaud, S.J. Obesity-induced changes in human islet G protein-coupled receptor expression: Implications for metabolic regulation. Pharmacol. Ther. 2021, 228, 107928. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Kryukova, Y.N.; Shyng, S.L. Leptin regulates KATPchannel trafficking in pancreatic β-cells by a signaling mechanism involving AMP-activated protein kinase (AMPK) and camp-dependent protein kinase (PKA). J. Biol. Chem. 2013, 288, 34098–34109. [Google Scholar] [CrossRef]
- Okamoto, M.; Ohara-Imaizumi, M.; Kubota, N.; Hashimoto, S.; Eto, K.; Kanno, T.; Kubota, T.; Wakui, M.; Nagai, R.; Noda, M.; et al. Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia 2008, 51, 827–835. [Google Scholar] [CrossRef]
- Ford, S.P.; Zhang, L.; Zhu, M.; Miller, M.M.; Smith, D.T.; Hess, B.W.; Moss, G.E.; Nathanielsz, P.; Nijland, M.J. Maternal obesity accelerates fetal pancreatic β-cell but not α-cell development in sheep: Prenatal consequences. Am. J. Physiol. Integr. Comp. Physiol. 2009, 297, R835–R843. [Google Scholar] [CrossRef]
- Nicholas, L.M.; Nagao, M.; Kusinski, L.C.; Fernandez-Twinn, D.S.; Eliasson, L.; Ozanne, S.E. Exposure to maternal obesity programs sex differences in pancreatic islets of the offspring in mice. Diabetologia 2020, 63, 324. [Google Scholar] [CrossRef]
- Shah, N.; Rocha, J.P.; Bhutiani, N.; Endashaw, O.; Omer, E. Nonalcoholic Fatty Pancreas Disease. Nutr. Clin. Pr. 2019, 34, S49–S56. [Google Scholar] [CrossRef]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2020, 110, 921–937. [Google Scholar] [CrossRef]
- Liu, Y.; Vu, V.; Sweeney, G. Examining the Potential of Developing and Implementing Use of Adiponectin-Targeted Therapeutics for Metabolic and Cardiovascular Diseases. Front. Endocrinol. 2019, 10, 842. [Google Scholar] [CrossRef]
- Poetsch, M.S.; Strano, A.; Guan, K. Role of Leptin in Cardiovascular Diseases. Front. Endocrinol. 2020, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Simoniene, D.; Velickiene, D.; Platukiene, A. Insulin resistance in diabetes mellitus type 1, and its association with cardiovascular disease, sex hormones. Endocr. Abstr. 2018, 56. [Google Scholar] [CrossRef]
- Ng, J.C.M.; Schooling, C.M. Effect of Glucagon on Ischemic Heart Disease and Its Risk Factors: A Mendelian Randomization Study. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Van Bunderen, C.C.; Oosterwerff, M.M.; van Schoor, N.M.; Deeg, D.J.; Lips, P.; Drent, M.L. Serum IGF1 metabolic syndrome, and incident cardiovascular disease in older people: A population-based study. Eur. J. Endocrinol. 2013, 168, 393–401. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zweck, E.; Roden, M. GLP-1 receptor agonists and cardiovascular disease: Drug-specific or class effects? Lancet Diabetes Endocrinol. 2019, 7, 89–90. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Pereira, S.P.; Tavares, L.C.; Duarte, A.I.; Baldeiras, I.; Cunha-Oliveira, T.; Martins, J.D.; Santos, M.S.; Maloyan, A.; Moreno, A.J.; Cox, L.A.; et al. Sex-dependent vulnerability of fetal nonhuman primate cardiac mitochondria to moderate maternal nutrient reduction. Clin. Sci. 2021, 135, 1103–1126. [Google Scholar] [CrossRef]
- Samuelsson, A.M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.; et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, X.; Zhao, J.X.; Zhu, M.J.; McCormick, R.J.; Ford, S.P.; Nathanielsz, P.; Ren, J.; Du, M. Maternal obesity induces fibrosis in fetal myocardium of sheep. Am. J. Physiol. Metab. 2010, 299, E968–E975. [Google Scholar] [CrossRef]
- Ghnenis, A.B.; Odhiambo, J.F.; McCormick, R.J.; Nathanielsz, P.; Ford, S.P. Maternal obesity in the ewe increases cardiac ventricular expression of glucocorticoid receptors, proinflammatory cytokines and fibrosis in adult male offspring. PLoS ONE 2017, 12, e0189977. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, C.; Sun, M.; Maimaiti, R.; Ford, S.P.; Nathanielsz, P.W.; Ren, J.; Guo, W. Maternal obesity impairs fetal cardiomyocyte contractile function in sheep. FASEB J. 2018, 33, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, H.L.; Niu, Y.; Fernandez-Twinn, D.; Tarry-Adkins, J.L.; Giussani, D.; Ozanne, S. Maternal Diet-induced Obesity Programs Cardiovascular Dysfunction in Adult Male Mouse Offspring Independent of Current Body Weight. Endocrinology 2014, 155, 3970–3980. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, J.; Sormunen-Harju, H.; Girchenko, P.V.; Huvinen, E.; Stach-Lempinen, B.; Kajantie, E.; Villa, P.M.; Reynolds, R.M.; Hämäläinen, E.K.; Lahti-Pulkkinen, M.; et al. Longitudinal metabolic profiling of maternal obesity, gestational diabetes, and hypertensive pregnancy disorders. J. Clin. Endocrinol. Metab. 2021, 106, e4372–e4388. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Allan, K.M.; A Raja, E.; Bhattacharya, S.; McNeill, G.; Hannaford, P.C.; Sarwar, N.; Lee, A.J.; Norman, J. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: Follow-up of 1 323 275 person years. BMJ 2013, 347, f4539. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Llorens, S.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Talebi, M.; Shakibaei, M.; Samarghandian, S. An Overview of the Role of Adipokines in Cardiometabolic Diseases. Molecules 2020, 25, 5218. [Google Scholar] [CrossRef]
- Leyva, F.; Anker, S.D.; Egerer, K.; Stevenson, J.C.; Kox, W.J.; Coats, A.S. Hyperleptinaemia in chronic heart failure Relationships with insulin. Eur. Hear. J. 1998, 19, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Rzucidlo, E.M.; Davey, J.C.; Xie, Y.; Liu, R.; Jin, Y.; Stavola, L.; Martin, K.A. Adiponectin in the Heart and Vascular System. Vitam. Horm. 2012, 90, 289–319. [Google Scholar] [CrossRef]
- El-Mottaleb, N.A.A.; Galal, H.M.; El Maghraby, K.M.; Gadallah, A.I. Serum irisin level in myocardial infarction patients with or without heart failure. Can. J. Physiol. Pharmacol. 2019, 97, 932–938. [Google Scholar] [CrossRef]
- Senesi, P.; Luzi, L.; Terruzzi, I. Adipokines, Myokines, and Cardiokines: The Role of Nutritional Interventions. Int. J. Mol. Sci. 2020, 21, 8372. [Google Scholar] [CrossRef]
- Huby, A.C.; Otvos, L.; De Chantemèle, E.J.B. Leptin induces hypertension and endothelial dysfunction via aldosterone-dependent mechanisms in obese female mice. Hypertension 2016, 67, 1020–1028. [Google Scholar] [CrossRef]
- Liao, Y.; Takashima, S.; Maeda, N.; Ouchi, N.; Komamura, K.; Shimomura, I.; Hori, M.; Matsuzawa, Y.; Funahashi, T.; Kitakaze, M. Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovasc. Res. 2005, 67, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Dhandapany, P.S.; Kang, S.; Kashyap, D.K.; Rajagopal, R.; Sundaresan, N.R.; Singh, R.; Thangaraj, K.; Jayaprakash, S.; Manjunath, C.N.; Shenthar, J.; et al. Adiponectin receptor 1 variants contribute to hypertrophic cardiomyopathy that can be reversed by rapamycin. Sci. Adv. 2021, 7, eabb3991. [Google Scholar] [CrossRef]
- Park, M.; Sweeney, G. Direct effects of adipokines on the heart: Focus on adiponectin. Hear. Fail. Rev. 2012, 18, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Qu, S.; Tang, L.-X.; Li, L.-P.; He, D.-F.; Zeng, C.Y.; Wang, W.E. Irisin exerts a therapeutic effect against myocardial infarction via promoting angiogenesis. Acta Pharmacol. Sin. 2019, 40, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, K.; Han, Y.; Zhu, H.; Zhou, X.; Tan, T.; Zeng, J.; Zhang, J.; Liu, Y.; Li, Y.; et al. Irisin Protects Heart Against Ischemia-Reperfusion Injury Through a SOD2-Dependent Mitochondria Mechanism. J. Cardiovasc. Pharmacol. 2018, 72, 259–269. [Google Scholar] [CrossRef] [PubMed]
- González, G.L.R.; Reyes-Castro, L.A.; Bautista, C.J.; Beltrán, A.A.; Ibáñez, C.A.; Vega, C.C.; Lomas-Soria, C.; Castro-Rodríguez, D.C.; Elías-López, A.L.; Nathanielsz, P.W.; et al. Maternal obesity accelerates rat offspring metabolic ageing in a sex-dependent manner. J. Physiol. 2019, 597, 5549–5563. [Google Scholar] [CrossRef]
- Martens, D.S.; Plusquin, M.; Gyselaers, W.; De Vivo, I.; Nawrot, T.S. Maternal pre-pregnancy body mass index and newborn telomere length. BMC Med. 2016, 14, 1–10. [Google Scholar] [CrossRef]
- Kajantie, E.; Osmond, C.; Barker, D.J.P.; Forsén, T.; Phillips, D.I.W.; Eriksson, J. Size at birth as a predictor of mortality in adulthood: A follow-up of 350 000 person-years. Int. J. Epidemiology 2005, 34, 655–663. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grilo, L.F.; Diniz, M.S.; Tocantins, C.; Areia, A.L.; Pereira, S.P. The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities 2022, 2, 236-255. https://doi.org/10.3390/obesities2030019
Grilo LF, Diniz MS, Tocantins C, Areia AL, Pereira SP. The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities. 2022; 2(3):236-255. https://doi.org/10.3390/obesities2030019
Chicago/Turabian StyleGrilo, Luís F., Mariana S. Diniz, Carolina Tocantins, Ana L. Areia, and Susana P. Pereira. 2022. "The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming?" Obesities 2, no. 3: 236-255. https://doi.org/10.3390/obesities2030019
APA StyleGrilo, L. F., Diniz, M. S., Tocantins, C., Areia, A. L., & Pereira, S. P. (2022). The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities, 2(3), 236-255. https://doi.org/10.3390/obesities2030019