Abstract
The design of a 10 cm2 (3.4 cm by 3.4 cm) and a 100 cm2 (10 cm by 10 cm) anion exchange membrane (AEM) water electrolyser cell for hydrogen production are described. The AEM cells are based on a zero-gap configuration where the AEM is sandwiched between the anode and cathode so as to minimise voltage drop between the electrodes. Nonprecious nickel-based metal alloy and metal oxide catalysts were employed. Various experiments were carried out to understand the effects of operating parameters such as current densities, electrolyte concentrations, and testing regimes on the performance of both 10 cm2 and 100 cm2 AEM electrolyser cells. Increasing electrolyte concentration was seen to result in reductions in overpotentials which were proportional to current applied, whilst the use of catalysts improved performance consistently over the range of current densities tested. Extended galvanostatic and intermittent tests were demonstrated on both 10 cm2 and 100 cm2 cells, with higher voltage efficiencies achieved with the use of electrocatalysts. Stability tests in the 100 cm2 AEM electrolyser cell assembled with catalyst-coated electrodes demonstrated that the cell voltages remained stable at 2.03 V and 2.17 V during 72 h operation in 4 M KOH and 1 M KOH electrolyte, respectively, at a current density of 0.3 A cm−2 at 323 K. The inclusion of cycling load tests in testing protocols is emphasized for rational evaluation of cell performance as this was observed to speed up the rate of degradation mechanisms such as membrane degradation.
1. Introduction
Hydrogen is not a natural resource, unlike fuel sources, such as coal or natural gas, and it must be produced and stored for use. Currently, approximately 96% of hydrogen is produced by the reformation of fossil fuels and the remaining 4% is from electrolysis, where hydrogen is generated as a byproduct [1]. These industrial processes for hydrogen production use large amounts of energy and generate CO2 emissions, both of which are unfavourable in the shift towards a low-carbon clean hydrogen supply.
Water electrolysis has garnered much attention since the 1980s as an alternative method of obtaining hydrogen from water [2]. As a result of this, substantial advancements in proton exchange membrane (PEM) electrolysers have elevated its status to that of a commercially available and mature technology [3]. The PEM system’s heavy dependence on precious metal catalysts and costly proton exchange membranes [4,5], however, has encouraged the emergence of anion exchange membrane (AEM) water electrolysers in the last decade or so. In an alkaline environment, the utilisation of cheaper nonprecious metals as catalysts and low-cost anion exchange membranes [6,7] are feasible, giving AEM electrolysers a considerable advantage over PEM electrolysers to be more cost-effective. Together with the falling cost of renewables, AEM water electrolysers enable hydrogen production from renewables to be more economically competitive, thereby supporting the global energy challenge of reaching net-zero emissions by 2070 [8].
In alkaline conditions, the half-cell reactions in an electrolyser cell are as follows:
Hydrogen evolution reaction (HER) at the cathode:
4H2O + 4e− → 2H2 + 4OH− E0 = −0.83 V vs. SHE
Oxygen evolution reaction (OER) at the anode:
4OH− → O2 + 2H2O + 4e− E0 = 0.40 V vs. SHE
The overall cell reaction is:
2H2O → 2H2 + O2 Ecell0 = 1.23 V vs. SHE
The overall reaction has a theoretical cell voltage of 1.23 V at standard temperature and pressure (298 K, 1 atm). Hydrogen and oxygen gas are produced at the cathode and anode, respectively, in a 2:1 molar ratio, as described by Equation (3).
Similar to PEM electrolysers, AEM electrolysers are based on a zero-gap design [9] where the anion exchange membrane is sandwiched between the anode and cathode to ensure that the contribution of cell resistance from both the electrolyte and bubble formation from oxygen evolution and hydrogen evolution is minimised. This is because the voltage drop through the cell is dependent on the interelectrode spacing between electrodes—a shorter interelectrode gap reduces voltage drop through the cell.
Research efforts on the AEM electrolyser largely involve oxygen evolution and hydrogen evolution reaction catalysts and membrane development, as well as cell design and construction [10,11]. These factors contribute to the practical cell voltage described by
where Ecell0 is the theoretical cell voltage, ηa and ηc are overpotentials at the anode and cathode, respectively, I is cell current, and Rcell is cell resistance.
Typically, oxygen evolution, which takes place at the anode, has a significantly greater overpotential, ηa, than ηc of hydrogen evolution at the cathode. However, as both reactions contribute to the overall cell voltage, extensive research has been carried out on both anodic and cathodic electrocatalysts [12,13,14,15] that assist in reducing ηa and ηc, respectively. Improvements in physicochemical and mechanical properties of AEM, including OH− ionic conductivity, alkali stability, water uptake rate and swelling ratio, permeability to gas diffusion, and membrane thickness [6,16], can also help to reduce IRmem. Furthermore, a suitable electrolyte flow system, which encourages an even flow distribution and minimises pressure drop through the cell, is crucial for the cell activity, long-term stability, and successful scale-up [17,18].
In general, the design of electrochemical cells requires the consideration of certain criteria for reliable and economically viable operation [19], such as:
- Selection of appropriate materials for cell components (e.g., corrosion resistant, low-cost, good mechanical and chemical stability, selectivity etc.).
- Secure seals to avoid electrolyte leakages, which has an impact on device lifetime.
- The distribution of current and potential, as well as mass transport of reactants and products, which influence the cell performance as current density is proportional to the rate of reaction.
- Modularity of the design which facilitates scale-up.
A comprehensive study on the development of a lab-scale alkaline water electrolysis system consisting of five subsystems: electrolysis cell, H2 and O2 separation and analysis, liquid electrolyte circulation, power supply, and data acquisition, was carried out by Ju et al. [20]. The cell was assembled with circular (⌀ 5 cm) Ni-plate electrodes at both the anode and cathode (approximately 20 cm2 active area) and a Zirfon separator. It was found that increasing temperature and electrolyte concentration improved voltage efficiency, and operating at higher pressures reduced electrical resistance due to smaller bubble sizes. Start-up shut-down cycling of the cell was also observed to have an impact on hydrogen purity. Hnat et al. [21] fabricated an alkaline zero-gap electrolyser stack of three repeating unit cells. Ni foam (5 cm by 5 cm) was used at both the anode and cathode without additional catalyst coating together with either commercial membrane, Ralex® (Czech Republic), or an experimental anion-selective low-density polyethylene (LDPE) membrane. The influence of flow rate, electrode feed arrangement, and electrolyte concentration on hydrogen purity was explored in the stack. A mathematical model was then applied to identify the limitations of the cell design to facilitate scale-up of future stacks.
The design of a modular laboratory scale AEM cell that can be arranged in a stack for scale-up was reported by Rearden et al. [22]. They employed stainless-steel electrodes (36 cm2 active area) and a Zirfon membrane in this design. The cell width was optimised from 12 mm in the initial design to 2 mm, which resulted in a reduction in cell resistance, and manifolds were incorporated into the cell wall as they were found to assist with removal of product gases. More recently, Faqeeh and Symes [23] constructed and optimized an electrolyser test cell (13 cm2 active area) as a standardized example for evaluating electrocatalysts. They tested the performance of stainless-steel fibre paper at the anode and Pt/C at the cathode in combination with commercial membranes Fumasep FAA-3-50 and Sustainion X37-50, reporting 1.4 A cm−2 and 2.74 A cm−2, respectively at 2.0 V in 1 M KOH, 60 °C. The effect of using different gas diffusion layers and membranes on performance was investigated with polarisation curves, chronopotentiometric holds, and electrochemical impedance spectroscopy.
In these studies, scale-up is often accomplished by creating a cell stack of unit cells; however, scale-up can also be achieved by increasing the electrode active area. An important condition for scaling-up of electrochemical devices is maintaining electrical similarity, such as keeping a constant interelectrode gap [24]. Other important factors to consider are kinematic similarity, which relates to the gas or liquid flows in the system, and thermal similarity, which refers to the temperature profiles in corresponding parts of the system. In this paper, we describe the design and scale-up of AEM cells from an active electrode area of 10 cm2 to 100 cm2. The electrolyser cells were tested under different operational parameters, including current densities, electrolyte concentrations, and testing regimes, as part of an investigation of the influential factors of AEM water electrolyser performance. Both 10 cm2 and 100 cm2 electrolyser cells were designed with a flow-by electrolyte pathway, enabling the use of thinner mesh electrodes.
2. Experimental
2.1. Catalyst Preparation
Nickel (II) sulphate hexahydrate (Sigma Aldrich (St. Louis, MO, USA), ACS ≥ 98.0%), iron (II) sulphate heptahydrate (Sigma Aldrich, ≥99%), cobalt (II) sulphate heptahydrate (Sigma Aldrich, ≥99%), sodium molybdate dihydrate (ACS reagent ≥99%), sodium citrate dihydrate (Sigma Aldrich, ≥99%), and ammonium sulphate (ACS ≥99%) were used as received for catalyst synthesis. Both OER and HER catalysts were prepared by electrodeposition, and the experimental procedures of catalyst preparation were described in detail previously [12,25]. In this study, cathodic electrodeposition of OER catalyst, Ni–Fe–Co hydroxide, and HER catalyst, Ni–Mo alloy, was conducted at 295 K under galvanostatic control from their respective aqueous sulphate-based solutions onto expanded Ni mesh (DeXmet, 4Ni 6-050FA). Ni–Fe–Co hydroxide was cathodically deposited at 12 mA cm−2 for 6 h from a solution of 36 mM Ni–Fe–Co transition metal sulphates in a 4:1:1 molar ratio, and 12 mM ammonium sulphate. Ni–Mo alloy was cathodically deposited at 12 mA cm−2 for 22 h from a solution of 11 mM nickel (II) sulphate hexahydrate + 9 mM sodium molybdate dihydrate + 30 mM sodium citrate dihydrate. These OER and HER catalyst-coated mesh electrodes were positioned in the AEM cells at the anode and cathode, respectively.
2.2. 10 cm2 and 100 cm2 AEM Cell Design
Both the 10 cm2 and 100 cm2 AEM cells were designed to allow for variable interelectrode gaps and for interchangeable current collectors with different flow-field patterns. The design also ensured a leak-free operation, and the selection of clear polymethyl methacrylate (PMMA) end-cases allowed in situ observation of the electrode reactions. For the 10 cm2 AEM cell, an area of 3.4 cm by 3.4 cm was localised in the centre of the current collectors to accommodate a 10 cm2 electrode area (Figure 1a), whilst in the 100 cm2 AEM cell, a 10 cm by 10 cm area was localised in the centre of the current collectors to accommodate a 100 cm2 electrode area (Figure 1b).

Figure 1.
Photographs of the (a) 10 cm2 and (b) 100 cm2 cell in operation and the top-down internal view of their respective cell components.
In general, the electrode assembly consisted of an anion exchange membrane (Fumatech, FAA-3-50) with a mesh electrode on either side sandwiched between two stainless-steel current collectors (316 L, 3 mm thick, 2B finish), sealed with a 0.25 mm thick nitrile-coated nylon gasket (RH Nuttall) with electrode areas cut out from the centre. The 10 cm2 and 100 cm2 active areas were controlled by two PMMA end-plates (10 cm2: 100 by 100 mm and 100 cm2: 180 by 180 mm; 2 mm thick) positioned on either side of the electrode assembly. The end-plate was designed to allow electrolyte inlet and outlet fittings (Legris pneumatic fittings, LIQUIfit series; 10 cm2: 1/8”, 6 mm push in, 100 cm2: 1/4”, 8 mm push in) to be inserted in the through-plane direction. For the 10 cm2 cell, the electrode assembly was aligned with eight nylon spacers (Vital Parts) of 10 mm O.D. and 6.2 mm I.D., length 40 mm, which also served as electrical insulators, preventing short circuits within the cell. In the design of the 100 cm2 cell, however, the electrode assembly was located between the end-plates with acetyl washers. The 10 cm2 and 100 cm2 cells were held together by eight and twelve M6 304 stainless-steel screws, respectively, which slotted through the nylon spacers of the 10 cm2 cell and acetyl washers of the 100 cm2 cell. The screws were tightened to a torque of 4 Nm.
Electrolyte was supplied to both the anode and cathode through Marprene tubing (Watson Marlow, 10 cm2: 3.2 mm bore, 1.6 mm wall thickness; 100 cm2: 6.4 mm bore, 1.6 mm wall thickness) from a 3 L electrolyte tank immersed in a bath of heated recirculating fluid (Lauda, Ultra 350). The electrolyte was pumped through the cells at a constant flow rate with the assistance of a peristaltic pump (Watson Marlow, 323S).
2.3. Electrochemical Measurements
Electrochemical measurements of the 10 cm2 cell were carried out by a Biologic potentiostat/galvanostat SP-300 with 10 A booster whilst measurements of the 100 cm2 cell were carried out by a Biologic BCS-815 cycler in parallel mode. EC-lab software was used to collect and analyse the experimental results. Effects of varying operating parameters such as flow-field designs, current density, electrolyte concentration and testing regime on the AEM cell performance were studied using galvanostatic techniques. As the operating temperature is largely dictated by the requirements of the membrane, temperatures at which the membrane was stable in were selected and this parameter was not explored further. I–V plots of the AEM cells were obtained by stepping current densities from 0.01 A cm−2 to 1 A cm−2 in increments of 0.025 A cm−2 up to 0.1 A cm−2 and in increments of 0.05 A cm−2 up to 1 A cm−2. The cells were held at each current density for 5 min to give the cell voltage sufficient time to stabilise, with a rest time of 60 s at open-circuit potential between each current density.
3. Results and Discussion
3.1. Flow Channel Designs
Four flow-field designs, which are commonly employed in water electrolysers, fuel cell, and flow battery technologies, i.e., parallel, single serpentine, double serpentine, and triple serpentine [26], were tested in the 10 cm2 cell, as shown in Figure 2. In the parallel flow channel design, the electrolyte flows across the electrode surface in the same direction through several flow channels, while in the serpentine flow designs, the electrolyte travels in a continuous zigzag path across the electrode surface. Angular bends, each consisting of three straight paths connected by two approximately 90° elbow bends, were adopted in the serpentine channel designs. All flow-field areas were equivalent to the active electrode area, with channels approximately 2 mm wide and 3 mm deep.
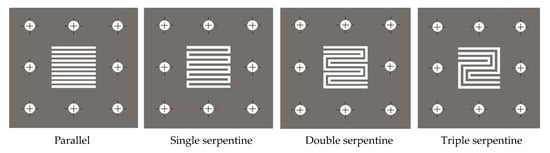
Figure 2.
Illustration of flow-field designs.
The performance of the cell assembled with different flow channel designs (same design at both electrodes) is reported in Figure 3a,b as I–V plots featuring current densities up to 1 A cm−2 and galvanostatic tests carried out at 0.5 A cm−2 and 1 A cm−2, respectively. The flow channel designs can be ranked based on ascending cell voltages from best to worst performance: single serpentine < double serpentine < triple serpentine < parallel. It should be noted that the reported cell performance does not directly correlate to the electrode area exposed to the electrolyte flow (Table S1), which indicates that performance is more reliant on flow channel design, and hence electrolyte flow distribution, across the electrode.
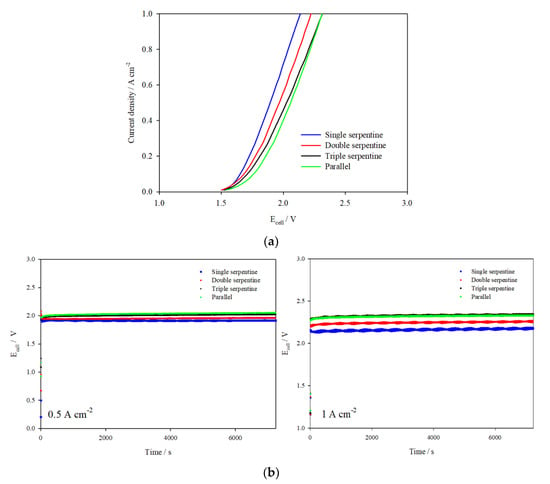
Figure 3.
(a) I–V curves and (b) Ecell vs. time plots for the 10 cm2 AEM cell assembled with various flow-field channels and catalyst-coated electrodes tested in 1 M KOH at 333 K, flow rate 270 mL min−1.
As seen in Figure 3a,b, the electrolyser assembled with parallel flow channels consistently reported the largest cell voltage at low and high current densities, demonstrating poorer cell performance. This can be explained by the flow of electrolyte across the electrode which is only in one direction, making it liable to nonuniform flow distribution and uneven distribution of temperature and pressure [27]. In contrast, the electrolyser assembled with serpentine flow channels reports lower cell voltages, attributing to the continuous flow paths which provide better flow distribution over the same electrode area and thus ensures better management of hydrogen and oxygen bubble production and temperature distribution.
It is apparent that the cell performance decreases when the number of serpentine channels in the flow-field design increases, particularly at higher current densities where overpotential becomes more dependent on mass transfer. As the pump speed used was constant for the experiments, it is likely that the electrolyte mass flow rate through the channels decreases as the electrolyte flow is split across more channels. This was confirmed by the observation of hydrogen gas bubbles accumulating in the double and triple serpentine channel designs during the galvanostatic tests. The removal of bubbles in the single serpentine channel design is therefore more efficient and the effect on performance is especially significant at higher current densities where the rate of reaction, and hence bubble production rate, is faster. As the mass flow rate of electrolyte through the single serpentine flow channel is higher, reactant utilisation and product removal are improved, which results in better cell performance.
Based on the findings, the single serpentine flow design was scaled up for the 100 cm2 electrode area AEM cell. The number of bends in the 100 cm2 serpentine flow design was kept the same as in the 10 cm2 AEM cell; however, the channel width was increased from 2 mm to 6 mm to accommodate the larger area. This was to ensure that the loss coefficient of the flow channel, KL, would remain the same and a similar pressure drop over the larger electrode area could be achieved in the 100 cm2 cell as in the 10 cm2 cell, since pressure drop increases with increasing path length.
3.2. Electrolyte Concentration
Figure 4 shows the I–V plots for the 10 cm2 cell operated in different KOH concentrations from 1 M to 8 M. The experimental results indicate that the cell operated in 4 M KOH delivered the lowest cell voltage for current densities up to 1 A cm−2. This was followed closely by the cell operated in 6 M KOH < 8 M KOH < 2 M KOH < 1 M KOH. One reason for the reduction in cell voltage as KOH concentration increases from 1 M to 4 M is the steady increase in specific KOH conductivity (Figure S2). This is determined from an empirical correlation developed for various molarities of KOH at 333 K: 0.32 S cm−2 for 1 M KOH, 0.58 S cm−2 for 2 M KOH, and 0.92 S cm−2 for 4 M KOH [28].
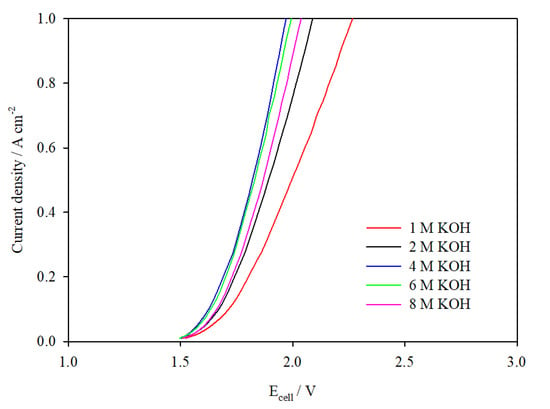
Figure 4.
I–V curves of the 10 cm2 AEM cell assembled with catalyst-coated Ni mesh electrodes operated in electrolytes of various concentrations at 333 K, flow rate 270 mL min−1.
Although the specific conductivities of 6 M and 8 M KOH are higher, the water available for electrolysis is considerably lower, which shifts the chemical equilibrium of Equation (1), slowing down the rate of H2 evolution and increasing the viscosity of the electrolyte solution [29]. This, in combination with the substantial decrease in solubility and diffusivity of both H2 and O2 in the more concentrated electrolyte [30] (Figure S3), hinders the rate of removal of gas bubbles produced, thereby affecting the cell performance, as seen in the I–V plots (Figure 4). Operation of the cell at high KOH concentrations is also not expected to be feasible over extended lengths of time as FAA-3-50 is not recommended to be used at > pH 14 [31]. Equally, it is widely understood that AEM stability is poor in alkaline environments due to nucleophilic substitution by hydroxide ions on the fixed cation charge sites, the loss of which has a negative impact on OH− ion conductivity over time [32,33].
Subsequent experiments in the 100 cm2 cell were carried out in either 1 M or 4 M KOH. The 1 M KOH was selected even though it reported the poorest performance because it is less corrosive and therefore has the least detrimental effect on the cell components. The 4 M KOH was selected for comparison as it yields the best performance over the other KOH concentrations. Figure S4 shows FAA-3-50 as received in dry, bromide form as a clear and flexible sheet (50 µm thick). Pretreatment of the membrane was performed by immersing in 1 M KOH overnight, followed by a 1 h treatment in 1 M KOH or 4 M KOH (depending on test conditions) at 330 K before assembly into the cell. After the first pretreatment step in 1 M KOH, the membrane took on a bright yellow colour, still remaining flexible, and the membrane area increased by approximately 10–20%. Subsequent heat treatment in 1 M KOH resulted in a further expansion of the membrane area by another 10–15%. However, heat treatment in 4 M KOH caused the membrane to become rigid and shrink in area by approximately 10–15% as a result of a reversal of the ion exchange process and loss of hydration in the more concentrated electrolyte. The Ecell vs. time plot given in Figure S5a of the 10 cm2 cell assembled with pretreated membrane and noncatalyst-coated Ni mesh electrodes tested in 0.5 A cm−2, 323 K, indicates that the pretreatment is effective in ensuring that adequate hydroxyl ion exchange takes place.
Following this, the 100 cm2 cell was assembled with noncatalyst-coated Ni mesh electrodes to establish the performance and stability of the cell in 1 M and 4 M KOH without the influence of catalysts. I–V curves in Figure 5a show that the Ecell at 0.5 A cm−2 is reduced by 380 mV when the cell is operated in 4 M KOH versus 1 M KOH. As the reduction in cell voltage increases proportionally with current density, it is clear that the improvement in performance in 4 M KOH is due to the increase in OH− conductivity. To further determine the stability of the cell in these electrolytes, galvanostatic tests were carried out at 0.3 A cm−2 for 72 h in both 1 M and 4 M KOH. Figure 5b illustrates the cell voltage vs. time response, which is stable in both electrolyte concentrations: 2.48 V in 1 M KOH and 2.25 V in 4 M KOH. The I–V curves collected before and after the galvanostatic tests (Figure 5a) show minimal increase in overpotentials, indicating little loss in performance. No visible changes were observed in the membrane, either, when the cells were disassembled, indicating that all cell components were stable under these conditions.
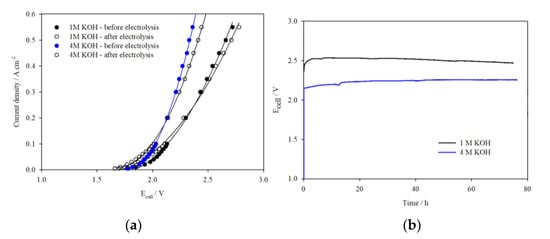
Figure 5.
(a) I–V curves of the 100 cm2 AEM cell assembled with uncoated Ni mesh electrodes tested in 1 M KOH and 4 M KOH before and after galvanostatic test; (b) Ecell vs. time plots at 0.3 A cm−2.
3.3. Catalyst-Coated Electrodes
In this system, where overpotential largely arises from the oxygen evolution and hydrogen evolution reactions, the application of active electrocatalysts is significant in reducing the overall cell voltage, thereby increasing voltage efficiency, which is defined as the thermal neutral voltage divided by the cell operating voltage [34]. Figure 6a,b display the I–V plots for the cell assembled with Ni mesh electrodes with and without catalyst coating. The reduction in Ecell with the application of catalysts over the range of current densities remains consistent between 310–360 mV, increasing only slightly with current density. This trend is observed in both 1 M KOH and 4 M KOH, indicating that the catalysts’ activity and ability to reduce OER and HER overpotentials is unaffected by electrolyte concentration.
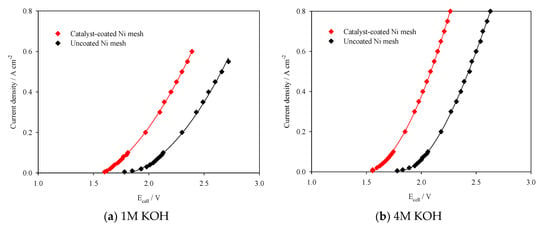
Figure 6.
I–V curves of the 100 cm2 AEM cell assembled with Ni mesh electrodes with and without catalyst coating in (a) 1 M KOH and (b) 4 M KOH.
Galvanostatic tests at 0.3 A cm−2 were carried out to test the catalysts’ stability in 1 M KOH and 4 M KOH over 72 h, as shown in Figure 7a,b. These results are compared to experiments where no catalyst was used. In both electrolyte concentrations, the cell voltage was reasonably stable over the test duration and voltage efficiency improved by 5–7% with the application of catalysts. The I–V curves presented in Figure 7c indicate little change in cell performance before and after electrolysis at 0.3 A cm−2, confirming the stability of the catalysts. Additional galvanostatic tests were carried out in the 100 cm2 cell assembled with catalyst-coated electrodes at 0.5 A cm−2 over 20 h (Figure 8), which deliver steady cell voltages of 2.27 V in 1 M KOH and 2.16 V in 4 M KOH, demonstrating stable performance of the catalysts at higher current densities.
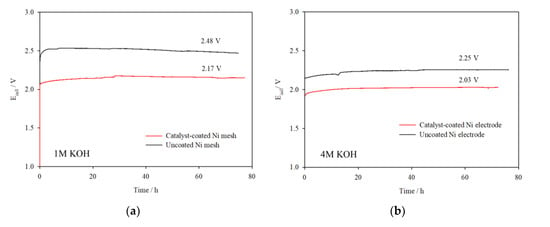
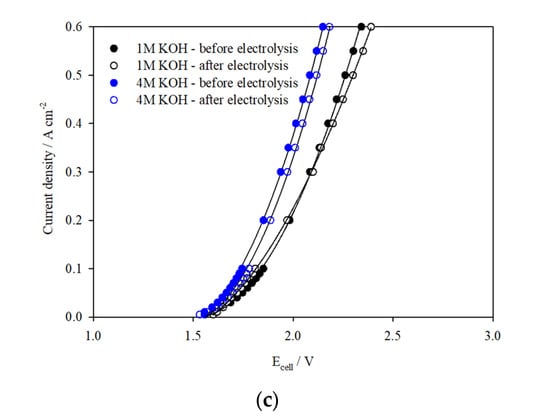
Figure 7.
Ecell vs. time plots for the 100 cm2 AEM cell assembled with uncoated and catalyst-coated Ni mesh electrodes (NiFeCo(OH)2 on Ni (anode) and NiMo alloy on Ni (cathode) operated at 0.3 A cm−2 in (a) 1 M KOH and (b) 4 M KOH, 323 K, flow rate 1100 mL min−1; (c) I–V curves before and after galvanostatic tests.
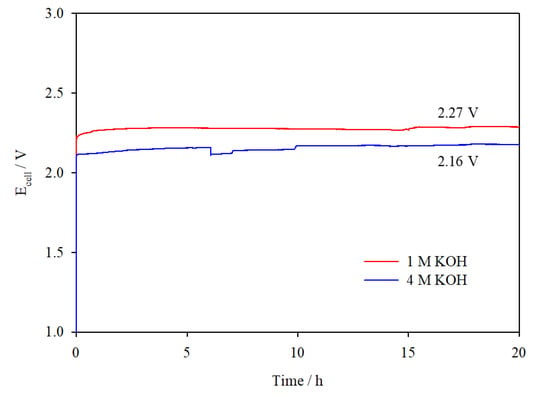
Figure 8.
Ecell vs. time plots for the 100 cm2 AEM cell assembled with catalyst-coated Ni mesh electrodes operated at 0.5 A cm−2 in 1 M and 4 M KOH, 323 K, flow rate 1100 mL min−1.
To examine the effect of the scale-up, Figure S5b displays comparison I–V plots of catalyst-coated Ni mesh electrodes assembled in the 10 cm2 and 100 cm2 cells. The overpotentials in the 100 cm2 cell are greater than in the 10 cm2 cell, as expected, and this increases with current density. Since the catalyst activity was shown to be less affected by electrolyte concentration, the increase in overpotentials in the 100 cm2 cell is suggested to be a result of greater transport and electrical resistances through the cell which become more prominent at large scales, and further optimization of the 100 cm2 cell design is recommended to reduce these resistances.
3.4. Load Cycling
So far in this paper, only galvanostatic experiments have been used to demonstrate cell performance stability. On–off load cycling, which mimics the dynamic nature of renewable energy sources, is equally important in assessing the overall stability as well as determining the potential for coupling the AEM cell with renewables. Tests where the current is varied intermittently with rest periods were therefore carried out with the AEM cells. In the 10 cm2 cell (Figure 9), the cell was held at 0.5 A cm−2 for 2 h, followed by a rest period of 20 min, then at a 1 A cm−2 for 2 h, followed by another 20 min rest period. This sequence was repeated throughout the duration of the test, >40 h.
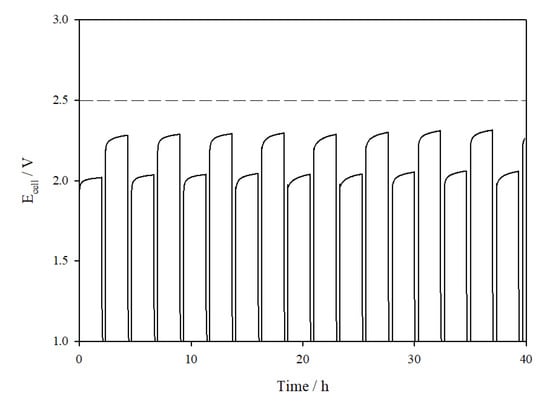
Figure 9.
Ecell vs. time plot for the 10 cm2 AEM cell assembled with catalyst-coated Ni mesh electrodes, operated intermittently at 0.5 A cm−2 and 1 A cm−2 with brief rest periods at 10 µA cm−2 in between.
Figure S6a presents the results of the 100 cm2 cell assembled with noncatalyst-coated electrodes, cycled between 0.1 A cm−2 and 0.3 A cm−2. The cell voltage was initially stable for 40 h (based on repeated tests), indicating the stability of the Ni mesh electrodes. However, continuous cycling led to degradation of the membrane and eventual shorting, as seen from the oscillations in cell voltage. This was confirmed upon disassembly of the cell, from the observation of small holes in the membrane (Figure S6b). The breakdown of the membrane during the intermittent test also appeared to corrode the stainless-steel current collectors, as pitting was seen on the surface where the holes in the membrane had formed. In the following experiment, the cell was assembled with catalyst-coated Ni mesh electrodes and subjected to the same intermittent conditions. The cell showed steady performance throughout the duration of the test (Figure 10a), delivering 160 mV and 180 mV less overpotentials at 0.1 A cm−2 and 0.3 A cm−2, respectively, compared to uncoated mesh electrodes over the first 40 h. There was no evidence of shorting or voltage oscillations in the cell voltage versus time plots; however, the voltage efficiency declined by 1–3%, particularly after the first three cycles. It is evident that the catalyst-coated mesh prolongs the cell performance by reducing the overpotentials at the electrodes, thereby slowing down the rate of membrane degradation. However, the decrease in voltage efficiency suggests that other irreversible modes of degradation are taking place. For instance, black precipitate was visually observed in the electrolyte reservoir after testing, suggesting that the catalyst layer faced mechanical degradation over time. This is seen in the SEM images (Figure S7), where it is clear that the HER catalyst layer is detaching from the electrode support after dynamic load cycling. The precipitate in the electrolyte was collected, filtered, and dried before analysing with EDX to determine elements present. The EDX spectrum presented in Figure S8a of the precipitate collected after galvanostatic hold at 0.5 A cm−2 shows the presence of Ni, Fe, and Co, indicating degradation of the OER catalyst layer. The EDX spectrum (Figure S8b) of the precipitate collected after dynamic load cycling contains Mo on top of Ni, Fe, and Co, signifying degradation of both the OER and HER catalyst layer. The presence of other elements was also seen in the spectrum at very low quantities, such as K from the electrolyte and Cr likely from the stainless-steel current collectors. This agrees well with the visual observation of discolouration of the stainless-steel current collectors on the anode side when the cell was opened for inspection, due to corrosion at >2.0 V [35] which causes degradation of contact between the current collector and catalyst layer.
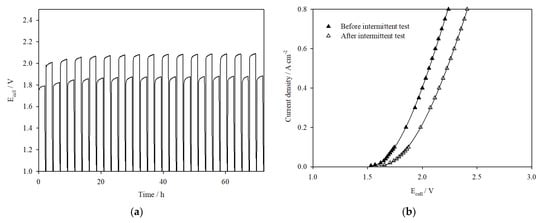
Figure 10.
(a) Ecell vs. time plot for the 100 cm2 AEM cell assembled with catalyst-coated Ni mesh electrodes operated intermittently at 0.1 A cm−2 and 0.3 A cm−2. Operating conditions: 4 M KOH, 323 K, 1100 mL min−1. (b) I–V curves of cell assembled with catalyst-coated Ni mesh electrodes before and after intermittent test in 4 M KOH, 323 K, flow rate 1100 mL min−1.
The voltage versus time plot in Figure 10a also shows that the voltage returns to a lower value at the beginning of each cycle when either 0.1 A cm−2 or 0.3 A cm−2 is applied. This reversible degradation is suggested to be related to ohmic resistance due to mass transport within the cell [36], such as the gradual build-up or entrapment of oxygen or hydrogen bubbles in the cell during operation which brings about an increase in resistance over time. The pump is left on during the rest periods so the recirculating electrolyte removes any hydrogen or oxygen trapped at the electrodes, which “resets” the cell voltage at the beginning of each current cycle. The I–V plots in Figure 10b display the cell performance over a range of current densities before and after intermittent cycling. The cell performance is observed to degrade more markedly after the intermittent test than the continuous galvanostatic test, with a consistent increase in overpotential at all current densities, even though the testing durations of the intermittent experiment and the galvanostatic experiment are the same. It is likely that the fluctuations in operating conditions during the on–off cycling regime speed up the rate of irreversible degradation of the cell components and should be further investigated in future work.
4. Conclusions
We demonstrated that extended galvanostatic and intermittent tests are possible on both the 10 cm2 and 100 cm2 in-house-designed AEM cells, with higher voltage efficiencies achieved with the use of electrocatalysts. Results from the galvanostatic tests at 0.5 A cm−2 in the 100 cm2 cell assembled with catalyst-coated electrodes showed steady cell voltages of 2.27 V in 1 M KOH and 2.16 V in 4 M KOH over 20 h.
From investigating the effects of varying electrolyte concentrations on the performance of the 100 cm2 cell, it was found that increasing the electrolyte concentrations, and therefore the conductivity, resulted in a reduction in overpotential which was proportional to current, whilst with the application of catalysts, the reduction in overpotential remained consistent between 310–360 mV and was unaffected by electrolyte concentrations. Although increasing the electrolyte concentrations does assist in reducing overpotentials, this is only significant at high current densities. Conversely, the application of catalysts improves performance consistently over a range of current densities.
The selection and preparation of a suitable AEM is crucial to extending cell life since the operating conditions of the system, such as electrolyte concentrations and temperatures, are limited by the membrane requirements. For long-term stability tests, the FAA-3-50 membrane is only stable in pH 14 at a maximum of 323 K. The testing protocol that the cell undergoes also affects the rate of degradation of the membrane, which if accelerated can lead to shorting and imminent failure of the electrolyser. The application of electrocatalysts at the electrodes not only serves to reduce reaction overpotentials, but also assists in slowing down the rate of degradation of the cell components via reducing the overpotentials, thereby extending overall cell life. This was observed particularly during intermittent tests, thereby emphasizing the importance of intermittent testing in testing protocols for rational evaluation of the cell performance.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/hydrogen4020018/s1, Table S1. Estimated exposed surface area for each flow-field plate design. Figure S2. Comparison of the conductivities [28] and viscosities [37] of KOH over a range of electrolyte concentrations, at 298 K and 333 K. Figure S3. Diffusion coefficients of O2 and H2 in KOH solutions of different molarities, at 298 K and 333 K [30]. Figure S4. (a) Dry FAA-3-50 and (b) FAA-3-50 after being immersed in 1 M KOH overnight. Figure S5. (a) Ecell vs. time plot of 10 cm2 AEM cell assembled with uncoated Ni mesh electrodes tested in 1 M KOH at 0.5 A cm−2, 323 K. (b) I-V curves of 10 cm2 cell and 100 cm2 AEM cells assembled with catalyst-coated Ni mesh tested in 1 M KOH and 4 M KOH. Figure S6. (a) Intermittent test cycled alternately at 0.1 A cm−2 and 0.3 A cm−2 with brief rest periods in between. The 100 cm2 cell is assembled with uncoated Ni mesh electrodes and FAA-3-50 membrane. Operating conditions: 4 M KOH, 323 K, flow rate 1100 mL min−1. (b) FAA-3-50 taken from the disassembled cell after the test. Figure S7. SEM images of the anode and cathode before testing, after galvanostatic holds at 0.5 A cm−2, and after cycling between 0.1 A cm−2 and 0.3 A cm−2 in 4 M KOH. Figure S8. EDX spectrums of filtered and dried precipitate from electrolyte (a) after galvanostatic hold at 0.5 A cm−2 and (b) after dynamic load cycling between 0.1 A cm−2 and 0.3 A cm−2.
Author Contributions
Conceptualization, A.L. and X.L.; methodology, A.L., X.L., P.S.; validation, A.L., X.L.; formal analysis, A.L., X.L.; investigation, A.L.; resources, X.L.; data curation/interpretation, A.L., X.L., P.S.; writing—original draft preparation, A.L.; writing—review and editing, A.L., X.L., P.S., S.S., Q.L., Y.L.; visualization, X.L.; supervision, X.L.; project administration, S.S.; funding acquisition, X.L. All authors have read and agreed to the published version of the manuscript.
Funding
The authors gratefully acknowledge that this project received funding from the Interreg 2 Seas programme 2014–2020 co-funded by the European Regional Development Fund under subsidy contract No. 2S03-019 and the Royal Academy of Engineering through the UK–Germany Energy Systems Symposium programme.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanley, E.S.; Deane, J.; Gallachóir, B.Ó. The role of hydrogen in low carbon energy futures–A review of existing perspectives. Renew. Sus. Energy Rev. 2018, 82, 3027–3045. [Google Scholar] [CrossRef]
- Russell, J.H.; Chludzinski, P.J.; Gupta, D.K.; Smarz, G.A.; Sedlak, J.M. Solid Polymer Electrolyte Water Electrolysis Technology Development for Large-Scale Hydrogen Production (Design Phase); General Electric Co., Direct Energy Conversion Programs: Wilmington, MA, USA, 1981. [Google Scholar]
- Rasten, E.; Hagen, G.; Tunold, R. Electrocatalysis in water electrolysis with solid polymer electrolyte. Electrochim. Acta 2003, 48, 3945–3952. [Google Scholar] [CrossRef]
- Taleb, A.; Kjeang, E.; Maine, E. Cost Analysis for Durable Proton Exchange Membrane in PEM Fuel Cells in PICMET ‘12: Technology Management for Emerging Technologies 2012; IEEE: Vancouver, BC, Canada, 2012. [Google Scholar]
- Du, N.; Roy, C.; Peach, R.; Turnbull, M.; Thiele, S.; Bock, C. Anion-Exchange Membrane Water Electrolyzers. Chem. Rev. 2022, 122, 11830–11895. [Google Scholar] [CrossRef] [PubMed]
- Hagesteijn, K.F.L.; Jiang, S.; Ladewig, B.P. A review of the synthesis and characterization of anion exchange membranes. J. Mater. Sci. 2018, 53, 11131–11150. [Google Scholar] [CrossRef]
- Shirvanian, P.; Loh, A.; Sluijter, S.; Li, X. Novel components in anion exchange membrane water electrolyzers (AEMWE’s): Status, challenges and future needs. A mini review. Electrochem. Commun. 2021, 132, 107140. [Google Scholar] [CrossRef]
- IEA. International Energy Agency (IEA): Energy Technology Policy Division, Energy Technology Perspectives; IEA: Paris, France, 2020. [Google Scholar]
- Pletcher, D.; Li, X. Prospects for alkaline zero gap water electrolysers for hydrogen production. Int. J. Hydrogen Energy 2011, 36, 15089–15104. [Google Scholar] [CrossRef]
- Khan, M.A.; Zhao, H.; Zou, W.; Chen, Z.; Cao, W.; Fang, J.; Xu, J.; Zhang, L.; Zhang, J. Recent Progresses in Electrocatalysts for Water Electrolysis. Electrochem. Energy Rev. 2018, 1, 483–530. [Google Scholar] [CrossRef]
- Phillips, R.; Dunnill, C.W. Zero gap alkaline electrolysis cell design for renewable energy storage as hydrogen gas. RSC Adv. 2016, 6, 100643–100651. [Google Scholar] [CrossRef]
- Li, X.; Walsh, F.C.; Pletcher, D. Nickel based electrocatalysts for oxygen evolution in high current density, alkaline water electrolysers. Phys. Chem. Chem. Phys. 2011, 13, 1162–1167. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötz, R.; Schmidt, T.J. Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef]
- Grimaud, A.; May, K.J.; Carlton, C.E.; Lee, Y.-L.; Risch, M.; Hong, W.T.; Zhou, J.; Shao-Horn, Y. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 2013, 4, 2439. [Google Scholar] [CrossRef] [PubMed]
- Varcoe, J.R.; Atanassov, P.; Dekel, D.R.; Herring, A.M.; Hickner, M.A.; Kohl, P.A.; Kucernak, A.R.; Mustain, W.E.; Nijmeijer, K.; Scott, K.; et al. Anion-exchange membranes in electrochemical energy systems. Energy Environ. Sci. 2014, 7, 3135–3191. [Google Scholar] [CrossRef]
- Wang, J. Theory and practice of flow field designs for fuel cell scaling-up: A critical review. Appl. Energy 2015, 157, 640–663. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H. Discrete approach for flow field designs of parallel channel configurations in fuel cells. Int. J. Hydrogen Energy 2012, 37, 10881–10897. [Google Scholar] [CrossRef]
- Walsh, F.C.; Pletcher, C. Electrochemical Engineering and Cell Design. In Developments in Electrochemistry: Science Inspired by Martin Fleischmann; Pletcher, D., Tian, Z., Williams, D.E., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Ju, W.; Heinz, M.; Pusterla, L.; Hofer, M.; Fumey, B.; Castiglioni, R.; Pagani, M.; Battaglia, C.; Vogt, U.F. Lab-Scale Alkaline Water Electrolyzer for Bridging Material Fundamentals with Realistic Operation. ACS Sustain. Chem. Eng. 2018, 6, 4829–4837. [Google Scholar] [CrossRef]
- Hnát, J.; Kodým, R.; Denk, K.; Paidar, M.; Žitka, J.; Bouzek, K. Design of a Zero-Gap Laboratory-Scale Polymer Electrolyte Membrane Alkaline Water Electrolysis Stack. Chem. Ing. Tech. 2019, 91, 821–832. [Google Scholar] [CrossRef]
- Rearden, A.; Mandale, S.; Glover, K.; Phillips, R.; Dunnill, C.W. Optimizing the design of an alkaline water splitting device test cell for renewable energy storage as hydrogen. Arch. Chem. and Chem. Eng. 2020, 2, 1–9. [Google Scholar]
- Faqeeh, A.H.; Symes, M.D. A standard electrolyzer test cell design for evaluating catalysts and cell components for anion exchange membrane water electrolysis. Electrochim. Acta 2023, 444, 142030. [Google Scholar] [CrossRef]
- Sulaymon, A.H.; Abbar, A.H. Scale-Up of Electrochemical Reactors. Electrolysis. In Electrolysis; Linkov, V., Kleperis, J., Eds.; InTechOpen: London, UK, 2012. [Google Scholar]
- Loh, A.; Li, X.; Taiwo, O.O.; Tariq, F.; Brandon, N.P.; Wang, P.; Xu, K.; Wang, B. Development of Ni–Fe based ternary metal hydroxides as highly efficient oxygen evolution catalysts in AEM water electrolysis for hydrogen production. Int. J. Hydrogen Energy 2020, 45, 24232–24247. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H. Flow-Field Designs of Bipolar Plates in PEM Fuel Cells: Theory and Applications. Fuel Cells 2012, 12, 989–1003. [Google Scholar] [CrossRef]
- Toghyani, S.; Afshari, E.; Baniasadi, E.; Atyabi, S. Thermal and electrochemical analysis of different flow field patterns in a PEM electrolyzer. Electrochim. Acta 2018, 267, 234–245. [Google Scholar] [CrossRef]
- Gilliam, R.J.; Graydon, J.W.; Kirk, D.W.; Thorpe, S.J. A review of specific conductivities of potassium hydroxide solutions for various concentrations and temperatures. Int. J. Hydrogen Energy 2007, 32, 359–364. [Google Scholar] [CrossRef]
- Sipos, P.M.; Hefter, G.; May, P.M. Viscosities and densities of highly concentrated aqueous MOH solutions (M+ ) Na+, K+, Li+, Cs+, (CH3)4N+) at 25.0 °C. J. Chem. Eng. Data 2000, 45, 613–617. [Google Scholar] [CrossRef]
- Tham, M.J.; Walker, R.D.; Gubbins, K.E. Diffusion of oxygen and hydrogen in aqueous potassium hydroxide solutions. J. Phys. Chem. 1970, 74, 1747–1751. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, W.; Chen, G.; Huang, J.; Song, C.; Chu, S.; Zhang, R.; Wang, G.; Li, C.; Ostrikov, K.K. Bi-metallic nitroxide nanodot-decorated tri-metallic sulphide nanosheets by on-electrode plas-ma-hydrothermal sprouting for overall water splitting. Appl. Catal. B Environ. 2020, 261, 118254. [Google Scholar] [CrossRef]
- Arges, C.G.; Ramani, V.K.; Pintauro, P.N. The Chalkboard: Anion Exchange Membrane Fuel Cells. Electrochem. Soc. Interface 2010, 19, 31–35. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Slade, R.C.T. Prospects for Alkaline Anion-Exchange Membranes in Low Temperature Fuel Cells. Fuel Cells 2004, 5, 187–200. [Google Scholar] [CrossRef]
- Harrison, K.W.; Remick, R.; Hoskin, A.; Martin, G.D. Hydrogen Production: Fundamentals and Case Study Summaries. In Proceedings of the 18th World Hydrogen Energy Conference, Essen, Germany, 16–20 May 2010. [Google Scholar]
- Wu, X.; Scott, K. A Li-doped Co3O4 oxygen evolution catalyst for non-precious metal alkaline anion exchange membrane water electrolysers. Int. J. Hydrogen Energy 2013, 38, 3123–3129. [Google Scholar] [CrossRef]
- Rakousky, C.; Keeley, G.P.; Wippermann, K.; Carmo, M.; Stolten, D. The stability challenge on the pathway to high-current-density polymer electrolyte membrane water electrolyzers. Electrochim. Acta 2018, 278, 324–331. [Google Scholar] [CrossRef]
- Occidental Chemical Corporation. Caustic Potash Handbook; Occidental Chemical Corporation: Dallas, TX, USA, 2018. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).