
Is Water the Engine of Protein Folding?



Abstract
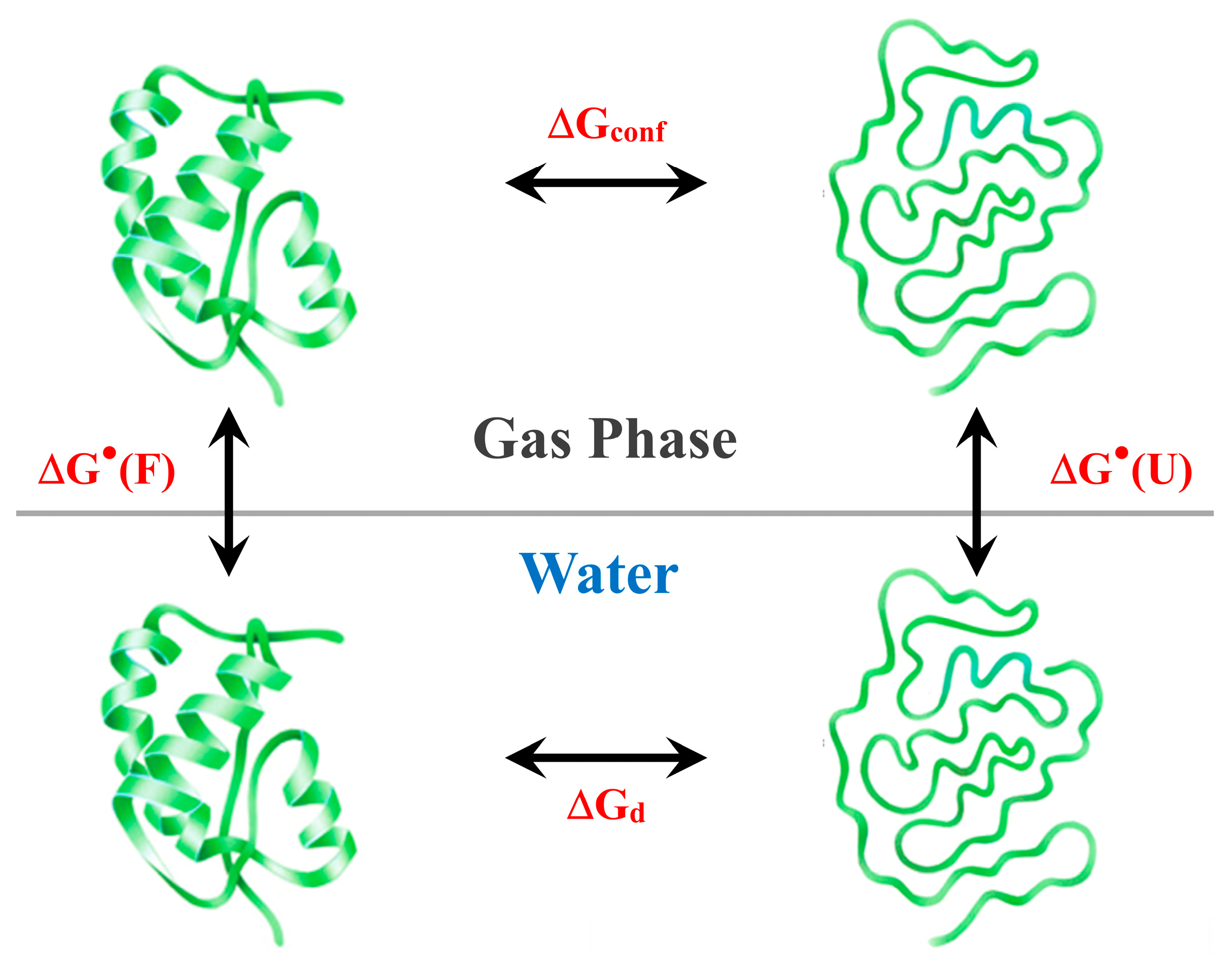
1. Introduction
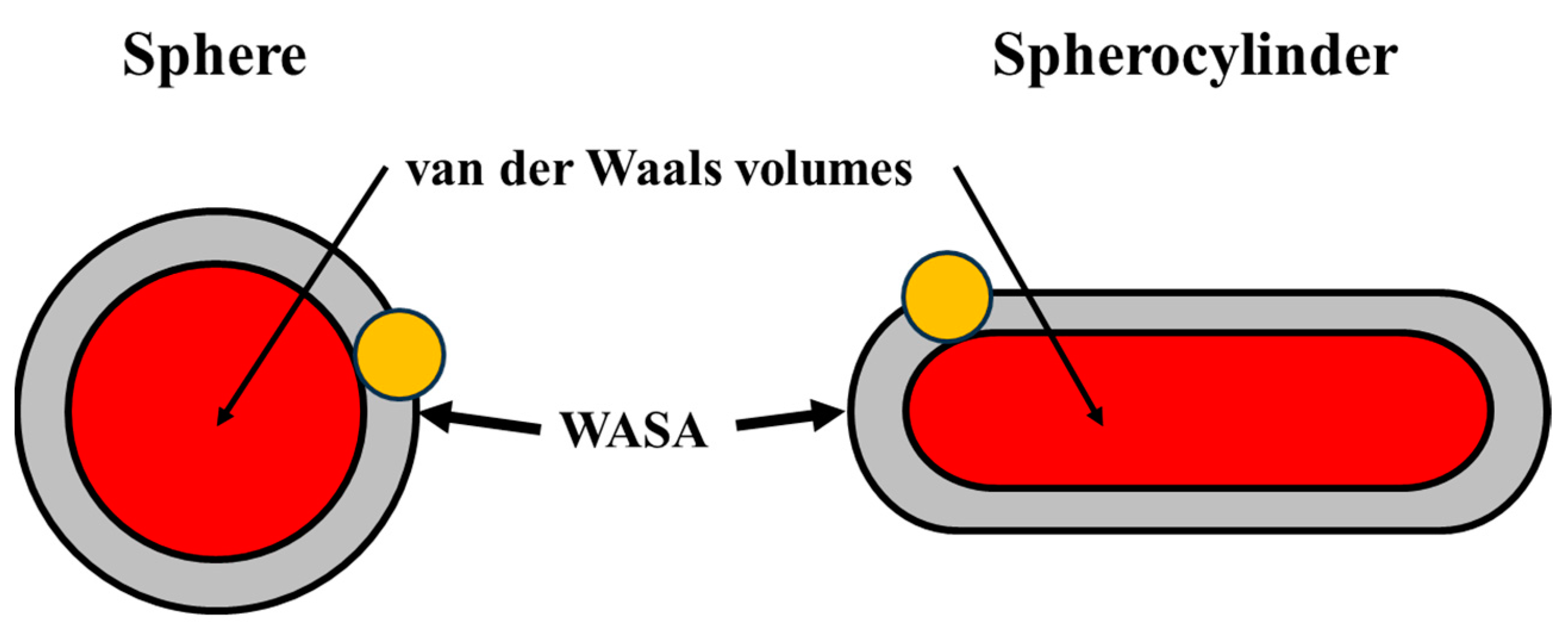
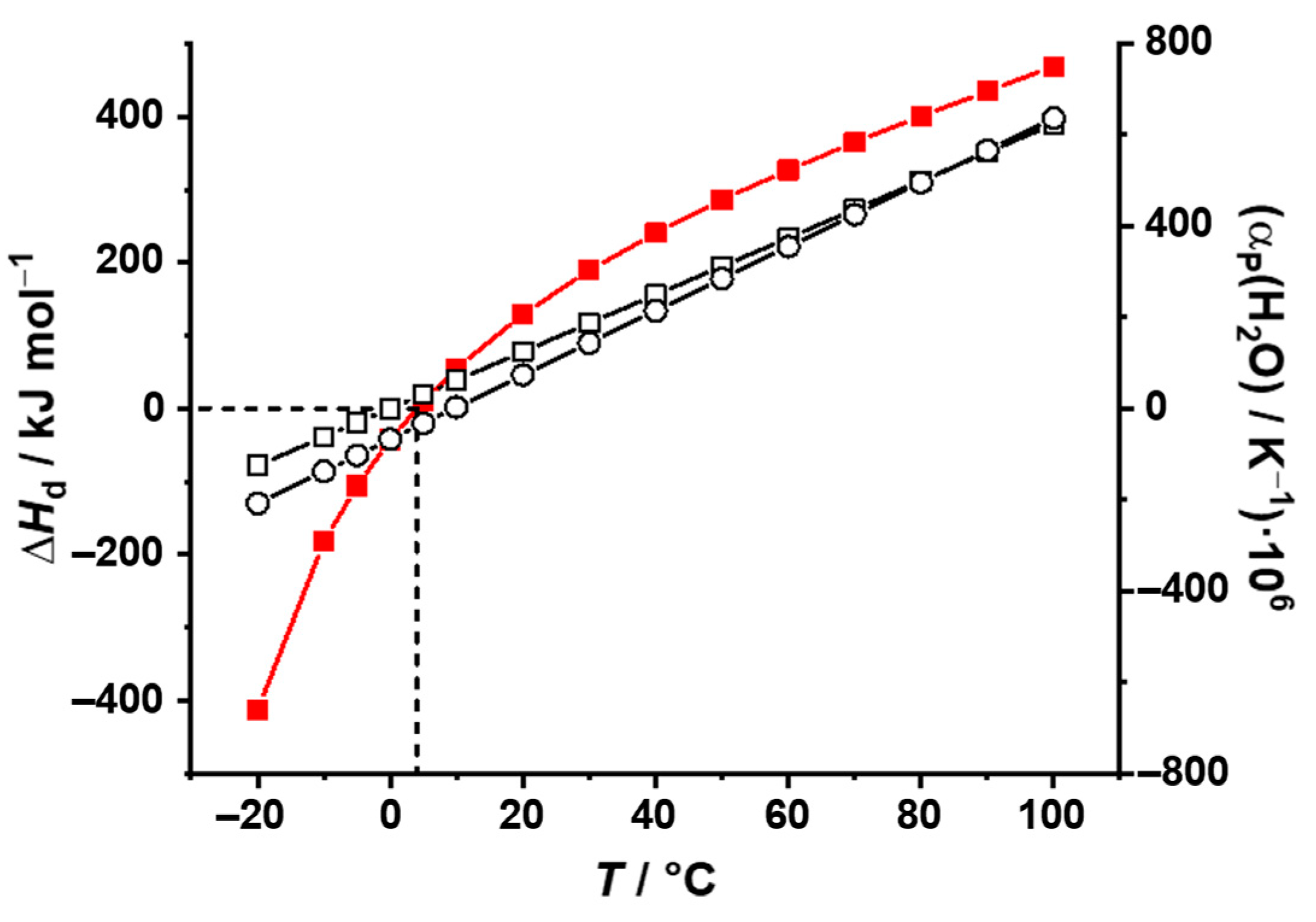
2. General Analysis
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1959; Volume 14, pp. 1–63. ISBN 978-0-12-034214-3. [Google Scholar]
- Dill, K.A. Dominant Forces in Protein Folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, W.; Engberts, J.B.F.N. Hydrophobic Effects. Opinions and Facts. Angew. Chem. Int. Ed. Engl. 1993, 32, 1545–1579. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Ball, P. Water as an Active Constituent in Cell Biology. Chem. Rev. 2008, 108, 74–108. [Google Scholar] [CrossRef]
- Sumi, T.; Imamura, H. WATER-MEDIATED Interactions Destabilize Proteins. Protein Sci. 2021, 30, 2132–2143. [Google Scholar] [CrossRef]
- Murphy, K.P.; Privalov, P.L.; Gill, S.J. Common Features of Protein Unfolding and Dissolution of Hydrophobic Compounds. Science 1990, 247, 559–561. [Google Scholar] [CrossRef]
- Herzfeld, J. Understanding Hydrophobic Behavior. Science 1991, 253, 88. [Google Scholar] [CrossRef]
- Muller, N. Does Hydrophobic Hydration Destabilize Protein Native Structures? Trends Biochem. Sci. 1992, 17, 459–463. [Google Scholar] [CrossRef]
- Oobatake, M.; Ooi, T. Hydration and Heat Stability Effects on Protein Unfolding. Prog. Biophys. Mol. Biol. 1993, 59, 237–284. [Google Scholar] [CrossRef]
- Makhatadze, G.I.; Privalov, P.L. Energetics of Protein Structure. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1995; Volume 47, pp. 307–425. ISBN 978-0-12-034247-1. [Google Scholar]
- Smith, P.E.; Van Gunsteren, W.F. When Are Free Energy Components Meaningful? J. Phys. Chem. 1994, 98, 13735–13740. [Google Scholar] [CrossRef]
- Mark, A.E.; Van Gunsteren, W.F. Decomposition of the Free Energy of a System in Terms of Specific Interactions. J. Mol. Biol. 1994, 240, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Boresch, S.; Karplus, M. The Meaning of Component Analysis: Decomposition of the Free Energy in Terms of Specific Interactions. J. Mol. Biol. 1995, 254, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F.M. The Interpretation of Protein Structures: Estimation of Static Accessibility. J. Mol. Biol. 1971, 55, 379-IN4. [Google Scholar] [CrossRef]
- Richards, F.M.; Lim, W.A. An Analysis of Packing in the Protein Folding Problem. Quart. Rev. Biophys. 1993, 26, 423–498. [Google Scholar] [CrossRef] [PubMed]
- Gekko, K.; Noguchi, H. Compressibility of Globular Proteins in Water at 25. Degree. C. J. Phys. Chem. 1979, 83, 2706–2714. [Google Scholar] [CrossRef]
- Kharakoz, D.P. Protein Compressibility, Dynamics, and Pressure. Biophys. J. 2000, 79, 511–525. [Google Scholar] [CrossRef]
- Cooper, A. Thermodynamic Fluctuations in Protein Molecules. Proc. Natl. Acad. Sci. USA 1976, 73, 2740–2741. [Google Scholar] [CrossRef]
- Cooper, A. Heat Capacity of Hydrogen-Bonded Networks: An Alternative View of Protein Folding Thermodynamics. Biophys. Chem. 2000, 85, 25–39. [Google Scholar] [CrossRef]
- Schrödinger, E.; Penrose, R. What Is Life?: With Mind and Matter and Autobiographical Sketches, 1st ed.; Cambridge University Press: Cambridge, UK, 1992; ISBN 978-0-521-42708-1. [Google Scholar]
- Liquori, A.M. The Stereochemical Code and the Logic of a Protein Molecule. Quart. Rev. Biophys. 1969, 2, 65–92. [Google Scholar] [CrossRef]
- Nozaki, Y.; Tanford, C. The Solubility of Amino Acids and Two Glycine Peptides in Aqueous Ethanol and Dioxane Solutions. J. Biol. Chem. 1971, 246, 2211–2217. [Google Scholar] [CrossRef]
- Damodaran, S.; Song, K.B. The Role of Solvent Polarity in the Free Energy of Transfer of Amino Acid Side Chains from Water to Organic Solvents. J. Biol. Chem. 1986, 261, 7220–7222. [Google Scholar] [CrossRef] [PubMed]
- Andrew Karplus, P. Hydrophobicity Regained. Protein Sci. 1997, 6, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Janin, J.; Lesk, A.M.; Chothia, C. Interior and Surface of Monomeric Proteins. J. Mol. Biol. 1987, 196, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Harpaz, Y.; Gerstein, M.; Chothia, C. Volume Changes on Protein Folding. Structure 1994, 2, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Merlino, A.; Pontillo, N.; Graziano, G. A Driving Force for Polypeptide and Protein Collapse. Phys. Chem. Chem. Phys. 2017, 19, 751–756. [Google Scholar] [CrossRef]
- Cooper, A. Protein Heat Capacity: An Anomaly That Maybe Never Was. J. Phys. Chem. Lett. 2010, 1, 3298–3304. [Google Scholar] [CrossRef]
- Dill, K.A. Additivity Principles in Biochemistry. J. Biol. Chem. 1997, 272, 701–704. [Google Scholar] [CrossRef]
- Dill, K.A. Polymer Principles and Protein Folding. Protein Sci. 1999, 8, 1166–1180. [Google Scholar] [CrossRef]
- Ben-Naim, A. Solvation Thermodynamics; Springer: Boston, MA, USA, 1987; ISBN 978-1-4757-6552-6. [Google Scholar]
- Lee, B. A Procedure for Calculating Thermodynamic Functions of Cavity Formation from the Pure Solvent Simulation Data. J. Chem. Phys. 1985, 83, 2421–2425. [Google Scholar] [CrossRef]
- Pohorille, A.; Pratt, L.R. Cavities in Molecular Liquids and the Theory of Hydrophobic Solubilities. J. Am. Chem. Soc. 1990, 112, 5066–5074. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Graziano, G. Scaled Particle Theory Study of the Length Scale Dependence of Cavity Thermodynamics in Different Liquids. J. Phys. Chem. B 2006, 110, 11421–11426. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. Dimerization Thermodynamics of Large Hydrophobic Plates: A Scaled Particle Theory Study. J. Phys. Chem. B 2009, 113, 11232–11239. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Statistical Mechanics; Harper’s Chemistry Series; Harper & Row: New York, NY, USA, 1976; ISBN 978-0-06-044366-5. [Google Scholar]
- Graziano, G. Contrasting the Hydration Thermodynamics of Methane and Methanol. Phys. Chem. Chem. Phys. 2019, 21, 21418–21430. [Google Scholar] [CrossRef] [PubMed]
- Bologna, A.; Graziano, G. Remarks on the Hydration Entropy of Polar and Nonpolar Species. J. Mol. Liq. 2023, 391, 123437. [Google Scholar] [CrossRef]
- Soper, A.K.; Finney, J.L. Hydration of Methanol in Aqueous Solution. Phys. Rev. Lett. 1993, 71, 4346–4349. [Google Scholar] [CrossRef]
- Buchanan, P.; Aldiwan, N.; Soper, A.K.; Creek, J.L.; Koh, C.A. Decreased Structure on Dissolving Methane in Water. Chem. Phys. Lett. 2005, 415, 89–93. [Google Scholar] [CrossRef]
- Graziano, G. Structural Order in the Hydration Shell of Nonpolar Groups versus That in Bulk Water. ChemPhysChem 2024, 25, e202400102. [Google Scholar] [CrossRef]
- Graziano, G. The Gibbs Energy Cost of Cavity Creation Depends on Geometry. J. Mol. Liq. 2015, 211, 1047–1051. [Google Scholar] [CrossRef]
- Pierotti, R.A. A Scaled Particle Theory of Aqueous and Nonaqueous Solutions. Chem. Rev. 1976, 76, 717–726. [Google Scholar] [CrossRef]
- Wallqvist, A.; Berne, B.J. Molecular Dynamics Study of the Dependence of Water Solvation Free Energy on Solute Curvature and Surface Area. J. Phys. Chem. 1995, 99, 2885–2892. [Google Scholar] [CrossRef]
- Patel, A.J.; Varilly, P.; Chandler, D.; Garde, S. Quantifying Density Fluctuations in Volumes of All Shapes and Sizes Using Indirect Umbrella Sampling. J. Stat. Phys. 2011, 145, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Sosso, G.C.; Caravati, S.; Rotskoff, G.; Vaikuntanathan, S.; Hassanali, A. On the Role of Nonspherical Cavities in Short Length-Scale Density Fluctuations in Water. J. Phys. Chem. A 2017, 121, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.A. Revisiting Volume Changes in Pressure-Induced Protein Unfolding. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzymol. 2002, 1595, 201–209. [Google Scholar] [CrossRef]
- Chalikian, T.V. Volumetric Properties of Proteins. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 207–235. [Google Scholar] [CrossRef]
- Chen, C.R.; Makhatadze, G.I. Molecular Determinant of the Effects of Hydrostatic Pressure on Protein Folding Stability. Nat. Commun. 2017, 8, 14561. [Google Scholar] [CrossRef]
- Graziano, G. On the Molecular Origin of Cold Denaturation of Globular Proteins. Phys. Chem. Chem. Phys. 2010, 12, 14245. [Google Scholar] [CrossRef]
- Graziano, G. On the Mechanism of Cold Denaturation. Phys. Chem. Chem. Phys. 2014, 16, 21755–21767. [Google Scholar] [CrossRef]
- Pica, A.; Graziano, G. Shedding Light on the Extra Thermal Stability of Thermophilic Proteins. Biopolymers 2016, 105, 856–863. [Google Scholar] [CrossRef]
- Maritan, A.; Micheletti, C.; Trovato, A.; Banavar, J.R. Optimal Shapes of Compact Strings. Nature 2000, 406, 287–290. [Google Scholar] [CrossRef]
- Banavar, J.R.; Maritan, A. Physics of Proteins. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Liquori, A.M.; Sadun, C. Close Packing of Amino Acid Residues in Globular Proteins: Specific Volume and Site Binding of Water Molecules. Int. J. Biol. Macromol. 1981, 3, 56–59. [Google Scholar] [CrossRef]
- Head-Gordon, T.; Hura, G. Water Structure from Scattering Experiments and Simulation. Chem. Rev. 2002, 102, 2651–2670. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Chan, H.S. From Levinthal to Pathways to Funnels. Nat. Struct. Biol. 1997, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Wolynes, P.G. Evolution, Energy Landscapes and the Paradoxes of Protein Folding. Biochimie 2015, 119, 218–230. [Google Scholar] [CrossRef]
- Baxa, M.C.; Haddadian, E.J.; Jumper, J.M.; Freed, K.F.; Sosnick, T.R. Loss of Conformational Entropy in Protein Folding Calculated Using Realistic Ensembles and Its Implications for NMR-Based Calculations. Proc. Natl. Acad. Sci. USA 2014, 111, 15396–15401. [Google Scholar] [CrossRef]
- Gong, H.; Porter, L.L.; Rose, G.D. Counting Peptide-water Hydrogen Bonds in Unfolded Proteins. Protein Sci. 2011, 20, 417–427. [Google Scholar] [CrossRef]
- Rose, G.D. Reframing the Protein Folding Problem: Entropy as Organizer. Biochemistry 2021, 60, 3753–3761. [Google Scholar] [CrossRef]
- Rose, G.D. Protein Folding—Seeing Is Deceiving. Protein Sci. 2021, 30, 1606–1616. [Google Scholar] [CrossRef]
- Privalov, P.L. Stability of Proteins Small Globular Proteins. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1979; Volume 33, pp. 167–241. ISBN 978-0-12-034233-4. [Google Scholar]
- Privalou, P.L. Thermodynamic Problems of Protein Structure. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 47–69. [Google Scholar] [CrossRef]
- Kaya, H.; Chan, H.S. Polymer Principles of Protein Calorimetric Two-State Cooperativity. Proteins 2000, 40, 637–661. [Google Scholar] [CrossRef] [PubMed]
- Becktel, W.J.; Schellman, J.A. Protein Stability Curves. Biopolymers 1987, 26, 1859–1877. [Google Scholar] [CrossRef]
- Kell, G.S. Density, Thermal Expansivity, and Compressibility of Liquid Water from 0.Deg. to 150.Deg.. Correlations and Tables for Atmospheric Pressure and Saturation Reviewed and Expressed on 1968 Temperature Scale. J. Chem. Eng. Data 1975, 20, 97–105. [Google Scholar] [CrossRef]
- Robertson, A.D.; Murphy, K.P. Protein Structure and the Energetics of Protein Stability. Chem. Rev. 1997, 97, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Sawle, L.; Ghosh, K. How Do Thermophilic Proteins and Proteomes Withstand High Temperature? Biophys. J. 2011, 101, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Lee, B. Enthalpy-Entropy Compensation in the Thermodynamics of Hydrophobicity. Biophys. Chem. 1994, 51, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D. Win Some, Lose Some: Enthalpy-Entropy Compensation in Weak Intermolecular Interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef]
- Harano, Y.; Kinoshita, M. Translational-Entropy Gain of Solvent upon Protein Folding. Biophys. J. 2005, 89, 2701–2710. [Google Scholar] [CrossRef]
- Kinoshita, M. A New Theoretical Approach to Biological Self-Assembly. Biophys. Rev. 2013, 5, 283–293. [Google Scholar] [CrossRef]
- Kinoshita, M. Importance of Translational Entropy of Water in Biological Self-Assembly Processes like Protein Folding. Int. J. Mol. Sci. 2009, 10, 1064–1080. [Google Scholar] [CrossRef]
- Maruyama, Y.; Harano, Y. Does Water Drive Protein Folding? Chem. Phys. Lett. 2013, 581, 85–90. [Google Scholar] [CrossRef]
- Kamo, F.; Ishizuka, R.; Matubayasi, N. Correlation Analysis for Heat Denaturation of Trp-cage Miniprotein with Explicit Solvent. Protein Sci. 2016, 25, 56–66. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Yamamori, Y.; Matubayasi, N. Probabilistic Analysis for Identifying the Driving Force of Protein Folding. J. Chem. Phys. 2018, 148, 125101. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
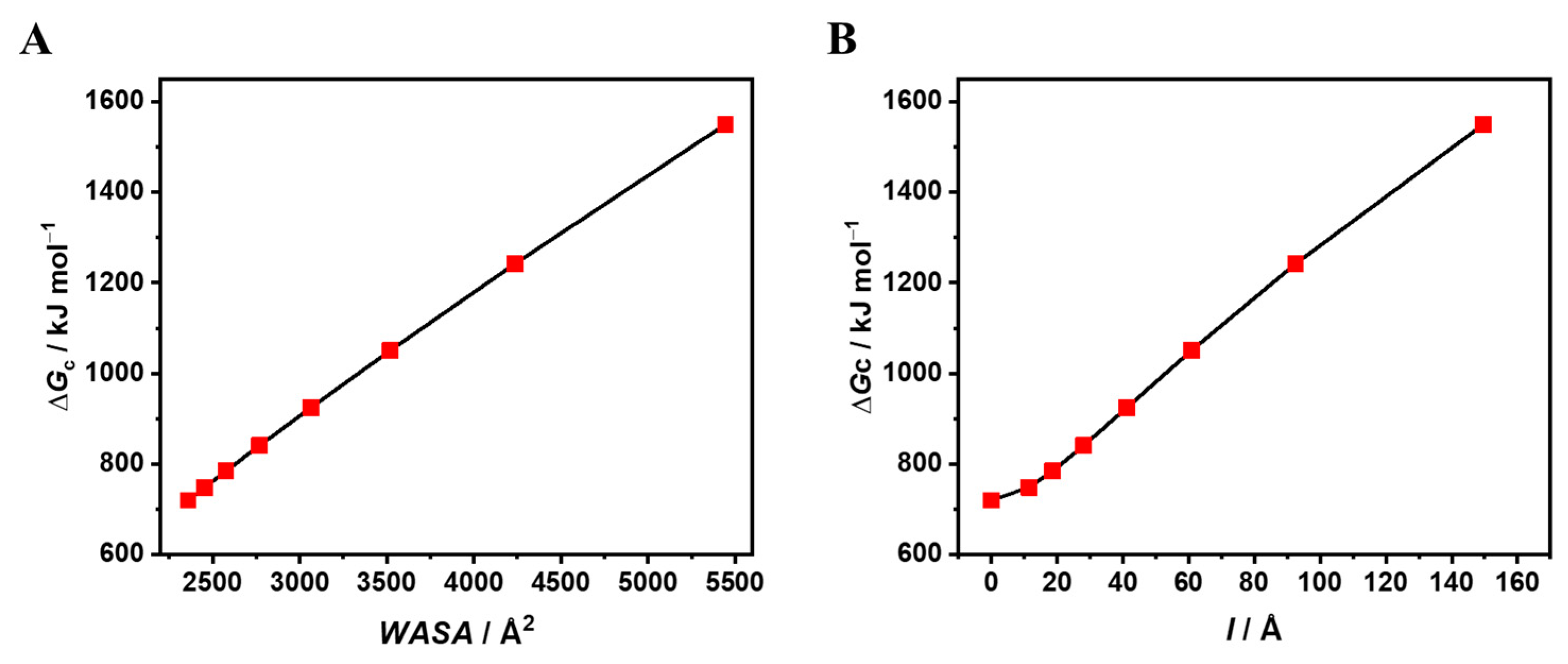
| a Å | l Å | VvdW Å3 | WASA Å2 | ΔGc kJ mol−1 | ΔGc/WASA J mol−1 Å−2 |
|---|---|---|---|---|---|
| 12.3 | / | 7790 | 2359 | 719.8 | 305.1 |
| 10.0 | 11.46 | 7790 | 2454 | 748.6 | 305.1 |
| 9.0 | 18.61 | 7790 | 2575 | 784.6 | 304.7 |
| 8.0 | 28.07 | 7790 | 2768 | 840.7 | 303.7 |
| 7.0 | 41.27 | 7790 | 3065 | 925.2 | 301.9 |
| 6.0 | 60.88 | 7790 | 3519 | 1050.9 | 298.6 |
| 5.0 | 92.51 | 7790 | 4235 | 1242.2 | 293.3 |
| 4.0 | 149.65 | 7790 | 5444 | 1550.1 | 284.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campanile, M.; Graziano, G. Is Water the Engine of Protein Folding? Biophysica 2024, 4, 507-516. https://doi.org/10.3390/biophysica4040033
Campanile M, Graziano G. Is Water the Engine of Protein Folding? Biophysica. 2024; 4(4):507-516. https://doi.org/10.3390/biophysica4040033
Chicago/Turabian StyleCampanile, Marco, and Giuseppe Graziano. 2024. "Is Water the Engine of Protein Folding?" Biophysica 4, no. 4: 507-516. https://doi.org/10.3390/biophysica4040033
APA StyleCampanile, M., & Graziano, G. (2024). Is Water the Engine of Protein Folding? Biophysica, 4(4), 507-516. https://doi.org/10.3390/biophysica4040033