Abstract
This study explored the inhibitory potential of gallium maltolate against severe acute respiratory syndrome coronavirus 2 and main and papain-like proteases. Computational methods, including density functional theory and molecular docking, were used to assess gallium maltolate reactivity and binding interactions. Density functional theory calculations revealed gallium maltolate’s high electron-capturing capacity, particularly around the gallium metal atom, which may contribute to their activity. Molecular docking demonstrated that gallium maltolate can form strong hydrogen bonds with key amino acid residues like glutamate-166 and cysteine-145, tightly binding to main and papain-like proteases. The binding energy and interactions of gallium maltolate were comparable to known SARS-CoV-2 inhibitors like N-[(5-methyl-1,2-oxazol-3-yl)carbonyl]-L-alanyl-L-valyl-N-{(2S,3E)-5-(benzyloxy)-5-oxo-1-[(3S)-2-oxopyrrolidin-3-yl]pent-3-en-2-yl}-L-leucinamide, indicating its potential as an antiviral agent. However, further experimental validation is required to confirm its effectiveness in inhibiting SARS-CoV-2 replication and treating COVID-19.
1. Introduction
Over the years, the pharmaceutical industry has been dominated mainly by organic chemistry; however, with the emergence of metallated drugs, a remarkable growth in inorganic medicinal chemistry has developed. In its beginnings, a well-known anticancer drug, cis-platinum (cis-[PtCl2(NH3)2]), a compound that forms adducts with DNA inhibiting its replication and transcription processes [1,2], leading to cell death by apoptosis, stood out [3]. However, this drug presented side effects such as nausea and neurotoxicity, which motivated different researchers to develop derivatives of this drug in order to improve its effectiveness and reduce its side effects [4,5]. Thus, over the years, different metal complexes have been designed that possess different ions, among which Pt(II) [6,7,8], Cu(II) [9,10], Pd(II) [6,11,12,13], Au(III) [6,14], Ag(I) [15,16], Ru(II) [17,18], and many others stand out [19,20,21].
Currently, different antiviral metal complexes have been developed that, before proceeding to the stage of their respective synthesis, are initially evaluated in vitro against different viruses, such as SARS-CoV-2, HIV, Zika virus, etc. [22,23,24]. Among the main antiviral complexes studied, cobalt complexes stand out, for which the synthesis of their Co(III) complexes with Schiff base derivatives of bis(acetone)ethylenediamine has already been described, the same that has succeeded in inhibiting the herpes virus (HSV-1) [25]. Other authors have obtained Pd(II) complexes from the addition of Michael, synthesizing three new ligands (SVSL1, SVSL2, and SVSL3), which were then reacted with [PdCl2(PPh3)2], forming Pd(II) complexes. Their cytotoxicity was evaluated against different human cancer cell lines (PANC-1, AsPC-1, MCF-7, and MDA-MB-231), showing high activity values compared to cisplatin, carboplatin, and gemcitabine (standard drugs) [26].
Among the elements of the periodic system, the Ga atom has attracted the attention of different groups of researchers and has been attractive for developing complexes with therapeutic activity since some simple gallium salts, such as nitrate or chloride, have been studied for a long time, showing moderate activity against tumors located in soft tissues [27]. Nowadays, gallium nitrate has been approved to treat hypercalcemia derived from tumors, and Ga(III) tris(8-hydroxyquinolinate) and Ga(III) tris(maltolate) have shown high success in animal models when administered orally, as well as superior antitumor activity to Ga(III) salts [28]. Additionally, other works have synthesized planar tetradentate Ga(III) complexes (2,3-bis[(4-dialkylamino-2-hydroxybenzylidene)amino]but-2-enodinitrile), together with labile solvent ligands, that have shown antitumor activity both in vitro and in vivo comparable to cisplatin and, aided by confocal fluorescence microscopy, Western transfer, mRNA profiling, and surface plasmon resonance (SPR), showed convincing evidence that PDIA3 (a protein disulfide isomerase (PDI) involved in the endoplasmic reticulum stress response) is a direct target of Ga-1 (a complex synthesized by refluxing cis-diaminomaleonitrile and salicylaldehydes with different steric substituents in the presence of excess GaCl3 in anhydrous acetonitrile) in tumor cells [29].
In light of the extensive body of research on gallium compounds and their diverse applications in pharmaceuticals and medical diagnostics, it is notable that their potential utility in combating the current COVID-19 pandemic has not been extensively investigated. This research gap, particularly pertinent during a global health crisis that demands innovative therapeutic strategies and diagnostic tools, has been the primary motivator for our computational study. Motivated by these findings, we have embarked on a computational exploration, evaluating various properties that might account for the activity of this compound. Our aim is to unveil valuable insights and potentially novel solutions through computational analyses, which could significantly contribute to the global fight against COVID-19.
In the context of the ongoing research and applications of gallium compounds, it is pertinent to note their significant contributions across various domains, particularly pharmaceuticals and medical diagnostics. Despite this extensive body of work, the exploration of gallium compounds in the context of the current COVID-19 pandemic remains relatively uncharted. This observation underscores a potential avenue for research, especially considering the urgent need for diverse therapeutic strategies and diagnostic tools to combat this global health crisis. Addressing this gap could provide valuable insights and possibly novel solutions in the fight against COVID-19. A notable example underscoring this potential is a recent study that demonstrated the efficacy of gallium maltolate (GaM) in inhibiting the replication of SARS-CoV-2 in cultured Vero E6 cells. This study found that GaM achieved a 50% effective concentration (EC50) of 14 mM for viral inhibition, with no cytotoxicity observed at concentrations up to at least 200 mM [30]. This finding serves as a pivotal reference in our computational study, guiding our evaluation of the properties that could account for GaM’s activity against this virus.
In this comprehensive research endeavor, we present an extensive in silico investigation focused on unraveling the inhibitory potential of gallium maltolate (GaM) against the SARS-CoV-2 main proteases, Mpro and PLpro. This multifaceted investigation was undertaken to shed light on the therapeutic prospects of GaM in combating the COVID-19 pandemic. This computational study was initiated by employing advanced reactivity descriptors, which are meticulously defined within the purview of the density functional theory (DFT) [31]. These descriptors were instrumental in scrutinizing and delineating the reactive attributes of the organometallic GaM molecule. It is worth noting that the use of DFT-based reactivity descriptors has been a proven and widely accepted methodology in the scientific community. It has been successfully utilized by numerous research groups, underscoring its efficacy in elucidating the reactivity patterns of various molecular systems. Following the rigorous assessment of GaM’s reactivity, we employed molecular docking techniques to assess its potential as an inhibitor of SARS-CoV-2. The results of this computational analysis yielded highly promising outcomes regarding GaM’s ability to inhibit the virus. Notably, the binding energies exhibited by GaM closely approximated those of N3, a well-established reference molecule known for its potent inhibitory activity against SARS-CoV-2. This suggests that GaM holds substantial promise as an antiviral agent. The basis for GaM’s remarkable inhibitory activity lies in its ability to forge strong interactions with the key amino acid residues Glu166 and Cys145, which are pivotal for the activity of the Mpro enzyme. These interactions are primarily facilitated by the formation of hydrogen bonds, underscoring the strategic significance of GaM in targeting the vital components of the virus’s replication machinery.
2. Materials and Methods
2.1. Reactivity Indexes and Molecular Electrostatic Potential (MEP) Analysis
The geometries of the GaM, TTT, and N3 molecules were optimized at the PBE0 [32]/Def2-TZVP [33] level using the Gaussian16 software suite [34]. Water was included in the simulations as a solvent with the SMD parametrization of the IEF-PCM [35]. To gain a better understanding of the reactivity of these molecules, we calculated various DFT-based global reactivity descriptors (refer to Table 1), including electronegativity (χ), global hardness (η), electrophilicity (ω), electrodonating (ω−), electroaccepting (ω+), and net electrophilicity (Δω±).

Table 1.
Summary of equations used to calculate various global reactivity indexes in the TAFF [36] pipeline. These indexes are calculated using conceptual density functional theory (DFT) descriptors derived from the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies.
Additionally, three-dimensional molecular electrostatic potential (MEP) surfaces were generated to depict the electronic charge distributions of GaM, TTT, and N3 molecules. These MEP surfaces provide a qualitative picture of chemical reactivity and site-specific interactions, as they provide a visual representation of electron density variations. Electrophilic (electron-deficient) and nucleophilic (electron-rich) attack sites are readily identified, enhancing our understanding of the potential noncovalent interactions, particularly within the active sites of protein–ligand complexes. The color-coded MEPs function as a predictive framework for electrostatic interaction potential: regions with a positive potential (shown in shades of blue) indicate likely targets for nucleophilic attacks, whereas areas with a negative potential (transitioning from green to red) are suggestive of electrophilic character, predisposing them to nucleophilic encounters. This nuanced visualization facilitates the discernment of key interactive sites that govern ligands’ binding affinity and specificity to their protein counterparts.
2.2. Molecular Docking
Molecular docking simulations were carried out employing AutoDock 4.0 [47,48], a widely recognized computational tool in medicinal chemistry for predicting the preferred orientation of small molecules to their protein targets. The coordination compound of interest, gallium maltolate (GaM), was procured from the Cambridge Crystallography Data Centre (CCDC) [49]. Benchmarking its interactions, we included two established co-crystallized ligands, specifically 5-amino-2-methyl-N-[(1R)-1-naphthalen-1-ylethyl] benzamide (TTT) and the complex N-[(5-Methylisoxazol-3-yl)Carbonyl]Alanyl-L-Valyl-N1-((1R,2Z)-4-(Benzyloxy)-4-Oxo-1-{[(3R)-2-Oxopyrrolidin-3-yl]methyl}But-2-Enyl)-L-Leucinamide (N3), both of which were retrieved from the Protein Data Bank (PDB) [50]. To tailor the computational environment for the unique characteristics of GaM, particularly the gallium ion, modifications were incorporated into the parameter file of AutoDock 4.0 using Autodock Tools (ADT) [51]. This step ensured accurate grid box delineation around the enzymatic binding site, as specified in the Grid Parameter File (GPF). Such precision in the computational setup is crucial for reflecting the physicochemical environment of the metal–ligand interaction within the active site. The ligands’ geometries, including those of GaM, TTT, and N3, were optimized at the PBE0/Def2-TZVP level using the Gaussian16 program, with Gasteiger charges applied to approximate the molecular electrostatic potential, informing the likely intermolecular interactions. For the receptor proteins, SARS-CoV-2 main protease Mpro (PDB code 6LU7) [52], and SARS-CoV-2 papain-like protease PLpro (PDB code 7CMD) [53], datasets were meticulously curated. A series of bioinformatics protocols were implemented to validate and refine the structural models using the Protein Preparation Wizard [54] from the Schrödinger suite, adopting the OPLS force field [55] to ensure computational modeling fidelity. The binding sites were inferred from the centroid of the co-crystallized ligands [56,57,58], which is a critical determinant for the generation of the grid maps by AutoGrid 4.0. These maps were constructed with a resolution of 0.375 Å across a 603-point grid, establishing the spatial context for the docking calculations. The Lamarckian Genetic Algorithm (LGA) underpinned the search strategy, simulating an evolutionary process to optimize the ligand conformations. Initial conditions included a diverse population of 150 random individuals, with the algorithm permitted to iterate over 2.5 × 107 energy evaluations and 27,000 generations, subjected to a mutation rate of 0.02 and a crossover rate of 0.80, to converge on the energetically favorable binding poses. The end-state of this computational endeavor was the elucidation of the lowest-energy bound conformations for GaM in complex with Mpro and PLpro, offering structural insights that are instrumental for the design and development of potential therapeutic agents against SARS-CoV-2.
2.3. Non-Covalent Interactions
To gain deeper insights into the intermolecular interactions, we probed the non-covalent binding between the gallium maltolate (GaM) ligand and the SARS-CoV-2 main protease (Mpro) as well as the papain-like protease (PLpro). The analysis was centered on the energetically most favorable GaM-protein complexes, employing the Non-Covalent Interaction (NCI) index [59] computed with the NCIPLOT software [60]. This methodological framework is based on the exploration of the electron density (ρ(r)) landscape and its gradient properties, principally through the reduced density gradient (RDG, represented as s). The RDG was computed through the following equation:
The visualization of NCI isosurfaces, corresponding to areas of low RDG values and low electron density, allows for the spatial localization of non-covalent interactions. Additionally, the qualitative nature of these interactions is discerned by examining the sign of the second eigenvalue of the electron density Hessian matrix (λ2) in conjunction with the electron density ρ(λ2). The resulting NCI isosurfaces are color-coded: blue indicates regions of strong and attractive interactions, typically hydrogen bonds; green denotes weaker interactions, such as van der Waals forces; and red highlights areas with strong, repulsive steric interactions. This chromatic coding provides a direct and interpretable mapping of interaction strengths and types, enhancing understanding of the molecular interplay at the binding interface.
3. Results
3.1. Reactivity Indexes and Molecular Electrostatic Potential Analysis
The therapeutic efficacy of a pharmacological agent is contingent upon its molecular affinity for binding to a biological target. This affinity can manifest as reversible or irreversible interactions based on the drug’s inherent chemical properties [61]. Reversible agents typically engage in non-covalent bonding with the target, encompassing a spectrum of weak intermolecular forces such as hydrogen bonds, ionic interactions, van der Waals interactions, and hydrophobic contacts. These interactions, although transient, are crucial for the reversible nature of the drug–target complex. In contrast, irreversible inhibitors form stable covalent bonds with specific amino acid residues within the target site, a process commonly facilitated by an electrophilic fragment within the molecule that reacts with a nucleophilic cysteine residue present in the enzyme’s active site.
The propensity of a molecule to form such bonds, indicative of its reactivity or binding potential, can be theoretically appraised through frontier molecular orbital (FMO) analysis. This entails an evaluation of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) [62]. The FMO theory suggests that a molecule’s chemical reactivity is established by the orbital interactions wherein a nucleophilic HOMO of the pharmacophore can interact with an electrophilic LUMO of the biological target. The HOMO–LUMO gap serves as an index of reactivity, with a smaller gap indicative of heightened reactivity due to more accessible orbital interactions. Further, FMOs provide insights into the potential electron flow from the drug to the target, aiding in the elucidation of reactivity through various chemical descriptors [62]. This analytical perspective is pivotal in guiding the structural optimization of both reversible and irreversible inhibitors. It enables the strategic positioning of noncovalent interaction points and the incorporation of electrophilic moieties designed to form covalent bonds with the biological target [63]. Therefore, integrating noncovalent interaction analyses and covalent bonding potential via a molecular orbital approach could substantially refine the process of rational drug design within the domain of medicinal chemistry.
The results in Table 2 provide insights into the electronic structure and reactivity of the GaM, TTT, and N3 compounds in vacuum and water environments. Overall, the data indicate that solvation in water leads to the stabilization of all three compounds, with the HOMO being stabilized more than the LUMO. This increases the HOMO-LUMO gap, reflecting greater stability in the solvent. When these compounds are placed in a water environment, the HOMO energy becomes more negative and the LUMO energy becomes more positive compared to their vacuum counterparts. For N3, the HOMO energy ranges from −7.0 to −7.3 eV, indicating that it readily donates electrons, making it an effective electron donor. On the other hand, GaM exhibits the least negative HOMO energy, ranging from −5.7 to −6.2 eV, suggesting that it is less prone to donating electrons. Regarding LUMO energy, N3 exhibits the lowest LUMO energy, ranging from −1.5 to −1.6 eV, indicating its high electron-accepting ability. GaM, with its higher LUMO energy ranging from −1.2 to −1.5 eV, is less effective as an electron acceptor. The fact that the GAP increases in water for all compounds suggests that water solvation enhances the stability of these compounds. N3, with the largest GAP ranging from 5.4 to 5.8 eV, should be the most stable among the three, i.e., less prone to undergoing chemical reactions. In contrast, GaM and TTT with smaller GAPs (4.5 to 4.7 eV) should be comparatively more reactive. Electronegativity (χ) represents a molecule’s ability to attract electrons, while global hardness (η) indicates its resistance to changes in electronic configuration. The slight increase in electronegativity and global hardness when the compounds are in a water environment suggests that they exhibit a stronger electron-attracting tendency and enhanced stability in water. N3, with the highest electronegativity (4.3 to 4.4 eV) and the highest global hardness range (2.7 to 2.9 eV), is the most electron-attracting and stable compound. In contrast, GaM, with the lowest electronegativity ranging from 3.5 to 3.9 eV and a global hardness of 2.3 eV, is less electron-attracting and less stable than the other compounds. The variations in electrophilicity (ω), electrodonating power (ω−), and electroaccepting power (ω+) in the presence of water are noteworthy. For N3, they slightly decrease in water, while GaM and TTT experience an increase in these descriptor values. N3 maintains the highest values for these parameters even after the slight decrease, signifying its strong reactivity and its ability to both donate and accept electrons. GaM exhibits the lowest ω− and ω+ in both solvents, indicating its reduced propensity to donate or accept electrons. The increase in net electrophilicity (Δω±) in the presence of water implies that water enhances the overall reactivity of these compounds. Once again, N3 stands out with the highest Δω±, indicating that it is the most reactive compound among the three. GaM, with the lowest Δω±, exhibits lower reactivity compared to the other compounds.
Table 2.
Global reactivity descriptors for GaM, N3, and TTT molecules optimized both in vacuum and water solvents at the PBE0/Def2-TZVP level. All units are measured in eV.
The results show N3 is the most reactive compound with the highest electronegativity, hardness, and electrophilic power, while GaM is the least reactive with the lowest values. Solvation in water stabilizes all three compounds.
According to the observed FMO calculations, GaM exhibits a reactivity profile quite comparable to that of the TTT molecule, a well-known reference compound. However, GaM distinguishes itself with a notably heightened capacity for electron capture, as indicated by its net electrophilicity. This unique ability to capture electrons seems to have its epicenter on the metal atom within GaM, but it does not confine itself to just the metal atom; instead, it extends its influence to neighboring atoms by drawing in the surrounding electronic cloud. In order to visualize and precisely quantify the magnitude of this electron-attracting effect, we employed molecular electrostatic potential (MEP) calculations for the three compounds under investigation. These MEP calculations provide a detailed map of the electron distribution around the molecules, shedding light on the areas where electrons are either drawn toward or repelled from the molecular structure. The insights gained from these MEP calculations will help us better understand the electron-capturing behavior of GaM and its implications for potential applications.
The electrostatic molecular potential serves as a valuable and insightful analytical instrument for evaluating drug reactivity when exposed to electrophilic and nucleophilic attacks. This assessment is crucial for understanding how pharmaceutical compounds interact with their target receptors and biological systems [64]. As depicted in Figure 1, the electrostatic molecular potential highlights specific charge distributions and regions of interest. Notably, GaM exhibits a distinctive charge distribution pattern. It is characterized by a pronounced positive charge (depicted in blue) concentrated around the methyl groups, indicating an electrophilic nature in these areas. Simultaneously, there is a negative charge predominantly localized on the metal-bound carboxylic groups, signifying susceptibility to nucleophilic attacks. This dual charge distribution suggests that GaM may engage in both electrophilic and nucleophilic interactions, making it a versatile candidate for pharmaceutical reactivity. In contrast, another compound, TTT, presents a distinct electrostatic profile. In Figure 1, TTT shows positive charge regions that surround its aromatic ring, depicted in green. These positive regions indicate a hydrophobic nature, which might suggest a preference for interactions in non-polar environments. Understanding these electrostatic characteristics is vital for predicting how TTT may interact with hydrophobic binding sites within biological systems or receptor proteins.
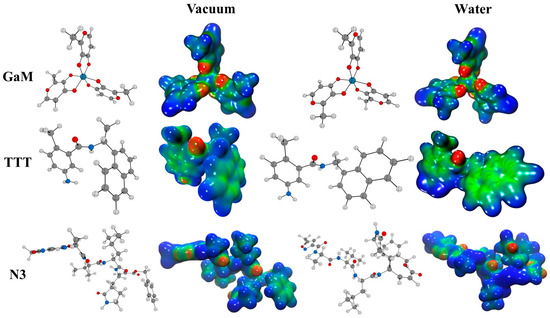
Figure 1.
A schematic representation of molecular electrostatic potential (MEP) maps (isovalue is 0.03 a.u.) for GaM, TTT, and N3 ais shown in both a vacuum and a water environment. In these representations, areas shaded in red, orange, and yellow denote electrophilic reactivity, while those in green and blue correspond to nucleophilic reactivity. The following elements have been highlighted in this figure: carbon (gray), hydrogen (light gray), oxygen (red), gallium (sapphire), and nitrogen (blue).
3.2. Molecular Docking Analysis
The structural features of two crucial enzymes associated with SARS-CoV-2 are the main protease (Mpro) and the papain-like protease (PLpro). These enzymes are pivotal in mediating viral replication and transcription [52,65], making them prime targets for drug development and therapeutic interventions. Mpro, also known as 3CLpro, is a dimeric enzyme with each protomer consisting of three distinct domains. The first two domains, Domain I and Domain II, form an antiparallel β-barrel structure, while Domain III is a globular cluster of five α-helices connected to Domain II by a long loop. The substrate binding site, where the catalytic action occurs, is between Domain I and Domain II and involves a catalytic dyad of Cys145 and His41 [52,65]. The substrate binding pocket of Mpro is highly conserved among coronaviruses, making it an attractive target for the development of broad-spectrum antiviral drugs. Inhibitors such as N3 and N1 covalently bind to Cys145, effectively occupying this pocket. The binding of inhibitors can induce significant conformational changes in Mpro, effectively sealing the entrance to the substrate binding pocket. This structural flexibility is crucial for accommodating different substrates and inhibitors [52,65,66]. PLpro is a monomeric enzyme characterized by four subdomains: the N-terminal ubiquitin-like domain (Ubl), the α-helical thumb domain, the β-stranded finger domain, and the palm domain. The catalytic triad responsible for PLpro’s enzymatic activity includes Cys111, His272, Tyr264, Tyr268, and Asp286 [53,56,67,68]. PLpro is highly similar to its counterpart in the SARS-CoV virus, sharing conserved structural features and an impressive 82% sequence identity. This similarity suggests that research on SARS-CoV PLpro can inform studies on SARS-CoV-2 PLpro. Crystal structures have shown that the binding of inhibitors causes the closure of the BL2 loop, sealing the cleft entrance where substrate cleavage occurs. The S3–S4 subsites within the substrate binding cleft are critical for accommodating key inhibitor groups, such as the zinc-binding moiety of TTT. Inhibitors targeting cysteine residues in the zinc-binding domain are particularly promising for drug development [53,56,67].
To investigate the possible mechanism by which selected GaM acts, molecular docking analysis of GaM was carried out at the active sites of SARS-CoV-2 Mpro and PLpro. The molecular docking scores are displayed in Table 3. The results of molecular docking studies are presented in terms of interaction energy (∆Ebinding), which reflects the strength of binding between the ligands and the target proteins. The ligands GaM, TTT, and N3 were evaluated for their binding interactions with two key proteins of the SARS-CoV-2 virus, Mpro and PLpro. GaM demonstrated moderate binding affinities with both Mpro and PLpro. With ∆Ebinding values of −6.98 kcal/mol for Mpro and −5.90 kcal/mol for PLpro, GaM appears to form reasonably favorable interactions with both proteins. TTT exhibited a significant interaction with SARS-CoV-2 PLpro, with an impressive ∆Ebinding value of −10.34 kcal/mol. This indicates a strong and favorable binding affinity. On the other hand, TTT has a higher ∆Ebinding value specified for its interaction with Mpro, which indicates that the calculations performed are correct since it is not its original protein. N3, showed an ∆Ebinding of −7.29 kcal/mol with Mpro, similar to the GaM interaction with Mpro. However, N3 has a higher ∆Ebinding value specified for its interaction with PLpro, which indicates that the calculations performed are correct since it is not its original protein.
Table 3.
Molecular docking study between selected ligands, GaM, TTT, and N3, and SARS-CoV-2 Mpro and PLpro. Intermolecular docking values (*), presented with their interaction energy (∆Ebinding). All units are measured in kcal/mol.
Table 4 presents a detailed overview of the interactions between GaM organometallic molecules and the amino acid residues of SARS-CoV-2 Mpro and PLpro proteases. This interaction data sheds light on the molecular mechanisms through which GaM interacts with these proteases, providing valuable insights into potential inhibitory effects. In the case of Mpro, several amino acid residues are involved in interacting with GaM. Notably, Cys145, Glu166, and Gln189 are identified as forming hydrogen bonds with GaM. These hydrogen bonds imply a strong and specific binding between GaM and Mpro, indicating a potentially robust inhibitory effect. Additionally, other amino acids like Phe140, His163, Ser144, and Ser305 participate in the interaction primarily through van der Waals forces and hydrophobic interactions. This diverse array of interactions suggests that GaM can engage with Mpro through multiple mechanisms, further reinforcing the likelihood of effective inhibition. In contrast, GaM interacts with PLpro through a different set of amino acid residues. Only two amino acids, Arg166 and Tyr264, form hydrogen bonds with GaM in the case of PLpro. This limited number of hydrogen bonding interactions in comparison to Mpro implies a comparatively weaker binding affinity for GaM with PLpro. Other amino acids in PLpro contribute to the interaction mainly through van der Waals forces and hydrophobic interactions, which is similar to the interaction pattern observed with Mpro. GaM forms a greater number of hydrogen bonds with Mpro as compared to PLpro. This difference in hydrogen bonding interactions suggests that GaM likely exhibits a stronger binding affinity for Mpro, making it a more potent inhibitor for this protease. The distinct sets of interacting residues also indicate that GaM binds to different regions or sites on the two proteases. These insights underscore the potential of GaM as an inhibitor of these proteases and suggest that it might exert a stronger inhibitory effect on Mpro due to a greater number of hydrogen bonding interactions. This knowledge can be instrumental in the development of therapeutic strategies targeting SARS-CoV-2 proteases for antiviral drug development.
Table 4.
Amino acid residues of SARS-CoV-2 Mpro and PLpro and hydrogen bonding (*) with the GaM molecule within a distance of 3.5 Å.
All these interactions are complemented by the analyses performed using the non-covalent interaction index (Figure 2). This index provides us with a deeper and more accurate view of the relationships that are established between the components (in terms of their non-covalent interactions). These relationships can include hydrogen bonds, dispersion forces, ionic interactions, and other types of non-covalent attractions. The results of these analyses allow for a more complete understanding of how the molecules or particles involved are intertwined and affect each other, which in turn sheds light on the properties and behavior of the system under study.
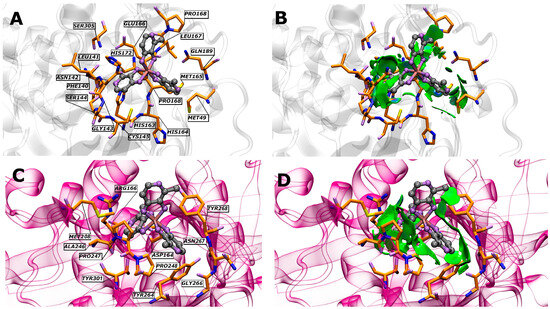
Figure 2.
Schematic representations of the NCIPLOT isosurface gradient (0.5 a.u.) of GaM on the structure of SARS-CoV-2 (A,B) Mpro and (C,D) PLpro. The sign of λ2 enables the identification of the interaction type. Attractive interactions appear at λ2 < 0, whereas in cases where λ2 is positive (as in rings or cages), usually several atoms interact but are not bonded, which corresponds to steric crowding according to classical chemistry. The blue regions indicate pronounced attractive interactions, whereas the green regions indicate relatively weaker interactions.
4. Conclusions
In conclusion, the computational studies conducted on gallium maltolate (GaM) demonstrate its promising potential as an antiviral agent against SARS-CoV-2. Density functional theory (DFT) calculations have revealed GaM’s distinct reactivity, showcasing its increased electron-accepting ability in comparison to reference inhibitors like N3. Moreover, molecular docking studies have indicated GaM’s effective binding to the active sites of SARS-CoV-2 proteases Mpro and PLpro, with binding energies comparable to established potent inhibitors such as N3. A detailed analysis has uncovered that GaM forms important bonding interactions, particularly hydrogen bonds, with crucial amino acid residues like Glu166 and Cys145 in Mpro. These interactions likely play a pivotal role in facilitating GaM’s ability to inhibit viral replication by targeting essential components of the viral machinery. Consequently, based on the wealth of computational evidence, GaM is supported as a promising candidate for further investigation as an antiviral therapeutic against COVID-19 or as a foundational basis for delving into the broader realm of metallodrugs, particularly those involving gallium.
Author Contributions
Investigation and writing, K.T.-H.; writing—review and editing, M.I.O.; investigation, D.I.; conceptualization, L.L.-P.; writing—original draft preparation, L.L.-P., L.R. and A.V.-N.; conceptualization, J.A.-H.; original draft preparation, writing—review and editing and investigation, O.Y. and W.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Agencia Nacional de Investigación y Desarrollo: 11241091.
Acknowledgments
This work was supported by the financial support of the National Agency for Research and Development (ANID) through Fondecyt projects 1211128 (W. T.) and National Agency for Research and Development (ANID)/Scholarship Program/Becas Doctorado Nacional/2019-21190427 (D. I.) and 2020-21201177 (L. L.-P.). O. Y. gratefully acknowledge the UDLA-PIR202409. Vicerrectorado de Investigación de la Universidad Nacional de Ingeniería FC-PF-01-2022 (K.T.-H) and (A.V.-N). Powered@NLHPC: This research waspartially supported by the supercomputing infrastructure of the NLHPC (ECM-02).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DFT | Density functional theory |
| COVID-19 | Coronavirus disease 2019 |
| GaM | Gallium maltolate |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| Mpro | Main protease |
| PLpro | Papain-like protease |
| FMO | Frontier molecular orbital |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| MEP | Molecular electrostatic potential |
| NCI | Non-covalent interaction |
| RDG | Reduced density gradient |
| TTT | 5-amino-2-methyl-N-[(1R)-1-naphthalen-1-ylethyl] benzamide |
| N3 | N-[(5-methyl-1,2-oxazol-3-yl)carbonyl]-L-alanyl-L-valyl-N-{(2S,3E)-5-(benzyloxy)-5-oxo-1-[(3S)-2-oxopyrrolidin-3-yl]pent-3-en-2-yl}-L-leucinamide |
| η | Global hardness |
| χ | Electronegativity |
| ω | Electrophilicity |
| ω+ | Electron acceptor |
| ω− | Electron donator |
| Δω± | Net electrophilicity |
| ΔEbinding | Binding energy from molecular docking calculations |
References
- Žákovská, A.; Nováková, O.; Balcarová, Z.; Bierbach, U.; Farrell, N.; Brabec, V. DNA Interactions of Antitumor Trans-[PtCl2(NH3)(Quinoline)]. Eur. J. Biochem. 1998, 254, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I. Platinum Complexes as Anticancer Agents. Recent Pat. Anti-Cancer Drug Discov. 2006, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Gómez, M. Uso de Cisplatino y Derivados de Platino En Quimioterapia. 2017. Available online: https://hdl.handle.net/20.500.14352/20714 (accessed on 24 January 2024).
- Liu, W.-P.; Ye, Q.-S.; Yu, Y.; Chen, X.-Z.; Hou, S.-Q.; Lou, L.-G.; Yang, Y.-P.; Wang, Y.-M.; Su, Q. Novel Lipophilic Platinum(II) Compounds of Salicylate Derivatives. Platin. Met. Rev. 2008, 52, 163–171. [Google Scholar] [CrossRef]
- Was, H.; Borkowska, A.; Bagues, A.; Tu, L.; Liu, J.Y.H.; Lu, Z.; Rudd, J.A.; Nurgali, K.; Abalo, R. Mechanisms of Chemotherapy-Induced Neurotoxicity. Front. Pharmacol. 2022, 13, 750507. [Google Scholar] [CrossRef] [PubMed]
- Eryazici, I.; Moorefield, C.N.; Newkome, G.R. Square-Planar Pd(II), Pt(II), and Au(III) Terpyridine Complexes: Their Syntheses, Physical Properties, Supramolecular Constructs, and Biomedical Activities. Chem. Rev. 2008, 108, 1834–1895. [Google Scholar] [CrossRef]
- Roberts, J.J.; Thomson, A.J. The Mechanism of Action of Antitumor Platinum Compounds. In Progress in Nucleic Acid Research and Molecular Biology; Cohn, W.E., Ed.; Academic Press: Cambridge, MA, USA, 1979; Volume 22, pp. 71–133. ISBN 0079-6603. [Google Scholar]
- Lippard, S.J. New Chemistry of an Old Molecule: Cis-[Pt(NH3)2Cl2]. Science 1982, 218, 1075–1082. [Google Scholar] [CrossRef]
- Kowol, C.R.; Heffeter, P.; Miklos, W.; Gille, L.; Trondl, R.; Cappellacci, L.; Berger, W.; Keppler, B.K. Mechanisms Underlying Reductant-Induced Reactive Oxygen Species Formation by Anticancer Copper(II) Compounds. J. Biol. Inorg. Chem. 2012, 17, 409–423. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Dong, S.; Li, X. DNa Breakage Induced by Piceatannol and Copper(II): Mechanism and Anticancer Properties. Oncol. Lett. 2012, 3, 1087–1094. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ulukaya, E.; Ari, F.; Dimas, K.; Ikitimur, E.I.; Guney, E.; Yilmaz, V.T. Anti-Cancer Activity of a Novel Palladium(II) Complex on Human Breast Cancer Cells in Vitro and in Vivo. Eur. J. Med. Chem. 2011, 46, 4957–4963. [Google Scholar] [CrossRef]
- Ferraz, K.S.O.; Ferandes, L.; Carrilho, D.; Pinto, M.C.X.; Leite, M.d.F.; Souza–Fagundes, E.M.; Speziali, N.L.; Mendes, I.C.; Beraldo, H. 2-Benzoylpyridine-N(4)-Tolyl Thiosemicarbazones and Their Palladium(II) Complexes: Cytotoxicity against Leukemia Cells. Bioorg. Med. Chem. 2009, 17, 7138–7144. [Google Scholar] [CrossRef]
- Abu-Surrah, A.S.; Abu Safieh, K.A.; Ahmad, I.M.; Abdalla, M.Y.; Ayoub, M.T.; Qaroush, A.K.; Abu-Mahtheieh, A.M. New Palladium(II) Complexes Bearing Pyrazole-Based Schiff Base Ligands: Synthesis, Characterization and Cytotoxicity. Eur. J. Med. Chem. 2010, 45, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Messori, L. Molecular Mechanisms and Proposed Targets for Selected Anticancer Gold Compounds. Curr. Top. Med. Chem. 2011, 11, 2647–2660. [Google Scholar] [CrossRef]
- Lin, I.W.-S.; Lok, C.-N.; Yan, K.; Che, C.-M. A Silver Complex of N,N′-Disubstituted Cyclic Thiourea as an Anti-Inflammatory Inhibitor of IκB Kinase. Chem. Commun. 2013, 49, 3297–3299. [Google Scholar] [CrossRef]
- Banti, C.N.; Giannoulis, A.D.; Kourkoumelis, N.; Owczarzak, A.M.; Poyraz, M.; Kubicki, M.; Charalabopoulos, K.; Hadjikakou, S.K. Mixed Ligand–Silver(i) Complexes with Anti-Inflammatory Agents Which Can Bind to Lipoxygenase and Calf-Thymus DNA, Modulating Their Function and Inducing Apoptosis†. Metallomics 2012, 4, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Gaiddon, C.; Schellens, J.H.M.; Beijnen, J.H.; Sava, G. Approaching Tumour Therapy beyond Platinum Drugs: Status of the Art and Perspectives of Ruthenium Drug Candidates. J. Inorg. Biochem. 2012, 106, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Sava, G. Ruthenium Anticancer Compounds: Myths and Realities of the Emerging Metal-Based Drugs. Dalton Trans. 2011, 40, 7817–7823. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.C.; Lage, D.P.; Ferreira, L.S.; Saboia-Vahia, L.; Coelho, E.A.F.; Belo, V.S.; Teixeira-Neto, R.G.; Soares, L.F.; Chagas, R.C.R.; da Silva, E.S. Leishmanicidal Activity of Ibuprofen and Its Complexes with Ni(II), Mn(II) and Pd(II). Inorg. Chem. Commun. 2020, 113, 107756. [Google Scholar] [CrossRef]
- Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Crisponi, G.; Zoroddu, M.A. Noble Metals in Medicine: Latest Advances. Coord. Chem. Rev. 2015, 284, 329–350. [Google Scholar] [CrossRef]
- Choroba, K.; Machura, B.; Szlapa-Kula, A.; Malecki, J.G.; Raposo, L.; Roma-Rodrigues, C.; Cordeiro, S.; Baptista, P.V.; Fernandes, A.R. Square Planar Au(III), Pt(II) and Cu(II) Complexes with Quinoline-Substituted 2,2′:6′,2″-Terpyridine Ligands: From in Vitro to in Vivo Biological Properties. Eur. J. Med. Chem. 2021, 218, 113404. [Google Scholar] [CrossRef]
- Karges, J.; Cohen, S.M. Metal Complexes as Antiviral Agents for SARS-CoV-2. ChemBioChem 2021, 22, 2600–2607. [Google Scholar] [CrossRef]
- Jaros, S.W.; Król, J.; Bażanów, B.; Poradowski, D.; Chrószcz, A.; Nesterov, D.S.; Kirillov, A.M.; Smoleński, P. Antiviral, Antibacterial, Antifungal, and Cytotoxic Silver(I) BioMOF Assembled from 1,3,5-Triaza-7-Phoshaadamantane and Pyromellitic Acid. Molecules 2020, 25, 2119. [Google Scholar] [CrossRef] [PubMed]
- Aprajita; Choudhary, M. Design, Synthesis and Characterization of Novel Ni(II) and Cu(II) Complexes as Antivirus Drug Candidates against SARS-CoV-2 and HIV Virus. J. Mol. Struct. 2022, 1263, 133114. [Google Scholar] [CrossRef]
- De Paiva, R.E.F.; Marçal Neto, A.; Santos, I.A.; Jardim, A.C.G.; Corbi, P.P.; Bergamini, F.R.G. What Is Holding Back the Development of Antiviral Metallodrugs? A Literature Overview and Implications for SARS-CoV-2 Therapeutics and Future Viral Outbreaks. Dalton Trans. 2020, 49, 16004–16033. [Google Scholar] [CrossRef] [PubMed]
- Haribabu, J.; Srividya, S.; Mahendiran, D.; Gayathri, D.; Venkatramu, V.; Bhuvanesh, N.; Karvembu, R. Synthesis of Palladium(II) Complexes via Michael Addition: Antiproliferative Effects through ROS-Mediated Mitochondrial Apoptosis and Docking with SARS-CoV-2. Inorg. Chem. 2020, 59, 17109–17122. [Google Scholar] [CrossRef]
- Rasey, J.S.; Nelson, N.J.; Larson, S.M. Tumor Cell Toxicity of Stable Gallium Nitrate: Enhancement by Transferrin and Protection by Iron. Eur. J. Cancer Clin. Oncol. 1982, 18, 661–668. [Google Scholar] [CrossRef]
- Baran, E.J. Metalofármacos: Una Nueva Perspectiva Para La Farmacología y La Medicina. Anales Acad. Nac. Cs. Ex. Fís. Nat. 2014, 66, 5–21. [Google Scholar]
- Yin, H.Y.; Gao, J.J.; Chen, X.; Ma, B.; Yang, Z.S.; Tang, J.; Wang, B.W.; Chen, T.; Wang, C.; Gao, S.; et al. A Gallium(III) Complex That Engages Protein Disulfide Isomerase A3 (PDIA3) as an Anticancer Target. Angew. Chem.-Int. Ed. 2020, 59, 20147–20153. [Google Scholar] [CrossRef]
- Bernstein, L.R.; Zhang, L. Gallium Maltolate Has in Vitro Antiviral Activity against SARS-CoV-2 and Is a Potential Treatment for COVID-19. Antivir. Chem. Chemother. 2020, 28, 1–4. [Google Scholar] [CrossRef]
- Mohapatra, R.K.; Perekhoda, L.; Azam, M.; Suleiman, M.; Sarangi, A.K.; Semenets, A.; Pintilie, L.; Al-Resayes, S.I. Computational Investigations of Three Main Drugs and Their Comparison with Synthesized Compounds as Potent Inhibitors of SARS-CoV-2 Main Protease (Mpro): DFT, QSAR, Molecular Docking, and in Silico Toxicity Analysis. J. King Saud. Univ. Sci. 2021, 33, 101315. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision B.01; EEUU: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Pino-Rios, R.; Yañez, O.; Inostroza, D.; Ruiz, L.; Cardenas, C.; Fuentealba, P.; Tiznado, W. Proposal of a Simple and Effective Local Reactivity Descriptor through a Topological Analysis of an Orbital-Weighted Fukui Function. J. Comput. Chem. 2017, 38, 481–488. [Google Scholar] [CrossRef]
- Koopmans, T. Über Die Zuordnung von Wellenfunktionen Und Eigenwerten Zu Den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Lewars, E.G. Computational Chemistry-Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Springer: New York, NY, USA, 2011; ISBN 978-90-481-3860-9. [Google Scholar]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; John Wiley & Sons: New York, NY, USA, 2002; ISBN 9780471333685. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2004; ISBN 978-0-470-09182-1. [Google Scholar]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness; Wiley: New York, NY, USA, 2005; ISBN 9783527294824. [Google Scholar]
- Gázquez, J.L.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Hou, X.; Du, J.; Zhang, J.; Du, L.; Fang, H.; Li, M. How to Improve Docking Accuracy of AutoDock4.2: A Case Study Using Different Electrostatic Potentials. J. Chem. Inf. Model. 2013, 53, 188–200. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Papain-like Protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2024-1; Canvas, Schrödinger, LLC: New York, NY, USA, 2024.
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Osorio, M.I.; Yáñez, O.; Gallardo, M.; Zuñiga-Bustos, M.; Mulia-Rodríguez, J.; López-Rendón, R.; García-Beltrán, O.; González-Nilo, F.; Pérez-Donoso, J.M. Search for Novel Potent Inhibitors of the SARS-CoV-2 Papain-like Enzyme: A Computational Biochemistry Approach. Pharmaceuticals 2022, 15, 986. [Google Scholar] [CrossRef] [PubMed]
- Yañez, O.; Osorio, M.I.; Uriarte, E.; Areche, C.; Tiznado, W.; Pérez-Donoso, J.M.; García-Beltrán, O.; González-Nilo, F. In Silico Study of Coumarins and Quinolines Derivatives as Potent Inhibitors of SARS-CoV-2 Main Protease. Front. Chem. 2021, 8, 1273. [Google Scholar] [CrossRef]
- Yañez, O.; Osorio, M.I.; Areche, C.; Vasquez-Espinal, A.; Bravo, J.; Sandoval-Aldana, A.; Pérez-Donoso, J.M.; González-Nilo, F.; Matos, M.J.; Osorio, E.; et al. Theobroma Cacao L. Compounds: Theoretical Study and Molecular Modeling as Inhibitors of Main SARS-CoV-2 Protease. Biomed. Pharmacother. 2021, 140, 111764. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Pichler, W.J. The Important Role of Non-Covalent Drug-Protein Interactions in Drug Hypersensitivity Reactions. Allergy 2022, 77, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Tandon, H.; Yadav, P.; Chakraborty, T.; Suhag, V. Can Chemical Reactivity Descriptors Explain Catalytic Reactivity? J. Organomet. Chem. 2022, 960, 122229. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent Inhibitors: A Rational Approach to Drug Discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID-19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 Main Protease as Drug Target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Ershov, P.V.; Yablokov, E.O.; Mezentsev, Y.V.; Chuev, G.N.; Fedotova, M.V.; Kruchinin, S.E.; Ivanov, A.S. SARS-COV-2 Coronavirus Papain-like Protease PLpro as an Antiviral Target for Inhibitors of Active Site and Protein–Protein Interactions. Biophysics 2022, 67, 902–912. [Google Scholar] [CrossRef]
- Patel, R.; Prajapati, J.; Rao, P.; Rawal, R.M.; Saraf, M.; Goswami, D. Repurposing the Antibacterial Drugs for Inhibition of SARS-CoV2-PLpro Using Molecular Docking, MD Simulation and Binding Energy Calculation. Mol. Divers. 2022, 26, 2189–2209. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).