Abstract
Past anti-bacterial use of bacteriophages (phage therapy) is already well reviewed as a potential therapeutic response to the emergence of multidrug-resistant, pathogenic bacteria. Phage therapy has been limited by the following. (1) The success rate is too low for routine use and Food and Drug Administration (FDA) approval. (2) Current strategies of routine phage characterization do not sufficiently improve the success rate of phage therapy. (3) The stability of many phages at ambient temperature is not high enough to routinely store and transport phages at ambient temperature. In the present communication, we present new and previous data that we interpret as introductory to biophysically and efficiently transforming phage therapy to the needed level of effectiveness. Included are (1) procedure and preliminary data for the use of native gel electrophoresis (a low-cost procedure) for projecting the therapy effectiveness of a newly isolated phage, (2) data that suggest a way to achieve stabilizing of dried, ambient-temperature phages via polymer embedding, and (3) data that suggest means to increase the blood persistence, and therefore the therapy effectiveness, of what would otherwise be a relatively low-persistence phage.
1. Introduction
The problem of multidrug-resistant (MDR) bacterial pathogens is now well documented and reviewed. In summary, antibiotic therapy has been compromised, and sometimes thwarted, by MDR pathogens. The reason is that the pathogens are resistant to either most or all antibiotics (reviews: [1,2,3,4,5,6,7,8,9,10]). The consequence is known to be lethal for roughly 48,000 citizens of the USA in 2019, when Clostridioides difficile (not typically pathogenic, but made pathogenic by long-term antibiotic use) is included [9,10]. Worldwide, the estimate is at least 4.95 million deaths in 2019, with the top six pathogens being Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Streptococcus pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa [10]. MDR bacterial infections in 2019 were the third leading cause of death worldwide, behind heart attacks and stroke [10]. However, ambiguity often exists for both in-hospital and outpatient cause of death. By one informed calculation, the number of deaths caused by MDR bacteria is higher, possibly as much as 7× higher in the US [11].
Therapeutic use of anti-bacterial viruses (bacteriophages or phages) is an obvious anti-bacterial measure (phage therapy) to counter infections by MDR pathogenic bacteria. However, as previously reviewed [12,13,14,15,16,17,18,19,20], phage therapy sometimes works but too often does not. Phage therapy is not yet an FDA-approved procedure in the US unless performed on a compassionate use basis, e.g., with a patient in an otherwise hopeless condition [17,18,19,20]. Nonetheless, clinical trials are in progress, with the goal of answering the question of whether or not phage therapy works [17,19,20,21,22]. The subtext of this question is, however, in conflict with the history of phage therapy. Our point here is that more appropriate is the question of which phages should be used and how. This point is emphasized by the spectacular successes that have occurred (recently: [17,23]) in the context of too many failures.
2. Phages to Use and Not to Use for Phage Therapy
2.1. Known Past Basics
A clear plaque is criterion #1 for choosing phages for phage therapy. The reason is that clear plaque-forming phages are the most likely to be lytic, i.e., bacterial lysis-inducing without capacity for lysogeny, i.e., without capacity to become a replicating component of the bacterial genome [12,13,14,15,16,17,18,19,20,24,25]. The preparation used for phage therapy is typically a mixture of several phages (phage cocktail); multi-phage cocktails appear to be the current standard [12,13,14,15,16,17,18,19,20].
Lysogenic phages are avoided because of the carrying, by some lysogenic phages, of genes encoding either bacterial toxins [24] or antibiotic resistance [24,25]. Verification of the absence of these latter genes can be achieved by whole-genome DNA sequencing [25]. This is criterion #2, which is used because of the potential of gene transfer to bacterial hosts.
Historically, other criteria appear to have been used but were not well documented. For example, one gets the impression from the second paragraph of [26] that, in the 1930s, the favored anti-typhoid fever phages made relatively small clear plaques. However, no definitive, rigorous statement was made. The early days of phage therapy appear to have been characterized by semi-empirical phage selection. One imagines that this is sometimes still occurring. Our data-generated opinion is that more precise criteria are needed for a next-generation, FDA-approved phage therapy.
2.2. Next-Generation Biophysical Screening of Phages: Average Electrical Surface Charge Density (σ)
Our main point (opinion) here is that the key objective is to develop additional phage screening criteria that bring phage therapy to the point that its success rate is at least as high as the success rate of the use of antibiotics with non-MDR bacterial pathogens. High speed is important because the observed [27,28] development of bacterial resistance to phages implies that phage therapy cocktails will periodically require new phages to keep the cocktail effective. In general, the relatively high speed and low cost of phage therapy-based response to increased bacterial resistance is anticipated to be a major advantage of phage therapy in relation to antibiotic therapy.
A data-suggested screening criterion is the rate of a patient’s innate immune clearance of the phage (reviews [16,29,30,31,32]). If one makes the pessimistic assumption of high clearance rate for all phages, one might conclude that phage therapy will never be routinely effective. In contrast, phages are found to sometimes enhance the innate immune clearing of bacteria, which would enhance phage therapy [31,32]. The complexity involved suggests an approach that does not depend on detailed knowledge of innate immunity pathways.
Specifically, a relatively optimistic assumption is that (1) the key parameter is indeed the rate at which inoculated phages are removed from circulation by innate immunity, and (2) this rate varies significantly among phages. If so, then lowering this rate is the key to improving the success rate of phage therapy.
To test the accuracy of this latter assumption in a controlled way, we [33] recently assembled (1) a collection of four phages and (2) bacterial hosts to selectively plate each of the phages. These phages and bacteria were used to perform the first test of the blood lifetime (to be called persistence) of four different phages in one mouse. The blood titers (plaque-forming units [PFU] per mL), normalized to the total PFU injected (approximately the same for all four phages), varied by about 10,000× at 2–4 h after intraperitoneal injection. Two phages, including phage T3, had relatively high persistence, and two, including the T3 relative, T7, had low persistence. We discuss more precise quantification of persistence in Section 5. Notably, the difference in persistence between T3 and T7, which are the same size and genetically related [34], indicates that phage size is not the only persistence-determining factor involved, although it might be one of several persistence-determining factors.
T3 and T7 differ in average electrical surface charge density (σ), as determined by extrapolation of gel electrophoretic migration to a gel concentration of 0 for both T3 and T7 [35]. In solution (without a gel), electrophoretic mobility depends only on σ for particles the size of the smallest and larger phages [36,37]. The σ of both T3 and T7 is negative, with T3 being 1.3× more negative than T7 [35]. Correlation of more negative σ with higher persistence has been observed for red blood cells. The sialyation of glycophorin, on the surface of red blood cells, generates a negative σ relatively high in magnitude [38]. Although more data are needed for phages, our working assumption is that, relative to phage T7, the co-existence of the more negative T3 σ and the higher T3 persistence are part of a σ/persistence correlative pattern. That is to say, rapid determination of σ is likely to be one of possibly several criteria useful to determine the probability that a newly isolated phage will have high persistence.
3. Biophysical Technique
3.1. Basics of Native Agarose Gel Electrophoresis (AGE)
For comparing σ values, a relatively rapid, inexpensive, and accurate procedure is to perform native (intact phage) agarose gel electrophoresis (AGE) of the two phages to be compared, according to the following protocol. (1) Embed agarose gels of different agarose percentage, A, (running gels) in a single more concentrated, physically supportive agarose frame (frame gel). Use an ultra-strong, ultra-low electro-osmosis agarose for the running gels (Electro-osmosis is electrophoresis-generated motion of water and will distort the results unless kept low.). (2) Determine distance migrated vs. A for both phages during electrophoresis. (3) Extrapolate distance migrated to an A of 0 and compare the result for the two phages [39]. That is how the data for T3 and T7 were obtained [35].
For any newly characterized phage, we have simplified the data processing by dividing this extrapolated distance by the extrapolated distance for phage T3, obtained in the same multigel. This is a process of normalization. A normalized σ will be called σN. We use σN, rather than the physical concept, σ, because (1) σ is an imprecise concept, complicated by lack of knowledge of the surface of slip, with some details implicit in the Debye–Hückel model [40], and (2) no reason exists to perform an analysis beyond σN at this point. We use T3 as the normalization standard because of the high persistence and high stability of T3. The stability includes stability in the presence of chelating agents and several buffers, which has made T3 a standard used for AGE of chromatin, for example [41,42].
That being said, we already know that persistence is dependent on more than σN. Specifically, phage G, a myophage, is a low-persistence phage [43]. The value of σN, however, is 1.20 (plot indicated by G in Figure 1; the T3 plot is indicated by T3 in Figure 1). This observation is in contradiction to the correlation of σN ≥ 1.0 with high persistence. Nonetheless, high-persistence phage T4 (also a myophage) has a σN of 0.99 (T4 plot in Figure 1), consistent with this correlation. Phage G is also the largest phage that has been laboratory-propagated; it has a genome about 3× the length of the T4 genome and a capsid about 60% larger. Correlation of large size with relatively high innate immune uptake by macrophages has been observed for latex spheres [44]. Thus, relatively large size might explain the relatively low persistence of phage G. Of course, the above analysis needs data from more phages. Moreover, other phage biophysical characteristics are likely to be needed for a complete analysis. These might include density of hydrophobic patches on the surface of the capsid, which can also be electrophoretically determined [45]. Further analysis by AGE is discussed in the next section.
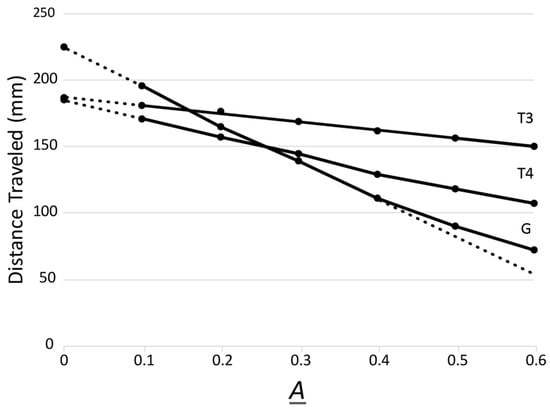
Figure 1.
Multigel AGE analysis of phages T4 and clear-plaque phage G, with a phage T3 standard. AGE was performed for 16.0 h at 1.0 V/cm and 25 °C, as described in [39], with Seakem Gold agarose (Lonza, Rockland, ME, USA) running gels of the following six A values: 0.1, 0.2, 0.3, 0.4, 0.5, and 0.6. The electrophoresis buffer was 0.09 M Tris-acetate, pH 8.4, 0.001 M MgCl2. All three phages were analyzed in the same 18-track multigel. The preparation of phages has been previously described: T3 [35], T4 [33], and G [43]. Clear-plaque phage G has a genomic sequence that differs from both the reference GenBank sequence (JN638751) and its parent at three single nucleotide locations. These changes, at co-ordinates 330,356 (T > C), 343,537 (G > A), and 454,601 (G > A), would produce non-synonymous changes to the (functionally uncharacterized) gene products G_424 (p.V1452A), G_428 (p.G35S), and G_625 (p.G64S), respectively. The dotted lines indicate linear extrapolations.
3.2. Further Phage Characterization by AGE
An advantage of multigel AGE is that one can use the sieving effects of the running gels to obtain information about the size and shape of particles being analyzed. Specifically, as the overall size of particles increases, the slope of the distance migrated vs. A plots increases at the lowest A values. A previous study of rod-shaped phages reveals that the effective radius (RE), as determined by this slope, is closest to the radius of a sphere that has the surface area of the non-spherical particle [46].
Thus, by use of AGE alone, one can estimate the RE of a phage for possible use in determining the probability of high persistence. AGE can be performed directly on phage plaques, without phage purification [47]. Thus, both RE and σN can be determined without purifying phage particles, which might be especially important for high-persistence screening in an emergency, such as a bacteremia projected to be otherwise rapidly lethal.
Furthermore, even some information about phage asymmetry can be obtained from multigel AGE. Specifically, the phage G plot in Figure 1 has upward curvature at the highest A values, as emphasized in Figure 1 by the dotted line that continues the straight line generated at the lowest A values. The opposite curvature is generated at the higher A values by more spherical particles, including podophages, such as T3 and T7 [46]. The upward curvature indicates either a long tail phage (myophage or siphophage) or a rod-shaped phage. Although not yet explored in detail, discrimination of these two can probably be made by determining the dependence on the magnitude of the electrical field used for AGE [48].
4. The Potential Future of Dry Phage Therapy Cocktails: An Improved Source of Phages
Phage storage inactivation is an obvious problem for liquid phage cocktails. This problem was recognized via (1) early phage characterizations [49] and (2) review of early phage therapy failures (review [50]). Typical long-term laboratory storage of phages manages this problem by freezing, typically at temperatures between −70 and −80 °C, in the presence of a cryo-protectant. The PS laboratory submerges a plaque in growth medium with 10% dextran 10 before freezing at −70 °C. Doing this in a biomedical context is expensive and potentially limiting to the use of phage therapy. A further complication is that phage G, for example, is not multi-year stable when stored this way. This may also be true for other phages.
A simpler, less expensive, less machine- and/or power failure-susceptible strategy would be storage of phages in dry conditions and at ambient temperature. The following observation indicates that one way to do this, for at least some phages, is to air-dry the phages in the presence of a phage-stabilizing agent. When a ranch animal proximal soil is dry enough so that molds do not grow during storage at room temperature, this soil is a prolific source of phages [51,52]. The key point here is the phage survival of drying. In our case, the dry environmental phages were (1) exposed to environmental temperatures above 50 °C and (2) air-dried (no vacuum). The pre-drying presence of phages is expected because non-dried samples of ranch animal excretions are found by others to be a prolific source of phages [53,54,55].
A new-phage yield of over 50 phages per week per person is a reasonable expectation for dry, ranch animal-proximal soil. Some of these soil-associated phages are so stabilized that they can be (have been) isolated 18 years after taking of the soil sample. The assumption is that soil-associated, phage-protective agents are involved. Candidates include the polymers that bacteria secrete to form biofilms (review [56]).
Moreover, in-liquid phage-stabilizing compounds exist and work on physical principles that should be applicable to all phages and in near-dry conditions. These compounds do not penetrate phages and thus generate a phage-stabilizing osmotic pressure across the DNA-containing protein shell of the capsid. This osmotic effect was first demonstrated for phages P22 and T7 [57]. It was later demonstrated in more quantitative detail for phage lambda. Its use was a critical part of the determination of the energetics of partial phage DNA packaging [58,59].
Studies in this general direction have been conducted for (1) stabilizing phages for aerosol delivery [60,61], (2) freeze drying, and (3) in-air spray drying [62,63,64,65]. Spray drying is closest to what happens in the environment. These studies appear to have been conducted without awareness of the points made in the previous paragraph, a condition that continues to the most recent review [65]. Nonetheless, when one averages a very large amount of data presented in [65], the conclusion is drawn that among the sugars tested as drying-stabilizers, trehalose and sucrose, both non-reducing disaccharides, are the most effective. Other sugars tested include two disaccharides, both reducing: maltose (α-linked) and lactose (β-linked). Given the above effect(s) of non-penetration of phages, the anomeric-to-anomeric carbon linkage of trehalose and sucrose may be the source of relatively high effectiveness via the provision of non-penetration. In any case, a uniform phage stabilization strategy best assists phage therapy. In our opinion, use of basic biophysical principles, such as those in the previous paragraph, is the best way to arrive at this condition.
5. Enhancing Phage Persistence: Future Quantification of Persistence
One can increase the murine blood persistence of phages lambda and P22 by serial exposure to murine blood, with each exposure followed by (mutagenic) phage propagation [66]. The higher persistence version of phage lambda is (1) a mutant with an identified amino acid change (glutamic acid to lysine) in the major protein of the DNA-encapsulating outer shell of the lambda capsid and (2) more effective in murine phage therapy [66]. This study represents the logical beginning of next-generation phage therapy.
Of course, one cannot be sure that the observed amino acid change is a direct cause of the high persistence. For example, mutations in other genes, not sequenced, may have been the cause via change in the conformation of the major capsid protein. Nonetheless, biomedically, this experiment reveals that directed evolution might be a way to increase phage persistence before the use of phages in phage therapy cocktails. However, biomedically using directed evolution introduces a time delay and a cost increase that makes phage therapy a less attractive prospect. A better approach would be introduction to existing phage therapy cocktails of an agent that increases the persistence of those phages already isolated but of low persistence.
We accidentally discovered a possible way to do this. We (primarily MA) subjected low-persistence phage G [43] to three serial passages through murine blood, with each passage interspersed with propagation of phage G, although propagation was in non-mutagenic conditions. High persistence was indeed achieved for partially purified (Legend to Figure 2) phages. The persistence progressively increased with passage number. The intermediate-persistence steps suggested the existence of several mutations. However, not a single mutation had occurred in the genome of the final product, as determined by whole-genome, Illumina sequencing (which was not practical at the time that the above work on phage lambda [66] was performed). The intermediate-persistence phage G was not sequenced. The initial phage G was a clear-plaque mutant, which had been selected to improve the accuracy of phage titers (Legend to Figure 1).
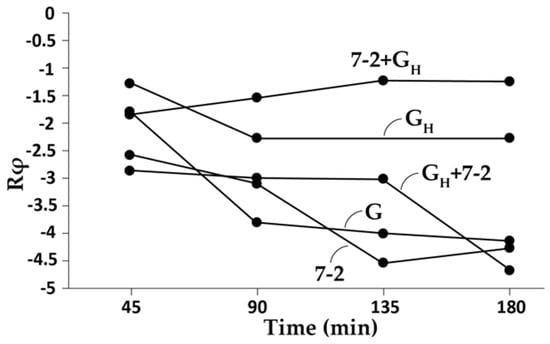
Figure 2.
Transfer of high persistence from phage G to phage 0105phi7-2. To increase phage G persistence, the clear-plaque mutant of phage G was passed through mouse blood three times, as previously described [43]. After each passage, a sample of blood was taken at 45 min after IP inoculation. Phages present were titered and then re-propagated in-gel (plate stock) as previously described [43]. After the final passage and plate stock production, the phages were partially purified by differential pelleting [43] (phage GH), which produced phages with an increase in persistence. Then, the phages were purified by rate zonal centrifugation through a sucrose gradient [43], which caused loss of the persistence increase. The persistence increase was restored by adding an extract of uninfected cells to the high-persistence selected phage G, but not to its unselected parent. In the current experiment, when GH was used, it was accompanied by 31.7 μL of uninfected cell extract (below). Phage 0105phi7-2 was obtained as described in [43]. Phages and phage mixtures indicated in the text were made and injected intraperitoneally into a 11–12-week-old female C57/BL6 mouse, as previously described [35,43]. The total PFU inoculated were: 0105phi7-2, 1.0 × 1010; G, 1.0 × 1010 in 180 μL final volume. At the times indicated on the horizontal axis, values of Rφ were determined by procedures previously described [43]. The extract of uninfected cells was made by harvesting an uninfected bacterial lawn from 4 × 10 cm Petri plates and 2 × 15 cm in diameter Petri plates. The top agarose layer was harvested and pelleted [43] and then resuspended in 1.0 mL of Milli-Q water (MilliporeSigma, Darmstadt, Germany). The cells were lysed by adding 1.7 mg lysozyme, followed by three cycles of freeze-thawing. This extract was clarified by pelleting at 10,000 rpm at 4 °C for 10 min (Beckman JLA 16.250 rotor, 15,000 g).
Furthermore, the high-persistence, phenotype-only variant of phage G, after the final repropagation and partial purification (to be called phage GH), depended on a host factor for its high persistence. When further purified by rate zonal centrifugation, phage GH lost its high persistence. However, the high persistence of phage GH was recovered by addition of an extract of uninfected host cells. Some details are in the Legend to Figure 2.
The key observation was that host extract-boosted, purified phage GH was found to transfer its high persistence to low-persistence phage 0105phi7-2. This is shown in Figure 2, where normalized in-blood phage titer (Rφ) is plotted vs. time; Rφ is the plaque-forming units (PFU) per ml of blood, divided by the total PFU inoculated intraperitoneally (IP). The persistence for unaltered phage G (G plot in Figure 2) confirmed the low phage G persistence. The plot for host extract-boosted phage GH (GH plot in Figure 2) showed a 10–100× increase in persistence depending on time. In addition, the low persistence of phage 0105phi7-2 (Figure 2; 7-2 plot) was increased when phage GH was mixed with phage 0105phi7-2 and then IP-inoculated (7-2 + GH plot in Figure 2). This increase was accompanied by a decrease in persistence of host extract-boosted phage GH (GH + 7-2 plot in Figure 2). The persistence-transferring agent is not yet known.
A precise definition of high and low persistence has not yet been presented, in part because of the large persistence differences of all comparisons made thus far. Moreover, this quantification will eventually be based on data beyond what we already have.
As a beginning in quantifying persistence, we define high persistence as maintaining a Rφ above 0.03 for at least 4 h. At 107 phages IP-inoculated for phage therapy and 107 pathogenic bacteria per ml of blood, this should be sufficient to clear a phage-susceptible bacteremia, assuming a latent period of less than 45 min and a burst size of at least 10, which are probably pessimistic assumptions for most lytic phages during phage therapy. More precise calculations, persistence definitions, and projections will be made as more data are obtained.
6. Implications for FDA Clearance: Safety and Effectiveness
The first point to note here is that phage therapy is biomedicine. Antibiotic therapy is what might be called chemo-medicine, even though many antibiotics were originally isolated from organisms. Thus, a complete re-think is needed to obtain FDA approval. To illustrate this point, think about the use of leeches to prevent and remove blood clots [67,68], which is also biomedicine and is FDA-approved [68]. No two leeches are exactly the same. Criteria to be used are (1) statistical in character and (2) include a range of possibilities, even after applying statistics. This problem exists and has indeed been solved, even for some complex chemo-medicines. These include tetanus toxoid-bacterial polysaccharide conjugate vaccines such as Prevnar and Pneumovax [69]. These vaccines are expected to have components that vary in size and even composition. Size variability has been found empirically by a procedure of two-dimensional (native) AGE; this procedure was developed for the characterization of phages [70].
In the case of phage therapy, we start from a good position. In the short term (weeks), no phage has ever been found to harm either a human or an animal (reviews [12,13,14,15,16,17,18,19,20,21]) at the levels used for phage therapy. These levels are typically 107–108 PFU. This non-toxicity is not surprising in that titers per ml of this magnitude are sometimes recorded for endogenous human phages in healthy people [71]. To test for possibly rare, short-term adverse reactions, a skin allergy-like test based on a dry phage therapy cocktail (above) might be developed and FDA-required. The long-term effects of phage therapy are not empirically known, and some data suggest that the possibility of negative long-term effects is significant (recent review [72]).
Of course, rapid phage-killing of Gram-negative bacteria potentially creates the problem of toxic levels of bacterial endotoxin (recent review [73]). Post-phage therapy, the occurrence of fever spikes, possibly endotoxin-caused, has been observed (e.g., [23,26,66]). Indeed, this occurrence is presumably a criterion for therapy success. A second FDA requirement might be standards for the time and characteristics of fever monitoring after phage therapy begins.
In the area of therapy effectiveness, dry phage therapy cocktails can be as easily screened as liquid phage preparations. If a cocktail is stabilization agent-embedded on, for example, a non-biological membrane, a small piece of this membrane can be cut away and placed on a Petri dish-contained agar layer that has confluent propagating pathogenic bacteria. Post-incubation, a clear ring, of size to be determined, around the membrane would be a criterion for cocktail effectiveness. This screening (1) is the way that disk-diffusion tests are performed for antibiotics [74,75] and (2) would be performed after previous screening for high persistence.
For extensive wounds, a piece of the phage cocktail membrane can be placed in one region of the wound to test for effectiveness. If effective, then more membrane pieces would be applied. Several different cocktails could be simultaneously tested. Here again, the procedure used could be regulated by the FDA. Apparently, a therapeutic procedure of this type is not practical for antibiotics, presumably because of diffusion-induced lowering of antibiotic concentration. However, the in-wound replication of phages should make this therapeutic procedure practical.
The problem of endotoxin removal from phage therapy cocktails is likely to be limiting for FDA approval. We recently found a reagent and method that can be used to remove over 99% of the in vitro endotoxin activity while also completely inactivating representative Gram-positive and Gram-negative bacteria [76]. Procedures of this type should simplify FDA approval of phage therapy cocktails. These procedures should also have application when removal of endotoxin is limiting for reducing inflammation and reducing pain, for example, after endodontic procedures [77].
7. Contextual Significance of Phage Therapy
Rather than developing new text to describe the origin of the problem of obtaining new antibiotics for MDR bacteria, we quote here the 2022 Annual Report of the drug company Pfizer [78], a company that produces multivalent vaccines for both Streptococcus pneumoniae and Neisseria meningitidis (in this quotation, AMR = antimicrobial resistance). “A robust pipeline of new antimicrobials is essential to restoring the balance against increasing rates of AMR. However, significant economic hurdles have made research and development in this area a challenge. No novel class of antibiotics has been launched for almost 40 years, and even when newly approved treatments come to market, they may be used sparingly to support good antimicrobial stewardship practices—making it difficult to recover the high cost associated with development. New reimbursement models that more fully reflect the complete value of antimicrobials are critical”.
The point missing in the above quotation is, obviously, the potential of covering the limitations of antibiotics with the use of phage therapy. Nonetheless, the therapeutic value of phages is a concept already embraced by Pfizer, although in the context of gene therapy [79]. Again, corporate development of anti-bacterial phage therapy appears to be limited by both phage isolation and phage characterization.
The following quotations make this point from the perspective of active phage therapy trials. “Currently, the therapeutic potential of phages is substantially limited by the great variation in phage susceptibility and a relatively small repertoire of therapeutically useful phages” [80]. “Although several scientific organizations/societies recognized that phage therapy could be of key value in modern wound care, specific aspects are critical for a burn surgeon and might represent pitfalls discouraging phage therapy adoption in burn wound management; in particular, the unavailability of consensual therapeutic guidelines/regulatory policies and the lack of laboratorial support that might be predictive of its efficacy” [13]. “One major limitation that we faced was the lack of available human pharmaceutical-grade phage preparations, which still remains a bottleneck in our system and a limiting step in each phage request timeline” [14]. “Phage treatment of Mycobacterium infections is challenging due to the limited repertoire of therapeutically useful phages, but favorable clinical outcomes in patients lacking any other treatment options support continued development of adjunctive phage therapy for some mycobacterial infections” [17].
8. Conclusions (Take-Home Lesson)
The data indicate that solving the MDR bacteria problem is possible with phage therapy. Doing this will take key problem-identifying/targeting research. This research is likely to (1) be enhanced by biological factors not necessarily anticipated in advance and (2) succeed via use of already established principles and techniques of biophysics and phage biology.
9. Patents
The procedure for persistence transfer in Figure 2 is the subject of a patent application in progress.
Author Contributions
The sequencing and sequence interpretation was performed by J.A.T. and R.A.W.III; the work on persistence was performed by M.A. and C.B.G.; the multi-gel analysis was performed by J.P.C. and P.S.; the work was directed by P.S.; conceptualization, J.P.C., M.A., and P.S.; methodology, J.P.C., M.A., C.B.G., and P.S.; software, J.A.T. and R.A.W.III; validation, J.P.C., M.A., J.A.T., R.A.W.III and P.S.; formal analysis, J.P.C., J.A.T., R.A.W.III and P.S.; investigation, J.P.C. and M.A.; resources, J.P.C., C.B.G., J.A.T., R.A.W.III and P.S.; data curation, J.P.C., M.A., and P.S.; writing—original draft preparation, P.S.; writing—review and editing, J.P.C., J.A.T., R.A.W.III and P.S.; supervision, J.P.C. and P.S.; project administration, P.S.; funding acquisition, J.P.C., R.A.W.III and P.S. All authors have read and agreed to the published version of the manuscript.
Funding
Research of the P.S. laboratory was supported by The Morrison Trust, grant numbers 2021–2022, and the San Antonio Medical Foundation, grant number 2022. Research of the J.P.C. laboratory was supported by the San Antonio Medical Foundation, grant number 2022. Research of the R.A.W. laboratory was supported by the UNC Charlotte Department Bioinformatics and Genomics start-up package from the North Carolina Research Campus in Kannapolis, NC, USA.
Data Availability Statement
The data presented in this study are all available within one or more of the following: Figures and Text.
Acknowledgments
We are grateful to Girish Kumar (Rochester institute of Technology) for his sequencing of the Phage G clear-plaque mutant. We thank Meagan S. Weaver-Rosen (UT Health) for help in assembling the final manuscript. We acknowledge the UNC Charlotte University Research Computing Community and the UNC Charlotte College of Computing and Informatics for computational and logistical support.
Conflicts of Interest
R.A.W.III is the CEO of RAW Molecular Systems (RAW) LLC. R.A.W.III received no financial support, IP, or other support from RAW LLC for this study. J.P.C. and P.S. are founding members of a company, Phage Refinery LLC, that has plans to use the data presented here for the purposes of the company. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Gajic, I.; Jovicevic, M.; Popadic, V.; Trudic, A.; Kabic, J.; Kekic, D.; Ilic, A.; Klasnja, S.; Hadnadjev, M.; Popadic, D.; et al. The emergence of multi-drug-resistant bacteria causing healthcare-associated infections in COVID-19 patients: A retrospective multi-centre study. J. Hosp. Infect. 2023, 137, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.P.F.; Rebelo, J.S.; Dionisio, F.; Nogueira, T. Multi-drug resistance in bacterial genomes—A comprehensive bioinformatic analysis. Int. J. Mol. Sci. 2023, 24, 11438. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; Manson, A.L.; Desjardins, C.A.; Abeel, T.; Earl, A.M. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: Progress, promise, and challenges. Genome Med. 2019, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Ntshanka, N.G.M.; Msagati, T.A.M. Trends and Progress on antibiotic-resistant mycobacterium tuberculosis and genes in relation to human immunodeficiency virus. Can. J. Infect. Dis. Med Microbiol. 2023, 2023, 6659212. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.J.K.; El Ghali, A.; Holger, D.; Rebold, N.; Rybak, M.J. Therapeutic Strategies for emerging multidrug-resistant pseudomonas aeruginosa. Infect. Dis. Ther. 2022, 11, 661–682. [Google Scholar] [CrossRef] [PubMed]
- Giovagnorio, F.; De Vito, A.; Madeddu, G.; Parisi, S.G.; Geremia, N. Resistance in Pseudomonas aeruginosa: A Narrative Review of Antibiogram Interpretation and Emerging Treatments. Antibiotics 2023, 12, 1621. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.R.; Shrivastava, P.S.; Ramasamy, J. Responding to the challenge of antibiotic resistance: World Health Organization. J. Res. Med Sci. 2018, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- AR Threats Report. Centers for Disease Control and Infection, US Department of Health and Human Services. Available online: www.cdc.gov/DrugResistance/Biggest-Threats.html (accessed on 26 January 2024).
- Centers for Disease Control and Infection, US Department of Health and Human Services. Antibiotic Resistance Threats in the United States. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 26 January 2024).
- Antimicrobial resistance collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655.
- Burnham, J.P.; Olsen, M.A.; Kollef, M.H. Re-estimating annual deaths due to multidrug-resistant organism infections. Infect. Control. Hosp. Epidemiol. 2019, 40, 112–113. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage therapy for antibiotic-resistant bacterial infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef]
- Azevedo, M.M.; Pina-Vaz, C.; Rodrigues, A.G. The role of phage therapy in burn wound infections management: Advantages and pitfalls. J. Burn. Care Res. 2022, 43, 336–342. [Google Scholar] [CrossRef]
- Onallah, H.; Hazan, R.; Nir-Paz, R.; Yerushalmy, O.; Rimon, A.; Braunstein, R.; Gelman, D.; Alkalay, S.; Abdalrhman, M.; Stuczynski, D.; et al. Compassionate use of bacteriophages for failed persistent infections during the first 5 years of the israeli phage therapy center. Open Forum Infect. Dis. 2023, 10, ofad221. [Google Scholar] [CrossRef]
- Diallo, K.; Dublanchet, A. A Century of Clinical Use of Phages: A Literature Review. Antibiotics 2023, 12, 751. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; E Smith, B.; Cristinziano, M.; Freeman, K.G.; Jacobs-Sera, D.; Belessis, Y.; Brown, A.W.; A Cohen, K.; Davidson, R.M.; van Duin, D.; et al. Phage therapy of Mycobacterium infections: Compassionate use of phages in 20 patients with drug-resistant mycobacterial disease. Clin. Infect. Dis. 2023, 76, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Adesanya, O.; Oduselu, T.; Akin-Ajani, O.; Adewumi, O.M.; Ademowo, O.G. An exegesis of bacteriophage therapy: An emerging player in the fight against anti-microbial resistance. AIMS Microbiol. 2020, 6, 204–230. [Google Scholar] [CrossRef]
- McCallin, S.; Sacher, J.C.; Zheng, J.; Chan, B.K. Current state of compassionate phage therapy. Viruses 2019, 11, 343. [Google Scholar] [CrossRef]
- Górski, A.; Borysowski, J.; Międzybrodzki, R. Phage therapy: Towards a successful clinical trial. Antibiotics 2020, 9, 827. [Google Scholar] [CrossRef]
- Ali, Y.; Inusa, I.; Sanghvi, G.; Mandaliya, V.B.; Bishoyi, A.K. The current status of phage therapy and its advancement towards establishing standard antimicrobials for combating multi drug-resistant bacterial pathogens. Microb. Pathog. 2023, 181, 106199. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P. Phages for phage therapy: Isolation, characterization, and host range breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef]
- Desranleau, J.M. Progress in the treatment of typhoid fever with Vi bacteriophages. Can. J. Public Health 1949, 40, 473–478. [Google Scholar] [PubMed]
- Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.E.; Costa, A.R.; Aparicio-Maldonado, C.; Haas, P.-J.; Brouns, S.J.J. Mechanisms and clinical importance of bacteriophage resistance. FEMS Microbiol. Rev. 2022, 46, fuab048. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Marchi, J.; Zborowsky, S.; Debarbieux, L.; Weitz, J.S. The dynamic interplay of bacteriophage, bacteria and the mammalian host during phage therapy. iScience 2023, 26, 106004. [Google Scholar] [CrossRef] [PubMed]
- Van Belleghem, J.D.; Dąbrowska, K.; Vaneechoutte, M.; Barr, J.J.; Bollyky, P.L. Interactions between bacteriophage, bacteria, and the mammalian immune system. Viruses 2018, 11, 10. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Santo, P.D.; Weitz, J.S.; Debarbieux, L.; Unit, I.I. Immunophage synergy is essential for eradicating pathogens that provoke acute respiratory infections. Cell Host Microbe 2017, 22, 38–47. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T.; De La Chapa, J.; Gonzales, C.B. Basics for improved use of phages for therapy. Antibiotics 2021, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Issinger, O.G.; Falk, H. Comparative studies on the structure proteins of T3 and T7 phages. Arch. Virol. 1976, 52, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Watson, R.H.; Hayes, S.J.; Allen, J.L. Comparison of the physical properties and assembly pathways of the related bacteriophages T7, T3 and II. J. Mol. Biol. 1983, 170, 447–469. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.J. Electrophoresis; Academic Press: London, UK, 1969; pp. 4–26. [Google Scholar]
- Stellwagen, E.; Lu, Y.; Stellwagen, N.C. Unified description of electrophoresis and diffusion for DNA and other polyions. Biochemistry 2003, 42, 11745–11750. [Google Scholar] [CrossRef] [PubMed]
- Jan, K.-M.; Chien, S. Role of surface electric charge in red blood cell interactions. J. Gen. Physiol. 1973, 61, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P. Improvements in procedures for electrophoresis in dilute agarose gels. Anal. Biochem. 1981, 112, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Yang, X.; Yang, F.; Zhang, C.; Zhang, Q.; Duan, G.; Jiang, S. Research progress of the ion activity co-efficient of polyelectrolytes: A review. Molecules 2023, 28, 2042. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Fu, Q.; Hayward, W.; Lindsay, S.M.; Fletcher, T.M. The Myb/SANT domain of the telomere-binding protein TRF2 alters chromatin structure. Nucleic Acids Res. 2009, 37, 5019–5031. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, T.M.; Serwer, P.; Hansen, J.C. Quantitative analysis of macromolecular conformational changes using agarose gel electrophoresis: Application to chromatin folding. Biochemistry 1994, 33, 10859–10863. [Google Scholar] [CrossRef]
- Roberts, S.M.; Aldis, M.; Wright, E.T.; Gonzales, C.B.; Lai, Z.; Weintraub, S.T.; Hardies, S.C.; Serwer, P. Sipho-phage 0105phi7-2 of Bacillus thuringiensis: Novel propagation, DNA, and genome-implied assembly. Int. J. Mol. Sci. 2023, 24, 8941. [Google Scholar] [CrossRef]
- Petithory, T.; Pieuchot, L.; Josien, L.; Ponche, A.; Anselme, K.; Vonna, L. Size-dependent internalization effi-ciency of macrophages from adsorbed nanoparticle-based monolayers. Nanomaterials 2021, 11, 1963. [Google Scholar] [CrossRef]
- Bastin, G.; Gantzer, C.; Sautrey, G. New method to quantify hydrophobicity of non-enveloped virions in aqueous media by capillary zone electrophoresis. Virology 2022, 568, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Griess, G.A.; Moreno, E.T.; Herrmann, R.; Serwer, P. The sieving of rod-shaped viruses during agarose gel elec-trophoresis. I. Comparison with the sieving of spheres. Biopolymers 1990, 29, 1277–1287. [Google Scholar] [CrossRef]
- Serwer, P.; Hayes, S.J.; Watson, R.H.; Khan, S.A. Gel electrophoretic analysis of bacteriophage assembly inter-mediates in bacteriophage plaques. Appl. Theor. Electrophor. 1995, 4, 211–217. [Google Scholar] [PubMed]
- Serwer, P.; Khan, S.A.; Griess, G.A. Non-denaturing gel electrophoresis of biological nanoparticles: Viruses. J. Chromatogr. A 1995, 698, 251–261. [Google Scholar] [CrossRef]
- d’Herelle, F.H.D. The Bacteriophage and Its Behavior; The Williams & Wilkins Company: Baltimore, MD, USA, 1926. [Google Scholar]
- Dublanchet, A.; Fruciano, E. Brève histoire de la phagothérapie A short history of phage therapy. Med. Mal. Infect. 2008, 38, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Hayes, S.J.; Zaman, S.; Lieman, K.; Rolando, M.; Hardies, S.C. Improved isolation of undersampled bacteriophages: Finding of distant terminase genes. Virology 2004, 329, 412–424. [Google Scholar] [CrossRef]
- Serwer, P.; Hayes, S.J.; Thomas, J.A.; Hardies, S.C. Propagating the missing bacteriophages: A large bacterio-phage in a new class. Virol. J. 2007, 4, 21. [Google Scholar] [CrossRef]
- Montso, P.K.; Mlambo, V.; Ateba, C.N. Characterization of lytic bacteriophages infecting multidrug-resistant shiga toxigenic atypical Escherichia coli O177 strains isolated from cattle feces. Front. Public Health 2019, 7, 355. [Google Scholar] [CrossRef]
- Letarov, A.; Kulikov, E. The bacteriophages in human- and animal body-associated microbial communities. J. Appl. Microbiol. 2009, 107, 1–13. [Google Scholar] [CrossRef]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial Biofilm: A Review on Formation, Infection, Antibiotic Resistance, Control Measures, and Innovative Treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; E Masker, W.; Allen, J.L. Stability and in vitro DNA packaging of bacteriophages: Effects of dextrans, sugars, and polyols. J. Virol. 1983, 45, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Evilevitch, A.; Fang, L.T.; Yoffe, A.M.; Castelnovo, M.; Rau, D.C.; Parsegian, V.A.; Gelbart, W.M.; Knobler, C.M. Effects of salt concentrations and bending energy on the extent of ejection of phage genomes. Biophys. J. 2008, 94, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Grayson, P.; Evilevitch, A.; Inamdar, M.M.; Purohit, P.K.; Gelbart, W.M.; Knobler, C.M.; Phillips, R. The effect of genome length on ejection forces in bacteriophage lambda. Virology 2006, 348, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Flint, R.; Laucirica, D.R.; Chan, H.-K.; Chang, B.J.; Stick, S.M.; Kicic, A. Stability considerations for bacteriophages in liquid formulations designed for nebulization. Cells 2023, 12, 2057. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Luscher, A.; Falconnet, L.; Resch, G.; McBride, R.; Mai, Q.A.; Simonin, J.L.; Chanson, M.; Maco, B.; Galiotto, R.; et al. Personalized aerosolised bacteriophage treatment of a chronic lung infection due to multidrug-resistant Pseudomonas aeruginosa. Nat. Commun. 2023, 14, 3629. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.A.; Geary, D. Proceedings: Preservation of bacteriophages by freezing and freeze-drying. Cryobiol 1973, 10, 351–360. [Google Scholar] [CrossRef]
- Jończyk-Matysiak, E.; Łodej, N.; Kula, D.; Owczarek, B.; Orwat, F.; Międzybrodzki, R.; Neuberg, J.; Bagińska, N.; Weber-Dąbrowska, B.; Górski, A. Factors determining phage stability/activity: Challenges in practical phage application. Expert Rev. Anti-Infect. Ther. 2019, 17, 583–606. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef]
- Wdowiak, M.; Paczesny, J.; Raza, S. Enhancing the stability of bacteriophages using physical, chemical, and nano-based approaches: A review. Pharmaceutics 2022, 14, 1936. [Google Scholar] [CrossRef]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Sig, A.K.; Guney, M.; Guclu, A.U.; Ozmen, E. Medicinal leech therapy—An overall perspective. Integr. Med. Res. 2017, 6, 337–343. [Google Scholar] [CrossRef]
- Hackenberger, P.N.B.; Janis, J.E.M. A Comprehensive review of medicinal leeches in plastic and reconstructive surgery. Plast. Reconstr. Surg.–Glob. Open 2019, 7, e2555. [Google Scholar] [CrossRef] [PubMed]
- El-Beyrouty, C.; Buckler, R.; Mitchell, M.; Phillips, S.; Groome, S. Pneumococcal vaccination—A literature review and practice guideline update. Pharmacotherapy 2022, 42, 724–740. [Google Scholar] [CrossRef]
- Tietz, D.; Aldroubi, A.; Schneerson, R.; Unser, M.; Chrambach, A. The distribution of particles characterized by size and free mobility within polydisperse populations of protein-polysaccharide conjugates, determined from two-dimensional agarose electropherograms. Electrophoresis 1991, 12, 46–54. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Żaczek, M.; Borysowski, J.; Górski, A. The presence of bacteriophages in the human body: Good, bad or neutral? Microorganisms 2020, 8, 2012. [Google Scholar] [CrossRef] [PubMed]
- Podlacha, M.; Węgrzyn, G.; Węgrzyn, A. Bacteriophages—Dangerous viruses acting incognito or underestimated saviors in the fight against bacteria? Int. J. Mol. Sci. 2024, 25, 2107. [Google Scholar] [CrossRef]
- Foster, D.M.; Kellum, J.A. Endotoxic septic shock: Diagnosis and treatment. Int. J. Mol. Sci. 2023, 24, 16185. [Google Scholar] [CrossRef]
- Gefen, O.; Chekol, B.; Strahilevitz, J.; Balaban, N.Q. TDtest: Easy detection of bacterial tolerance and persistence in clinical isolates by a modified disk-diffusion assay. Sci. Rep. 2017, 7, 41284. [Google Scholar] [CrossRef]
- Tumpa, A.; Štritof, Z.; Pintarić, S. Prevalence and antimicrobial susceptibility of Enterococcus spp. from urine of dogs and cats in northwestern Croatia. Res. Veter- Sci. 2022, 151, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.P.; Wright, E.T.; Hunter, B.; Serwer, P. Inactivating host bacteria for characterization and use of phages. Biophysica 2023, 3, 558–568. [Google Scholar] [CrossRef]
- Martinho, F.C.; de Rabello, D.G.D.; Ferreira, L.L.; Nascimento, G.G. Participation of endotoxin in root canal infections: A systematic review and meta-analysis. Eur. J. Dent. 2017, 11, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.pfizer.com/sites/default/files/investors/financial_reports/annual_reports/2022/files/Pfizer_Annual_Review.pdf (accessed on 29 December 2023).
- Available online: https://www.imperial.ac.uk/enterprise/business/industry-partnerships-and-commercialisation/industry-partnerships/featured-partnerships/pfizer/ (accessed on 29 December 2023).
- Hatfull, G.F. Phage therapy for nontuberculous mycobacteria: Challenges and opportunities. Pulm. Ther. 2023, 9, 91–107. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).