
Adsorption of Heparin-Binding Fragments of Fibronectin onto Hydrophobic Surfaces


Abstract
:1. Introduction
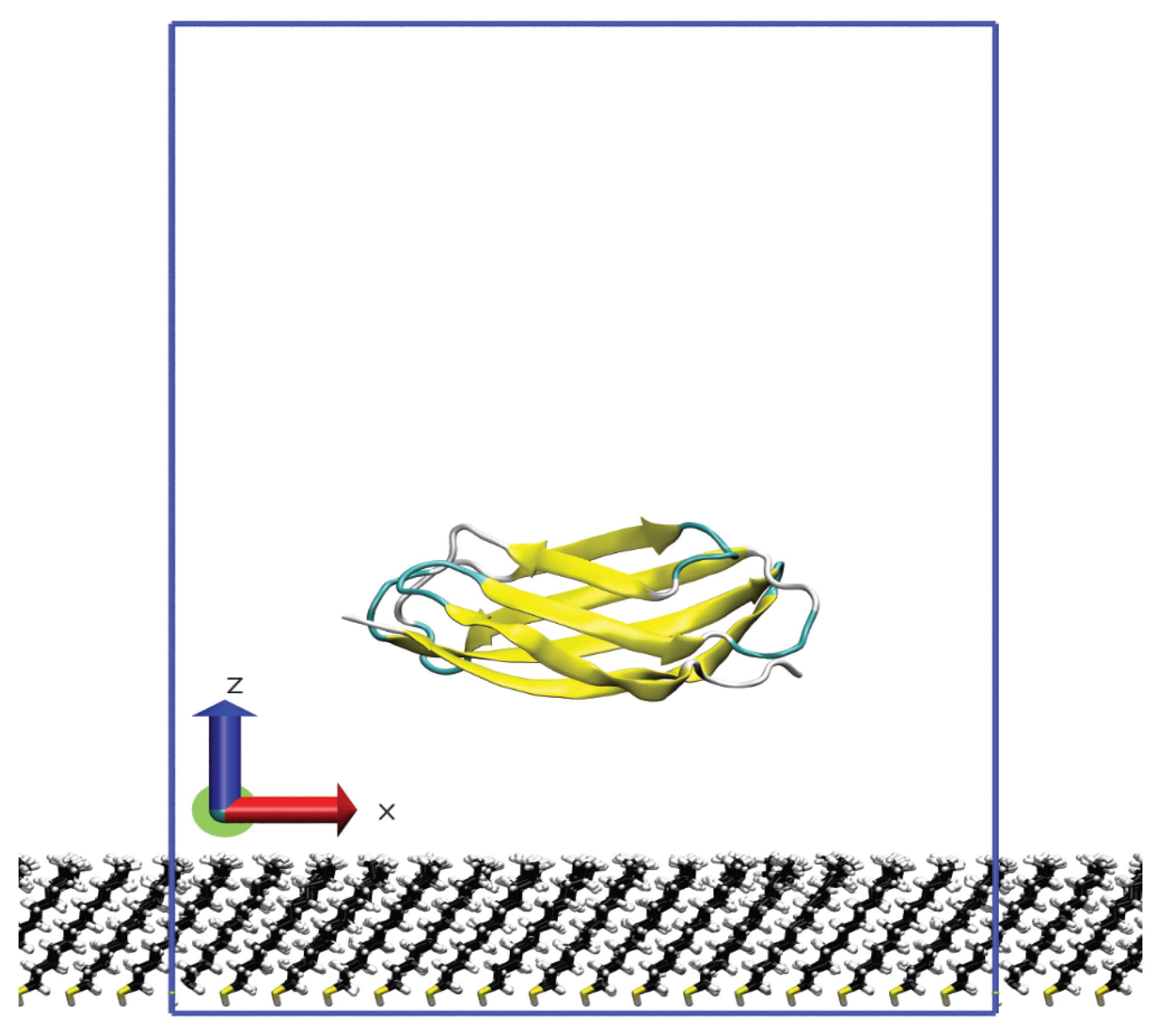
2. Model and Methodology
3. Results
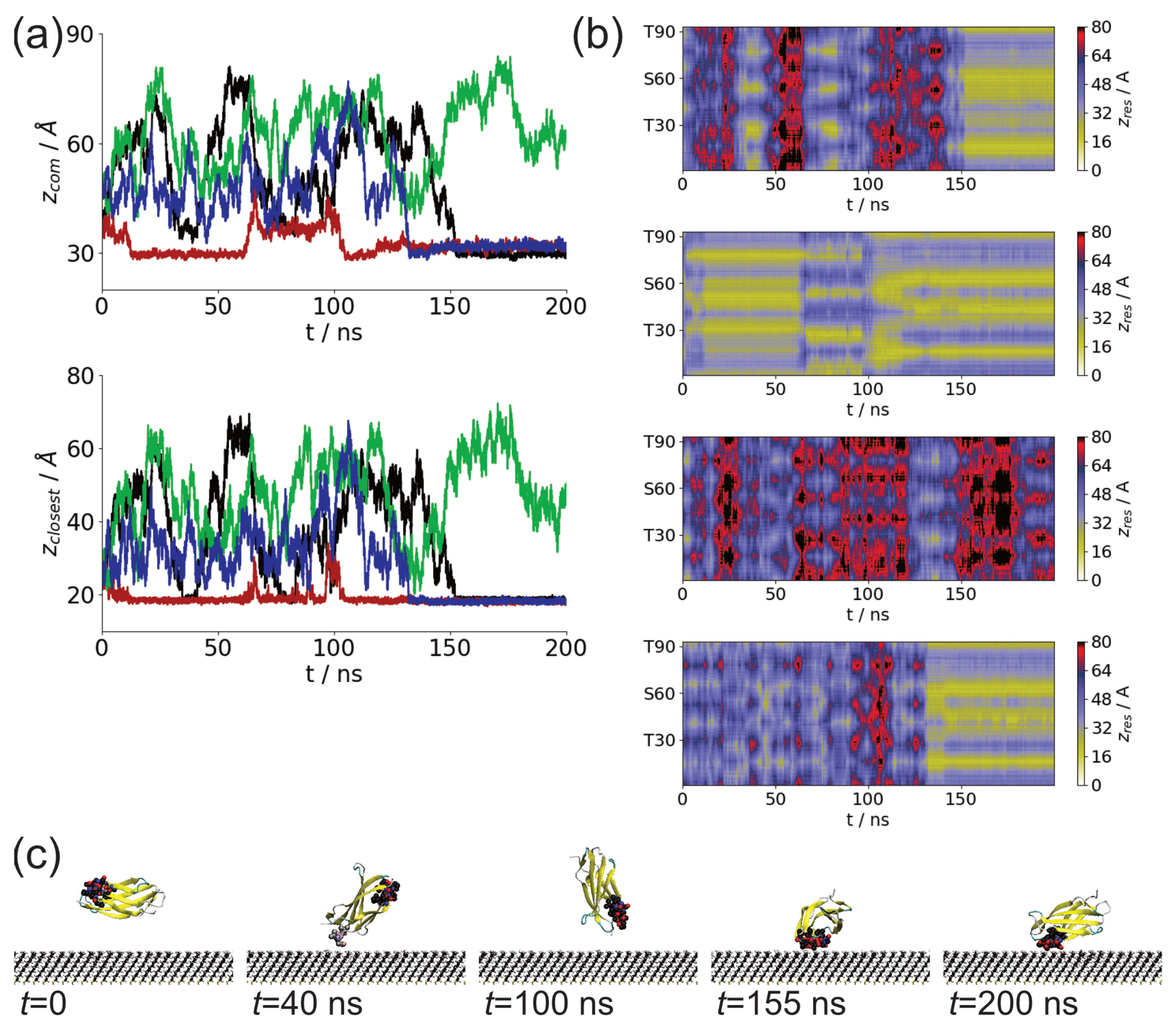
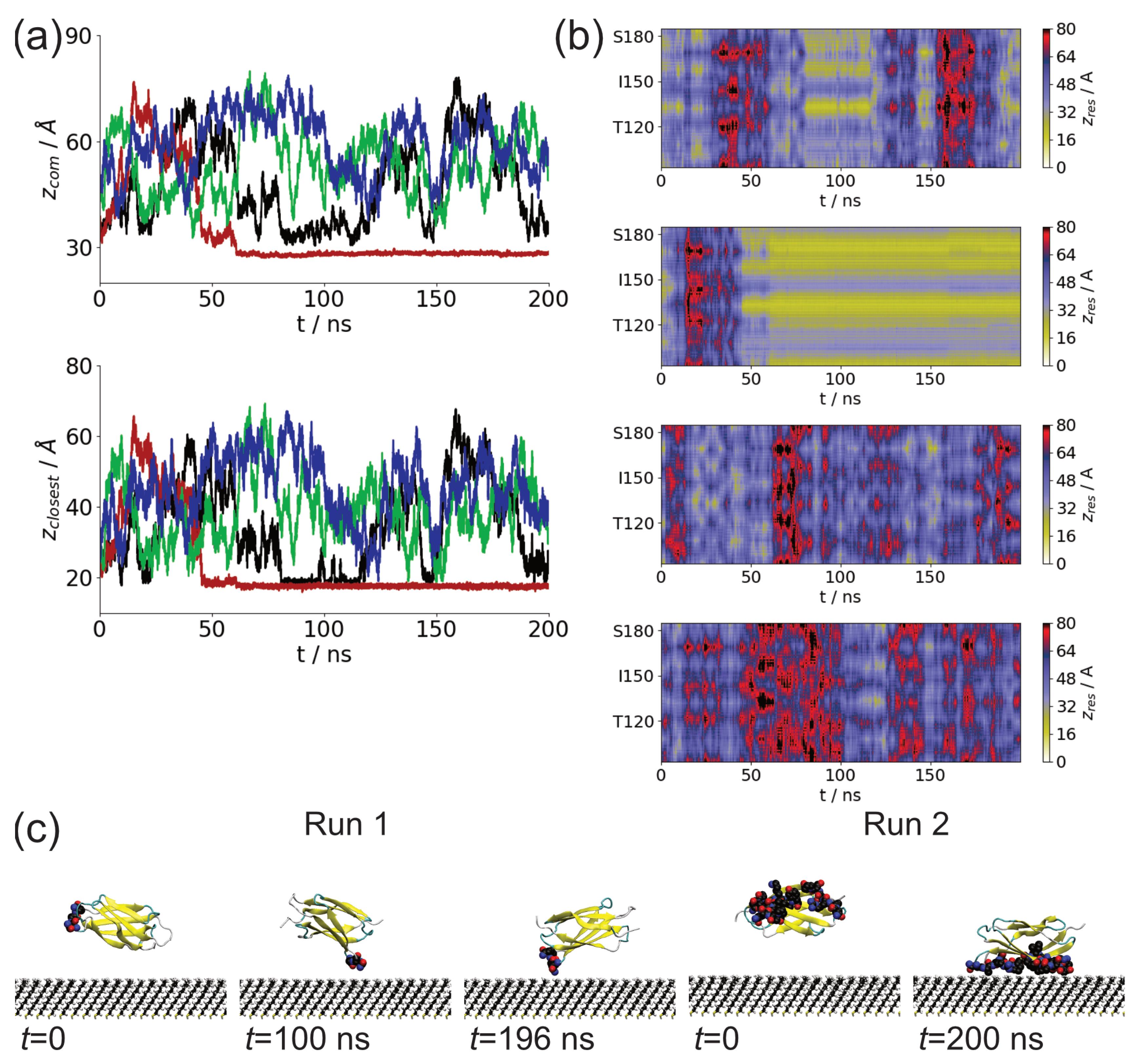
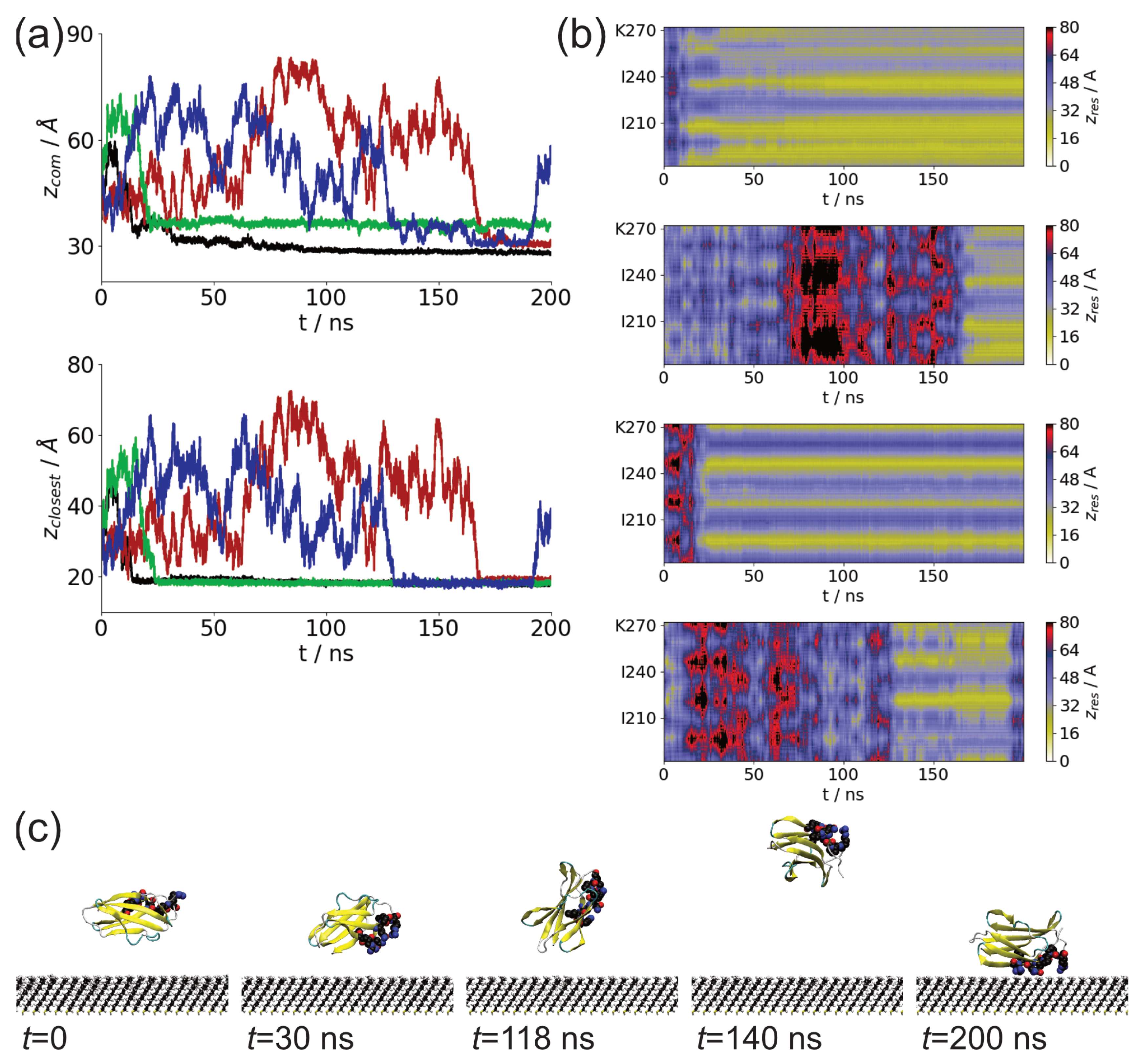
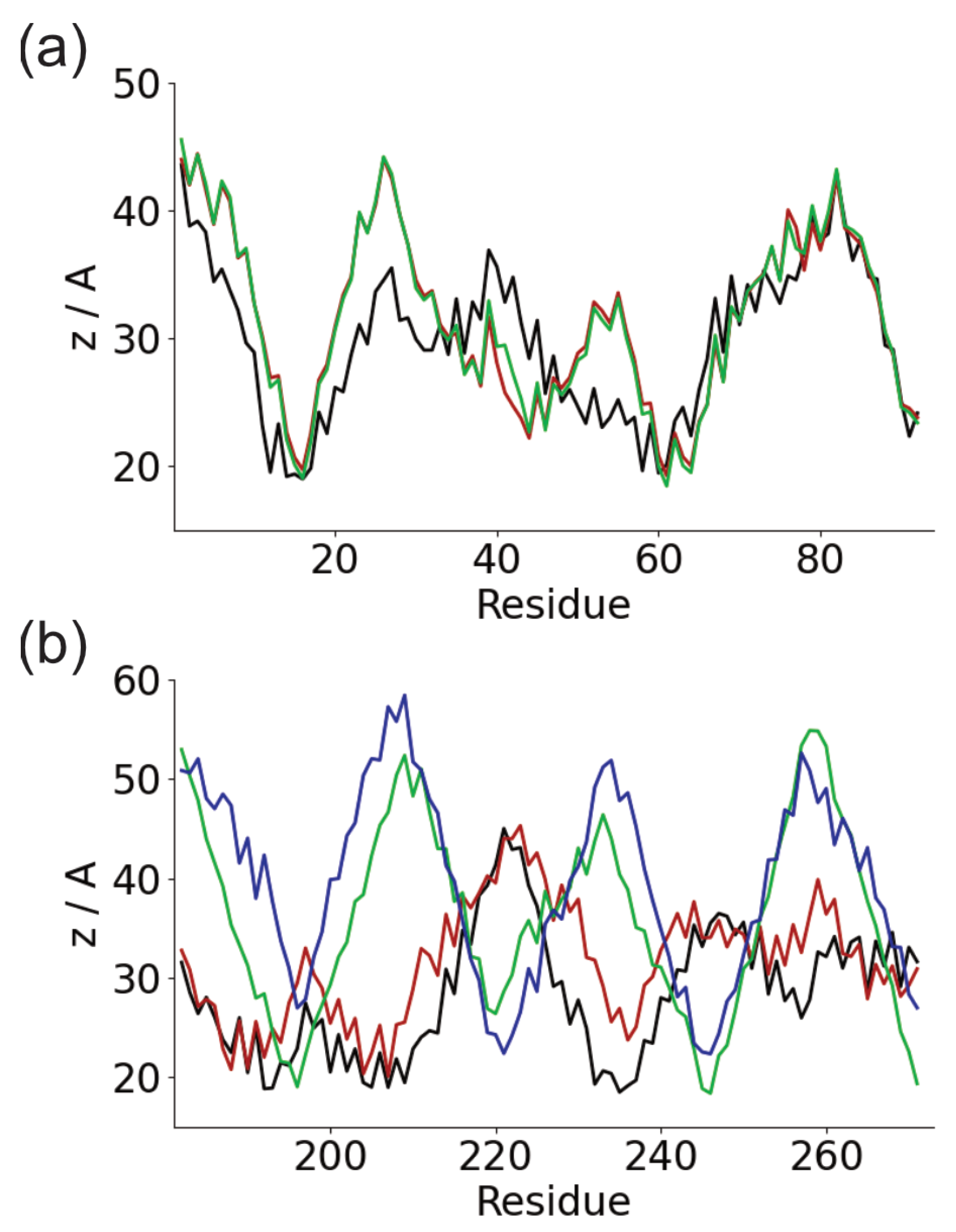
3.1. Adsorption of Fibronectin Domains onto Hydrophobic Surface
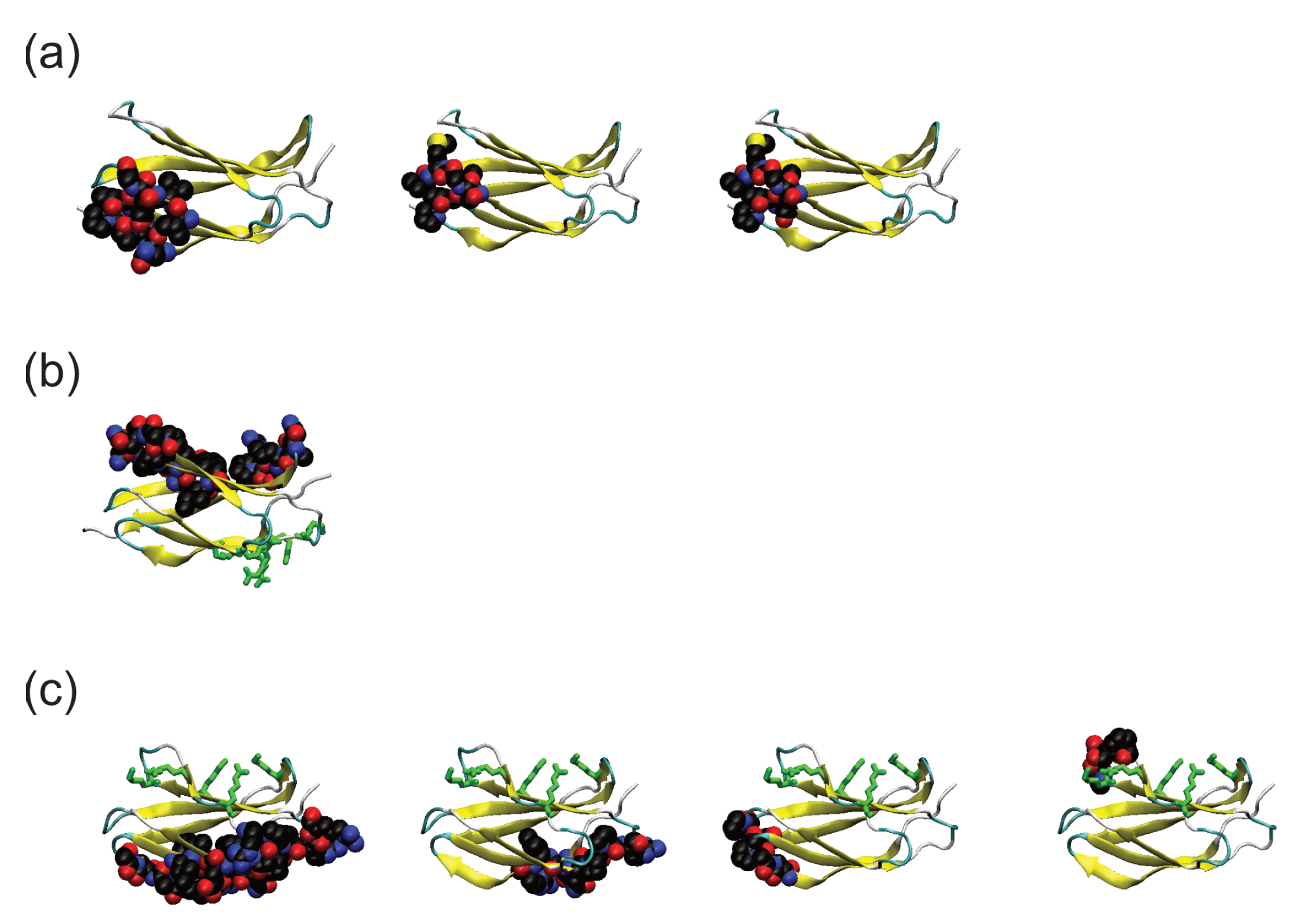
3.2. Comparison of Adsorbed Residues
3.3. Adsorption Free Energies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, I.; Pangule, R.C.; Kane, R.S. Antifouling coatings: Recent developments in the design of surfaces that prevent fouling by proteins, bacteria, and marine organisms. Adv. Mater. (Deerfield Beach Fla.) 2011, 23, 690–718. [Google Scholar] [CrossRef] [PubMed]
- Blaszykowski, C.; Sheikh, S.; Thompson, M. Surface chemistry to minimize fouling from blood-based fluids. Chem. Soc. Rev. 2012, 41, 5599. [Google Scholar] [CrossRef]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouberai, M.M.; Xu, K.; Welland, M.E. Effect of the interplay between protein and surface on the properties of adsorbed protein layers. Biomaterials 2014, 35, 6157–6163. [Google Scholar] [CrossRef] [Green Version]
- Abaricia, J.O.; Farzad, N.; Heath, T.J.; Simmons, J.; Morandini, L.; Olivares-Navarrete, R. Control of innate immune response by biomaterial surface topography, energy, and stiffness. Acta Biomater. 2021, 133, 58–73. [Google Scholar] [CrossRef]
- Brancolini, G.; Bellucci, L.; Maschio, M.C.; Di Felice, R.; Corni, S. The interaction of peptides and proteins with nanostructures surfaces: A challenge for nanoscience. Curr. Opin. Colloid Interface Sci. 2019, 41, 86–94. [Google Scholar] [CrossRef]
- Amani, H.; Arzaghi, H.; Bayandori, M.; Dezfuli, A.S.; Pazoki-Toroudi, H.; Shafiee, A.; Moradi, L. Controlling Cell Behavior through the Design of Biomaterial Surfaces: A Focus on Surface Modification Techniques. Adv. Mater. Interfaces 2019, 6, 1900572. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Liu, W.; Wu, Z.; Chen, H. Chemical Surface Modification of Polymeric Biomaterials for Biomedical Applications. Macromol. Rapid Commun. 2020, 41, 1900430. [Google Scholar] [CrossRef] [PubMed]
- Mantha, S.; Pillai, S.; Khayambashi, P.; Upadhyay, A.; Zhang, Y.; Tao, O.; Pham, H.M.; Tran, S.D. Smart Hydrogels in Tissue Engineering and Regenerative Medicine. Materials 2019, 12, 3323. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.F. Challenges with the Development of Biomaterials for Sustainable Tissue Engineering. Front. Bioeng. Biotechnol. 2019, 7, 127. [Google Scholar] [CrossRef] [Green Version]
- Dalton, C.J.; Lemmon, C.A. Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells 2021, 10, 2443. [Google Scholar] [CrossRef]
- Mezzenga, R.; Mitsi, M. The Molecular Dance of Fibronectin: Conformational Flexibility Leads to Functional Versatility. Biomacromolecules 2019, 20, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Salmerón-Sánchez, M.; Rico, P.; Moratal, D.; Lee, T.T.; Schwarzbauer, J.E.; García, A.J. Role of material-driven fibronectin fibrillogenesis in cell differentiation. Biomaterials 2011, 32, 2099–2105. [Google Scholar] [CrossRef]
- Sottile, J.; Hocking, D.; Langenbach, K. Fibronectin polymerization stimulates cell growth by RGD-dependent and -independent mechanisms. J. Cell Sci. 2000, 113, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Engler, A.J.; Slone, R.D.; Galante, L.L.; Schwarzbauer, J.E. Fibronectin Expression Modulates Mammary Epithelial Cell Proliferation during Acinar Differentiation. Cancer Res. 2008, 68, 3185–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, T.E.; Thøgersen, H.C.; Skorstengaard, K.; Vibe-Pedersen, K.; Sahl, P.; Sottrup-Jensen, L.; Magnusson, S. Partial primary structure of bovine plasma fibronectin: Three types of internal homology. Proc. Natl. Acad. Sci. USA 1983, 80, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, M.M.; Hubbell, J.A. The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010, 24, 4711–4721. [Google Scholar] [CrossRef] [Green Version]
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and simulation of protein-surface interactions: Achievements and challenges. Q. Rev. Biophys. 2016, 49, e4. [Google Scholar] [CrossRef] [Green Version]
- Ley, K.; Christofferson, A.; Penna, M.; Winkler, D.; Maclaughlin, S.; Yarovsky, I. Surface-water Interface Induces Conformational Changes Critical for Protein Adsorption: Implications for Monolayer Formation of EAS Hydrophobin. Front. Mol. Biosci. 2015, 2, 64. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Wu, C.; Chen, M.; Zheng, T.; Chen, N.; Cummings, P.T. Molecular modeling of fibronectin adsorption on topographically nanostructured rutile (110) surfaces. Appl. Surf. Sci. 2016, 384, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Penna, M.; Ley, K.; Maclaughlin, S.; Yarovsky, I. Surface heterogeneity: A friend or foe of protein adsorption – insights from theoretical simulations. Faraday Discuss. 2016, 191, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D. Effect of surface structure on peptide adsorption on soft surfaces. Chem. Phys. Lett. 2020, 758, 137929. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. Molecular Dynamics Simulation of the Adsorption of a Fibronectin Module on a Graphite Surface. Langmuir 2004, 20, 3371–3378. [Google Scholar] [CrossRef]
- Panos, M.; Sen, T.Z.; Ahunbay, M.G. Molecular Simulation of Fibronectin Adsorption onto Polyurethane Surfaces. Langmuir 2012, 28, 12619–12628. [Google Scholar] [CrossRef] [PubMed]
- Kubiak-Ossowska, K.; Mulheran, P.A.; Nowak, W. Fibronectin Module FNIII9 Adsorption at Contrasting Solid Model Surfaces Studied by Atomistic Molecular Dynamics. J. Phys. Chem. B 2014, 118, 9900–9908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallinson, D.; Cheung, D.L.; Simionesie, D.; Mullen, A.B.; Zhang, Z.J.; Lamprou, D.A. Experimental and computational examination of anastellin (FnIII1c)-polymer interactions. J. Biomed. Mater. Res. Part A 2017, 105, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liamas, E.; Kubiak-Ossowska, K.; Black, R.; Thomas, O.; Zhang, Z.; Mulheran, P. Adsorption of Fibronectin Fragment on Surfaces Using Fully Atomistic Molecular Dynamics Simulations. Int. J. Mol. Sci. 2018, 19, 3321. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Hao, L.; Li, J.; Du, C.; Wang, Y. Role of Ninth Type-III Domain of Fibronectin in the Mediation of Cell-Binding Domain Adsorption on Surfaces with Different Chemistries. Langmuir 2018, 34, 9847–9855. [Google Scholar] [CrossRef]
- Bieniek, M.K.; Llopis-Hernandez, V.; Douglas, K.; Salmeron-Sanchez, M.; Lorenz, C.D. Minor Chemistry Changes Alter Surface Hydration to Control Fibronectin Adsorption and Assembly into Nanofibrils. Adv. Theory Simul. 2019, 2, 1900169. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Li, T.; Wang, L.; Shi, X.; Fan, Y.; Du, C.; Wang, Y. Mechanistic insights into the adsorption and bioactivity of fibronectin on surfaces with varying chemistries by a combination of experimental strategies and molecular simulations. Bioact. Mater. 2021, 6, 3125–3135. [Google Scholar] [CrossRef]
- Basu, S.; Basu, B.; Maiti, P.K. A computational study on strontium ion modified hydroxyapatite–fibronectin interactions. Phys. Chem. Chem. Phys. 2022, 24, 27989–28002. [Google Scholar] [CrossRef] [PubMed]
- Leahy, D.J.; Aukhil, I.; Erickson, H.P. 2.0 Å Crystal Structure of a Four-Domain Segment of Human Fibronectin Encompassing the RGD Loop and Synergy Region. Cell 1996, 84, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A. Crystal structure of a heparin-and integrin-binding segment of human fibronectin. EMBO J. 1999, 18, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, C.A.; Ohashi, T.; Erickson, H.P. Probing the Folded State of Fibronectin Type III Domains in Stretched Fibrils by Measuring Buried Cysteine Accessibility. J. Biol. Chem. 2011, 286, 26375–26382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2009, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Ulman, A.; Eilers, J.E.; Tillman, N. Packing and molecular orientation of alkanethiol monolayers on gold surfaces. Langmuir 1989, 5, 1147–1152. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph-McCarthy, D.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the {CHARMM} Force Field in {GROMACS}: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; Mackerell, A.D. Charmm36M: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of MolecularDynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.; Mager, M.; Hembury, M.; Stellacci, F.; Stevens, M.M.; Yarovsky, I. Amphiphilic amino acids: A key to adsorbing proteins to nanopatterned surfaces? Chem. Sci. 2013, 4, 928–937. [Google Scholar] [CrossRef]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, C.M.; Ma, H.; Wei, T. Study of lysozyme mobility and binding free energy during adsorption on a graphene surface. Appl. Phys. Lett. 2015, 106, 153701. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N.; Wolynes, P.G. Fly-casting in protein-DNA binding: Frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 2007, 129, 738–739. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment | Run | Interval (ns) | Residues |
|---|---|---|---|
| FnIII-12 | 1 | 34.71–41.2 | 26–28: NVQ |
| 76.13–83.41 | 26–28: NVQ | ||
| 151.29–200 | 12–17: NVTPTS, 58–61: VVSG | ||
| 2 | 2.84–65.38 | 30: T, 49–52: NLAP, 77–78: DT | |
| 66.84–88.47 | 26–28: NVQ | ||
| 89.6–96.55 | 26–28: NVQ | ||
| 102.6–200 | 15–16: PT, 60–64: SGLMV | ||
| 4 | 131.12–200 | 15-17: PTS, 60-64: SGLMV | |
| FnIII-13 | 1 | 19.22–22.7 | 183: I |
| 80.76–98.51 | 131–134: ANGQ | ||
| 99.88–103.46 | 132–134: NGQ | ||
| 104.75–116.43 | 131–134: ANGQ | ||
| 143.84–148.73 | 169: N | ||
| 195.37–196.41 | 132–133: NG | ||
| 197.99–199.43 | 132: N | ||
| 2 | 45.42–200 | 129–136: VPANGQTP, 160–162: KIY, 169–174: NARSSP | |
| FnIII-14 | 1 | 13.59–200 | 190–195: RFLATT, 200–209: LVSWQPPRAR, 232–237: RPGVTE |
| 2 | 167.29–200 | 188–190: NLR, 204–207: QPPR | |
| 3 | 23.62–200 | 194–196: TTP, 245–246: PG, 271: T | |
| 4 | 130.04–192.84 | 220–223: PGSP |
| Fragment | Run | Interval (ns) | |||
|---|---|---|---|---|---|
| FnIII-12 | 1 | 34.71–41.2 | −6.9 ± 0.3 | −15.1 ± 1.0 | 8.3 ± 0.9 |
| 76.13–83.41 | −5.0 ± 0.3 | −16.2 ± 0.4 | 11.2 ± 0.3 | ||
| 151.29–200 | −12.2 ± 0.3 | −19.1 ± 0.5 | 6.9 ± 0.3 | ||
| 2 | 2.84–65.38 | −16.0 ± 0.3 | −18.9 ± 0.4 | 2.9 ± 0.3 | |
| 66.84–88.47 | −6.4 ± 0.2 | −10.2 ± 0.5 | 3.8 ± 0.4 | ||
| 89.6–96.55 | −4.7 ± 0.3 | −5.9 ± 1.3 | 1.3 ± 1.1 | ||
| 102.6–200 | −20.0 ± 0.2 | −32.6 ± 0.4 | 12.6 ± 0.2 | ||
| 4 | 131.12–200 | −10.1 ± 0.2 | −18.9 ± 0.3 | 8.8 ± 0.2 | |
| FnIII-13 | 1 | 19.22–22.70 | −5.4 ± 0.4 | −4 ± 1.4 | −1.4 ± 1.3 |
| 80.76–98.51 | −8.6 ± 0.2 | −17.5 ± 0.7 | 8.9 ± 0.6 | ||
| 99.88–103.46 | −7.4 ± 0.4 | −26.9 ± 0.8 | 19.5 ± 0.5 | ||
| 104.75–116.43 | −5.7 ± 0.2 | −12.2 ± 0.9 | 6.5 ± 0.9 | ||
| 143.84–148.73 | −2.9 ± 0.2 | −5.2 ± 0.7 | 2.3 ± 0.6 | ||
| 195.37–196.41 | −1.8 ± 0.3 | 7.3 ± 0.9 | −9.2 ± 0.9 | ||
| 197.99–199.43 | −3.7 ± 0.4 | 14.8 ± 0.7 | −18.5 ± 0.6 | ||
| 2 | 45.42–200 | −27.6 ± 0.2 | −51.7 ± 0.5 | 24.1 ± 0.4 | |
| FnIII-14 | 1 | 13.59–200 | −20.9 ± 0.2 | −54.1 ± 0.7 | 33.2 ± 0.5 |
| 2 | 167.29–200 | −10.8 ± 0.2 | −6.5 ± 0.7 | −4.4 ± 0.7 | |
| 3 | 23.62–200 | −14.4 ± 0.1 | −19.6 ± 0.3 | 5.3 ± 0.2 | |
| 4 | 130.04–192.84 | −11.9 ± 0.2 | −22.8 ± 0.6 | 10.9 ± 0.5 |
| Domain | GRAVY Value |
|---|---|
| FnIII-12 | −0.106 |
| FnIII-13 | −0.444 |
| FnIII-14 | −0.457 |
| FnIII-8 | −0.391 |
| FnIII-9 | −0.454 |
| FnIII-10 | −0.114 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vittaladevaram, V.; Cheung, D.L. Adsorption of Heparin-Binding Fragments of Fibronectin onto Hydrophobic Surfaces. Biophysica 2023, 3, 409-424. https://doi.org/10.3390/biophysica3030027
Vittaladevaram V, Cheung DL. Adsorption of Heparin-Binding Fragments of Fibronectin onto Hydrophobic Surfaces. Biophysica. 2023; 3(3):409-424. https://doi.org/10.3390/biophysica3030027
Chicago/Turabian StyleVittaladevaram, Viswanath, and David L. Cheung. 2023. "Adsorption of Heparin-Binding Fragments of Fibronectin onto Hydrophobic Surfaces" Biophysica 3, no. 3: 409-424. https://doi.org/10.3390/biophysica3030027
APA StyleVittaladevaram, V., & Cheung, D. L. (2023). Adsorption of Heparin-Binding Fragments of Fibronectin onto Hydrophobic Surfaces. Biophysica, 3(3), 409-424. https://doi.org/10.3390/biophysica3030027