Abstract
Deep eutectic solvents (DESs) and ionic liquids (ILs) are highly tailorable solvents that have shown a lot of promise for a variety of applications including cryopreservation, drug delivery, and protein stabilisation. However, to date, there is very limited information on the detailed interactions of these solvents with mammalian cells. In this work, we studied six DESs and one IL that show promise as cryoprotective agents, applying synchrotron macro–ATR–FTIR to examine their effects on key biochemical components of HaCat mammalian cells. These data were paired with resazurin metabolic assays and neutron reflectivity experiments to correlate cellular interactions with cellular toxicity. Stark differences were observed even between solvents that shared similar components. In particular, it was found that solvents that are effective cryoprotective agents consistently showed interactions with cellular membranes, while high toxicity correlated with strong interactions of the DES/IL with nucleic acids and proteins. This work sheds new light on the interactions between novel solvents and cells that may underpin future biomedical applications.
1. Introduction
Cryopreservation is the invaluable process of cooling biological samples to very low temperatures for long–term storage []. This technology has led to significant benefits including increasing the supply of medically relevant resources through blood and stem-cell banks [,,,]. Cryopreservation also is vital for assisted reproductive technologies, as well as species conservation [,,]. However, cryopreservation is limited to certain cell types and with current technology cannot be applied to organs [,,]. This leads to massive wastage of donated organs such as hearts and lungs due to inadequate storage methods [,,].
The primary factor limiting the wider application of cryopreservation is the ongoing reliance on predominantly just two cryoprotective agents (CPAs); dimethylsulfoxide (DMSO), and glycerol [,]. CPAs are added to biological samples prior to cooling in order to suppress intracellular ice formation, which is lethal [,]. However, DMSO and glycerol are ineffective for many cell types, and can be toxic [,,]. In fact, recent work has demonstrated that DMSO can cause genome-wide changes, which have significant implications for its use in assisted reproductive technologies []. Our recent research has demonstrated that neoteric solvents offer promising alternatives to existing CPAs [,].
Neoteric solvents are a broad classification that includes ionic liquids (ILs) and deep eutectic solvents (DESs) []. Protic ILs are synthesised by combining a Brønsted acid with a Brønsted base [], whereas DESs are made from mixtures of hydrogen bond donors and hydrogen bond acceptors [,]. In both cases, the ability to switch out components (either acid or base for ILs, or hydrogen bond donor or acceptor in the case of DESs) means that these solvents can be tailored to specific applications. This includes reducing toxicity to biological cells and promoting glass formation—both of which are favourable properties for CPAs. There is evidence to suggest that cold-resistant plant and animal species produce metabolites that form so-called ‘natural’ deep eutectic solvents, that help them resist freezing damage at sub-zero temperatures []. Despite their promise, only a handful of neoteric solvents have been explored as CPAs. We recently demonstrated that ethylammonium acetate (EAA) could act as a CPA for a human cell line [], whereas others have shown that zwitterion-type ILs are effective non-cell-permeable CPAs [,]. Similarly, our group recently demonstrated that a DES composed of proline and glycerol was an effective CPA for all four human cell lines tested []. Others have demonstrated the efficacy of glycerol- and glucose-containing DESs [], and trehalose/glycerol mixtures [].
In general, the success of neoteric solvents as CPAs is attributed to their suppression of ice formation by interacting with water (e.g., through hydrogen bonding) to prevent crystallisation and favour a glassy, amorphous solid state [,,,]. However, investigations into specific mechanisms and cellular interactions are limited, and to date only report broad findings—for example, that hydrophobic DES molecules interact more strongly with cellular membranes, leading to greater toxicity []. In the related field of lipid behaviour and self-assembly in neoteric solvents, there has been more fundamental research and some of the findings may be relevant. For example, long alkyl chains in protic ILs have been found to lead to protein unfolding [], whereas protic ILs with non-amphiphilic cations support more diverse lipid liquid crystal phases compared to amphiphilic ones [,]. Patterns within DESs are still emerging, although similar effects in regards to alkyl chain lengths have been observed [], and the ability of DESs to support lipid self-assembly is well established [,].
This work explores a suite of neoteric solvents including three glycerol-containing and three galactose-containing DESs and one protic IL (ethylammonium acetate), compared with DMSO, using toxicity assays, macro attenuated total reflection Fourier transform infrared (macro–ATR–FTIR) microspectroscopy, and neutron/X-ray reflectivity. These techniques enable chemical mapping of solvent-cell interactions as well as detailed insight into the location of solvent components in model lipid membranes. This research aims to shed light on why certain neoteric solvents are effective CPAs whereas others are not, despite sharing some components and properties. This information can be used to further design and tailor neoteric solvents to specific applications, including cryopreservation.
2. Materials and Methods
2.1. Neoteric Solvents
All chemicals were purchased from Merck (Darmstadt, Germany) unless stated otherwise. Deep eutectic solvents were synthesised as previously described []. Briefly, choline chloride (ChCl) (>98%), betaine (Bet) (>99%), or L-proline (Prol) (>99%) were combined with glycerol (Gly) (>99%) or D-galactose (Gal) (99%) in the following mol ratios: ChCl–Gly (1:2), Bet–Gly (1:2), Prol–Gly (1:3), ChCl-Gal (5:2), Bet-Gal (5:2), Prol–Gal (5:3). The molecular structures of each component are shown in Figure 1. These mixtures were heated to ~50 °C with stirring until they formed homogenous liquids. The galactose-based DESs required the addition of water to allow proper mixing. Each DES was then dried using rotoevaporation and freeze drying to obtain a water content below 2 wt% (based on Karl Fischer titration).
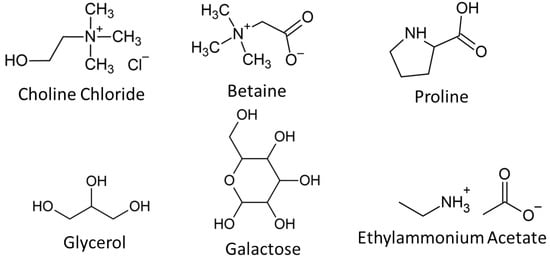
Figure 1.
Chemical structures of the DES components and ethylammonium acetate used in this study.
The ionic liquid ethylammonium acetate (EAA) was synthesised as previously described []. Briefly, acetic acid (99%) was added dropwise to ethylamine (66%) to achieve neutralisation. Excess water was removed using rotary evaporation and freeze drying to achieve a water content of less than 0.5 wt%. The chemical structure of EAA is shown in Figure 1.
2.2. Cell Culturing
Human skin keratinocyte (HaCaT) cells were maintained in Dulbecco’s modified Eagle medium (DMEM), containing high glucose, GlutaMAXTM supplement, and pyruvate (Gibco, New York, NY, USA) supplemented with 10% foetal bovine serum (Gibco, New York, NY, USA) and 1% of 5000 U/mL penicillin-streptomycin (Gibco, New York, NY, USA). Cells were grown in T25 or T75 culture flasks (Corning Inc., Corning, NY, USA) at 37 °C, with 5% CO2 and 95% humidified air. The cells were grown to ~80% confluency and then subcultured. This involved removing all media and adding several millilitres of 25% trypsin-EDTA (Merck, Darmstadt, Germany) to cover the cells. The flask was incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 5–10 min. Fresh DMEM media was added to neutralise the trypsin-EDTA and the suspension was centrifuged at 200× g for 5 min and the supernatant removed. The cell pellet was then resuspended in fresh DMEM and placed into a fresh culture flask.
Viability Assay: To assess the viability of the HaCaT cells, 10,000 cells/well were seeded into 96 well plates and allowed to grow for 24 h at 37 °C. Cells were treated with 10 wt% DMSO, DES, or EAA in complete media for 5 min, 1 h, 3 h, 6 h, and 24 h at 37 °C. Untreated cells were used as a control. Cell-free controls, containing only the various treatments in media were also included. After treatment, the cells were washed three times in phosphate-buffered saline (PBS) (Gibco, New York, NY, USA) and fresh cell culture media containing a final concentration of 15 µg/mL resazurin sodium salt (Merck, Darmstadt, Germany) was added to each well (including cell-free controls). Cells were allowed to incubate at 37 °C for 3 h before recording the fluorescence on a ClarioStar (BMG Labtech, Ortenberg, Germany) plate reader, using 560 nm excitation and 590 nm emission. Three replicates were used for each sample and three independent experimental repeats were performed.
2.3. Synchrotron Macro–ATR–FTIR Microspectroscopy
To prepare the HaCat cells for the synchrotron macro–ATR–FTIR measurements, silicon wafers were first sterilised using ethanol and ultraviolet light before being placed into the bottom of 6-well culture plates. A suspension of HaCat cells was added to the wells and grown for at least 24 h at 37 °C in a humidified atmosphere containing 5% CO2 to ensure proper attachment and growth on the silicon wafers. The media was then removed and replaced with media containing 10 wt% neoteric solvent (or purely fresh media in the case of the untreated control) and incubated for a further 1 h. The silicon wafers (with the cells attached) were then washed three times with fresh cell media to remove any residual neoteric solvent. The silicon wafers were covered with 4% formaldehyde solution and left to fix for 15 min before being dehydrated by exposure to solutions with gradually increasing ethanol concentrations. The wafers were allowed to air-dry for several minutes prior to the spectral data acquisition.
Synchrotron macro–ATR–FTIR experiments were then performed on the infrared microspectroscopy (IRM) beamline at the ANSTO—Australian Synchrotron (Victoria, Australia), using a Bruker Vertex 80v spectrometer coupled with a Bruker Hyperion 3000 FTIR microscope and a liquid-nitrogen cooled narrow-band mercury cadmium telluride (MCT) detector (Bruker Optik GmbH, Ettlingen, Germany). All spectra were recorded at room temperature over a spectral range of 3900–750 cm−1 using 4-cm−1 spectral resolution. Blackman-Harris 3-Term apodization, Mertz phase correction, and a zero-filling factor of 2 were set as default acquisition parameters using OPUS 8.0 software suite (Bruker Optik GmbH, Ettlingen, Germany).
To acquire the spectral data in macro–ATR–FTIR mapping mode, the fixed HaCat cells on silicon wafers were mounted on an aluminium disc and placed into the sample stage of the in-house developed macro–ATR–FTIR unit []. This macro–ATR–FTIR device utilises a 250 μm diameter facet germanium (Ge) ATR crystal (nGe = 4.0) and a 20× IR objective (NA = 0.60; Bruker Optik GmbH, Ettlingen, Germany). After that, the Ge crystal was brought to the focus of the synchrotron-IR beam and a background spectrum was recorded in air using a projected aperture of 3.13 μm in diameter and 256 co-added scans. The fixed HaCat cells on the silicon wafer were then brought into contact with the Ge ATR crystal. First, a low-resolution overview synchrotron macro–ATR–FTIR chemical map was acquired to determine the locations of the cells and quality of the contact, with a 5 μm step interval, using 8 co-added scans. Subsequently, high-resolution synchrotron macro–ATR–FTIR mapping measurements were performed on specific areas of interest using a smaller step interval of 1 μm and 16 co-added scans. Good contact with the Ge ATR crystal was achieved based on the intensity of the amide I band (i.e., the key characteristic of proteins inside the cells).
2.4. Spectral Pre-Processing and Multivariate Data Analysis
Following data acquisition, synchrotron macro–ATR–FTIR maps were corrected using the atmospheric compensation function, to remove atmospheric water vapour and CO2 interference from the spectra. Chemical maps were then generated by integrating the area under the relevant peaks using OPUS 8.0 software.
Multivariate data analysis, including hierarchical cluster analysis (HCA) and principal component analysis (PCA), was performed using CytoSpec™ v. 2.00.07 (Cytospec Inc., Boston, MA, USA) and The Unscrambler X® 11.1 software package (CAMO Software AS, Oslo, Norway), respectively. In particular, HCA was used to group spectra of similar spectral patterns into five distinct clusters based on Ward’s algorithm within two spectral regions that contain the key biochemical information of the cells (i.e., 3010–2820 cm−1 and 1780–1030 cm−1). As shown in Figure S1, the resultant HCA clusters were well matched to the physical features of the cells, as determined by the optical microscopic image. The average spectra extracted from each of the five HCA clusters (Figure S2) were compared to select the best spectral cluster with the strongest amide I intensity, which usually equated to the spectra obtained from the center of the cellular region (see Figure S1 for an example).
To examine changes in biochemical compositions as a result of the treatment, absorbance spectra were extracted from the selected HCA clusters of the treated and control cells. These spectra were then combined and converted to 2nd derivatives using a 25-point Savitzky–Golay algorithm, to remove the broad baseline offset and curvature []. In total 100–300 spectra were extracted from each cluster group. The resultant 2nd derivative spectra were then corrected using an extended multiplicative scatter correction (EMSC) method over two specific spectral ranges (i.e., 3010–2820 cm−1 and 1810–1000 cm−1). In principle, the EMSC pre-treatment was used to remove light scattering artifacts and normalize the spectra, accounting for path-length differences [,,]. After that, the PCA was performed on the combined EMSC-corrected 2nd derivative spectral datasets containing the treated cells and the control group (no treatment), using a cross-validation method and 7 principal components (PCs). The PCA results provide insights into the similarities and differences in biochemical compositions between cells that have been exposed to one of the solvents of interest and cells that have not.
2.5. X-ray and Neutron Reflectivity
X-ray reflectivity (XRR) and neutron reflectivity (NR) experiments were carried out at the 20 MW Opal Research reactor at ANSTO (Lucas Heights, NSW, Australia). XRR was performed at room temperature on a Panalytical X’Pert Pro (Malvern Panalytical, Malvern, UK) instrument with a copper X-ray source, giving a q range of 0.02–0.5 Å−1.
NR experiments were carried out at room temperature on Platypus—the time-of-flight reflectometer using neutrons with wavelength ranges of 2.55–18.0 and 4.0–18.5 Å for respective incident angles of 1.1 and 4.8°, which gives a total q range of 0.015–0.2 Å−1.
Reflectivity experiments were carried out with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) monolayers prepared using a 601 Langmuir trough (Nima Technology, Coventry, UK). Briefly, POPC in either a hydrogenated (XRR) or partially deuterated (d31, NR) form was dissolved in chloroform at 1 mg/mL. The Langmuir trough was washed several times with either MilliQ water or D2O and several isotherms were run to ensure removal of all contaminants. The trough was then filled with 50 mL of the subphase (either Milli-Q water, or D2O) and approximately 17 µL of the POPC solution was applied to the surface. Several isotherms were run to ensure lipid equilibration and evaporation of chloroform. The solvent of interest was then added to the non-compressed area of the trough to a final concentration of ~10% and allowed to equilibrate for 15 min. The lipid monolayer was then compressed to a surface pressure of ~20 mN/m, and XRR or NR measurements were taken.
The XRR data reduction was performed using ‘in-house’ macros within the Igor Pro environment. NR data reduction was undertaken using refnx [] running in a Jupyter notebook. All data underwent background subtraction and intensity normalisation at the critical edge; the time-of-flight NR data were also divided by a direct beam run to account for wavelength-dependent air transmission. Reflectivity data were modelled using the Motofit analysis program in Igor Pro (Wavementrics, version 8.0.4.2, Portland, UK). The system was modelled as a series of layers with a certain thickness, interfacial roughness, and scattering length density. The quality of fits was monitored using the χ2 value. Both simultaneous and individual fitting of XRR and NR data was tested, but in most cases, the two datasets could be fit using roughly the same parameters.
3. Results and Discussion
3.1. Viability
The viability of HaCat cells following treatment with each of the compounds was measured at multiple time points (Figure 2). Figure 2 demonstrates that after 1 h, only EAA has caused a significant decrease in viability (down to 8 ± 1%). Longer treatments with EAA resulted in complete cell death. After 3 h, all of the treatments caused a drop to below 80% viability, compared to the untreated control cells. Treatment of the cells for 6 h resulted in viabilities less than 20% for DMSO, ChCl–Gly, ChCl–Gal, Bet–Gal, and Prol–Gal. In contrast, Prol–Gly retained a viability of 55 ± 9% after 6 h of treatment. Treatment with any of the compounds of interest, including DMSO, for 24 h resulted in complete cell death (not shown).
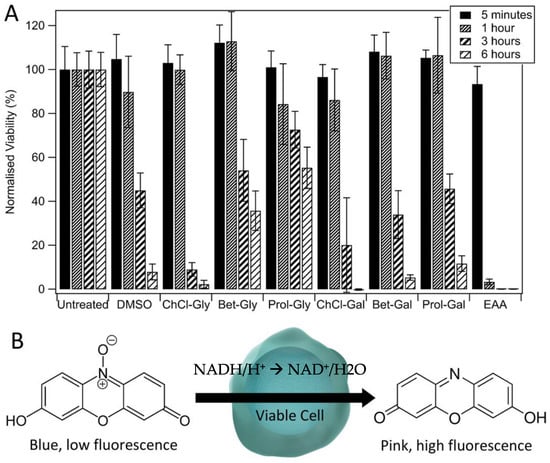
Figure 2.
(A) Viability of HaCat cells following exposure to DMSO, DESs, or EAA for 5 min, 1 h, 3 h, or 6 h based on resazurin assay. Errors are based on standard deviations of triplicate measurements of three repeats. (B) Schematic of the resazurin viability assay.
Prol–Gly had the lowest toxicity of the tested compounds, with HaCat cells retaining a viability of 55 ± 9% even with 6 h of treatment. As evidenced in Figure 2, the toxicity of the DESs seems to depend on both components. Therefore, although proline is an amino acid and was therefore expected to be non-toxic, Prol–Gal demonstrated quite high toxicity, possibly due to the galactose sugar. Similarly, ChCl–Gly had much higher toxicity than Prol–Gly, indicating that the choline chloride salt may be more toxic than either betaine or proline. The result for Prol–Gly aligns well with our previous work which demonstrated that this DES was an effective cryoprotective agent, even after incubating the cells with the DES for extended periods prior to freezing []. Our study also demonstrated that galactose-based DESs were more toxic to a human cell line than the glycerol-based ones, and that those containing choline chloride were more toxic than the others—both of these findings are further supported by the viability results shown in Figure 2. Galactose is known to be toxic to some cells, as well as more broadly to plants, animals, and fungi, though the mechanisms are still unclear [].
In comparison to the DESs, the IL tested here, EAA, was significantly more toxic, demonstrating significant cell death even after just 1 h of exposure. It is uncertain whether this toxicity arises from the ethylammonium or acetate ion, or some combination of the two. Our previous work demonstrated that EAA could act as a cryoprotectant for a human cell line, despite having some toxicity []. The toxicity found here is higher than previously reported; however, the different methods of measuring viability (trypan blue exclusion test, compared to resazurin assay), could account for that, as well as examining a different cell type.
In order to understand why there are vast differences in the toxicity of these solvents, and their capacity to act as cryoprotective agents, synchrotron macro–ATR–FTIR microspectroscopy and neutron reflectivity were performed. These allow detailed insight into the interactions of the solvents with specific cellular components and model membranes.
3.2. Synchrotron Macro–ATR–FTIR Microspectroscopy
High-resolution synchrotron macro–ATR–FTIR microspectroscopy was used to examine biochemical changes in mammalian cells after treatment with neoteric solvents for 1 h. Of particular interest are the methylene/methyl (CH2/CH3) region for lipids (3000–2800 cm−1), amide I and II bands for proteins (1705–1600 cm−1 and 1567–1448 cm−1, respectively), and fingerprint region for polysaccharides and nucleic acids (1200–1000 cm−1). Figure 3 shows the average synchrotron macro–ATR–FTIR spectra of the HaCat cells after different treatments, compared to that of the untreated control cells.
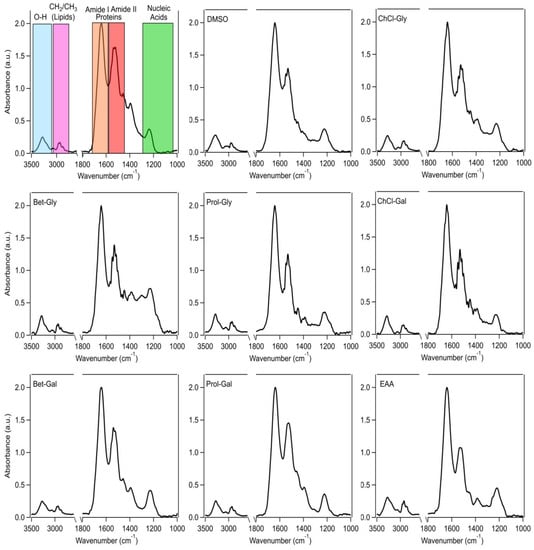
Figure 3.
Average synchrotron macro–ATR–FTIR spectra of HaCat cells from each treatment group with baseline correction and normalization to the amide I peak. The top left image shows the spectrum of an untreated control cell with spectral regions of interest highlighted.
Figure 4 shows spectral maps, integrated over the ranges of interest, for untreated cells, and cells treated with each of the neoteric solvents. The amide and lipid spectral maps showed clear alignment with cell structure, as visualized via optical microscopy, prior to taking the IR maps (Figure S1). More specifically, the lipid region tended to localize at the exterior of the cells forming a kind of ‘boundary’, consistent with the cell membrane. The amide I and amide II integrations showed the greatest absorbance in the center of the cells where proteins are expected to accumulate. In contrast, the nucleic acid region was less consistent, and more difficult to interpret. As shown in Figure 4, there was no clear localization of the nucleic acid region to the interior of the cells possibly due to polysaccharides being present in the external environment of the cells. HCA and PCA analysis was performed on spectra from the cells’ interior (Figure S1) which should include nucleic acids. Furthermore, PCA peak analysis included detailed assignment to specific functional groups to ensure that it was indeed the nucleic acid interactions being explored.
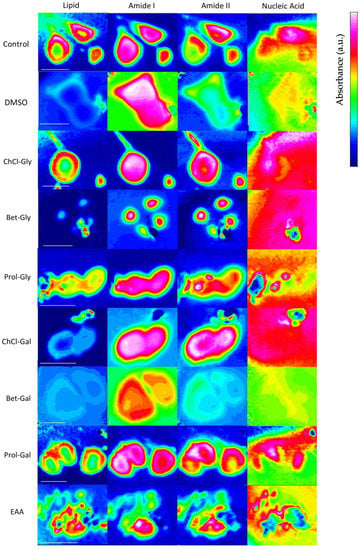
Figure 4.
Synchrotron macro–ATR–FTIR chemical maps of lipids (3010–2837 cm−1), amide I (1746–1563 cm−1), amide II (1567–1448 cm−1), and nucleic acid (1159–993 cm−1) (left to right) observed for untreated control HaCat cells (top row), and cells treated for 1 h with either DMSO or a neoteric solvent (subsequent rows). Scale bar = 20 μm.
Figure 5 shows PC–1, PC–2, and PC–3 loadings for DMSO-treated cells, with the key peaks identified. The key peaks were based on significance cutoffs whereby peaks above a certain loading (e.g., 0.7) were considered significant and used for subsequent analysis. Peaks below the significance threshold are attributed to either atmospheric noise or are not strong enough to confidently attribute to a real signal. The loading and score plots for the other treatments are given in the supplementary information (Figures S3–S10). In some cases, significant peaks were also identified based on the average 2nd derivative spectra of the treated cells in comparison to that of the untreated control (Figures S11 and S12).
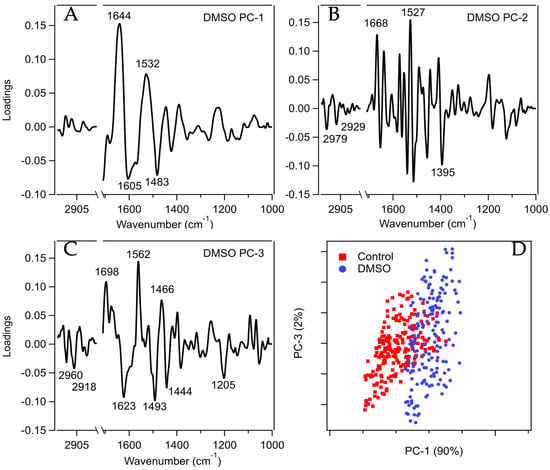
Figure 5.
(A–C) PCA loadings plots of HaCat cells treated with DMSO compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. A significance cut-off of 0.07 was applied to PC–1 and 3, and 0.1 was applied to PC–2. (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes.
Table S1 lists the peaks identified in the PCA loadings plots, along with their assignments based on existing literature. Where possible, these assignments have been broadly grouped into either ‘Lipids’, ‘Amide I (Proteins)’, ‘Amide II (Proteins)’, or ‘Nucleic Acids’. Some of the peaks fell into regions that are known to contain overlapping peaks of proteins, lipids, and polysaccharides, so these peaks were not used for further analysis. Similarly, the ‘Ring Base’ assignation appears frequently (1560–1566 cm−1), but the broad range of spectra and the non-specific allocation of these peaks means that these were also excluded from further comparisons.
Table 1 summarizes the broad assignations for up to PC–3 for each treatment. This table demonstrates key trends that provide insights into why some neoteric solvents may be better cryoprotectants than others, and also why some are more toxic to this particular cell line. The PCA results of the DMSO-treated cells exhibited significant loaded peaks in the lipid region in both PC–1 and PC–3, which agrees well with spectral differences observed in their average 2nd derivative spectra (Figure S11). These differences indicate that DMSO likely interacted with the lipid membranes of the cells and is consistent with previous fundamental studies which demonstrated that DMSO interacts with phospholipid headgroups and influences lipid phase behavior [,]. Membrane studies have shown that DMSO dehydrates lipid headgroups, reducing repulsion between lipids, whereas glycerol interacts with but does not necessarily dehydrate the lipids [].

Table 1.
Regions of variation based on PCA loading plots for each treatment compared to the control (green = peak in PC loading, pink = no peak in PC loading).
This hypothesis is further supported by the loading plots (Figures S4–S6), which showed peaks in the lipid region for all three of the glycerol-based DESs, but none was observed in the galactose-based ones (Figures S7–S9). The key influential peaks present in the loading plots are consistent with the average 2nd derivative spectra, showing a difference in cells treated with glycerol-based DESs, but not those treated with galactose-based DESs. Lipid interactions by both DMSO and glycerol could be one mechanism of cryoprotection [] and explain why the galactose-based DESs performed so poorly as cryoprotectants []. Similarly, EAA is an effective cryoprotective agent, and also resulted in a significant loading peak in the lipid region, further demonstrating the importance of lipid interactions for cryoprotection []. These PCA results are further supported by a detailed examination of the lipid region of the average spectra for cells treated with DMSO, EAA, Prol–Gly, and Prol–Gal in comparison to the control (Figure 6). The Prol–Gal spectra are almost identical to that of the control, whereas all of the other three show significant differences—further confirming interactions with the lipid region and highlighting the importance of lipid interactions for cryoprotective efficacy.
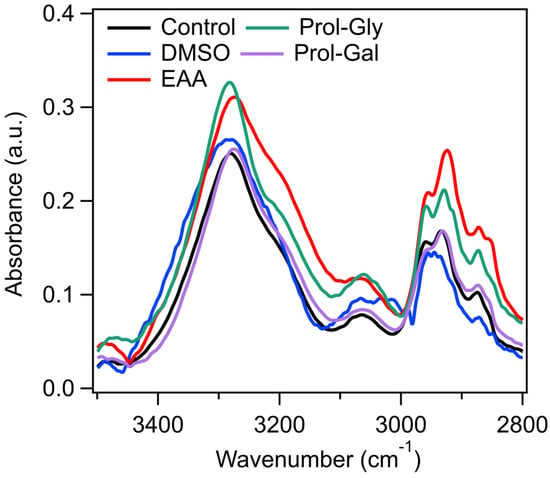
Figure 6.
Average spectra in the lipid region of control cells and those treated with either DMSO, EAA, Prol–Gly, or Prol–Gal.
Although all treatments resulted in changes to the amide I, amide II, and nucleic acid regions (Table 1), these were not all of the same magnitude (Figure 3). This further supports the growing literature that aqueous solutions of DESs do not behave as simple mixtures of their two components [,,,]. It is possible that some of the differences in the spectra observed in the amide I region are due to residual neoteric solvents, despite numerous washing and dehydration steps. As shown in Figure S13, these solvents have a peak in this region which could contribute to differences in the intensity of the peaks in this region. However, variations in the amide II and nucleic acid regions can shed new insights into possible toxicity mechanisms.
As discussed above, EAA showed the greatest toxicity to these cells, causing more than 90% cell death after just one hour of exposure based on the toxicity assay described in Section 3.1. Given that the cells were treated for one hour with the solvents prior to the synchrotron macro–ATR–FTIR measurements, it is likely that the majority of the cells exposed to EAA were dead when the macro–ATR–FTIR measurements were performed. Indeed, the integrated spectra in Figure 4 for the EAA-treated cells show a much less-defined cell shape for all the selected wavenumber regions compared to the other treatments. Comparison of the 2nd derivative spectra of EAA and the control cell (Figure S12) shows substantial differences, especially in the nucleic acid region. This suggests that EAA molecules may interact with the nucleic acid components inside the cells, ultimately causing extensive toxicity and cell death.
Previous studies have used FTIR to examine changes to cells with freezing/thawing in the presence of cryoprotective agents, including DMSO []. However, these have focused on cellular changes due to temperature and the freezing/thawing process, rather than due to the cryoprotective agent itself []. The synchrotron macro–ATR–FTIR results provide insights into possible mechanisms for both toxicity and cryopreservation of neoteric solvents toward a human cell line. The membrane interactions were further explored using neutron and X-ray reflectivity experiments.
3.3. Neutron and X-ray Reflectivity
Neutron and X-ray reflectivity measurements were carried out at the Lucas Heights campus of the Australian Nuclear Science and Technology Organisation (ANSTO). The experiments were performed on a POPC lipid monolayer with a varied subphase (that is, the solvent environment). These experiments compared DMSO to glycerol-containing DESs in an effort to understand why Prol–Gly and DMSO are effective CPAs, but ChCl–Gly and Bet–Gly are not. The reflectivity data along with best fits are shown in Figure S14. These data were fit using a slab model. For these fits different combinations of fixed and varied parameters were tested, but the most consistent results were found by fixing the headgroup thickness, and allowing tail thickness to vary. Table 2 shows the results of fitting these parameters for a POPC monolayer when exposed to a subphase containing 10 wt% of the compound of interest (DMSO, Prol–Gly, ChCl–Gly, or Bet–Gly).
Table 2.
Fitting results of reflectivity data of a POPC lipid monolayer with either pure water, or 10 wt% additive subphase.
To date, few studies have utilised reflectivity to examine the interactions of neoteric solvents with lipid layers. One study examined the interaction of DMSO with a DPPC monolayer but found no change in the bilayer thickness []. As shown in Table 2, the head and tail thicknesses of POPC monolayers changed little with exposure to different subphases. Of particular note is that good fits could only be achieved for XRR data for the DMSO-containing subphase when the SLDs of both the head and tail regions were allowed to vary. This is strongly indicative of DMSO penetration into the lipid region, consistent with previous studies [,] and the synchrotron macro–ATR–FTIR results discussed above. A DMSO subphase also appeared to cause a slight decrease in tail thickness of ~1 Å.
Neither ChCl–Gly nor Bet–Gly caused significant changes to the reflectivity profile of the POPC monolayer. This is somewhat counter to the PCA-loaded peaks observed in the synchrotron macro–ATR–FTIR spectra discussed above, which showed changes to the lipid region on treatment with these two solvents. There are several possible explanations for this. First, the cell membrane is made up of hundreds of different types of lipids, whereas the reflectivity measurements utilise a monolayer of just one type. In addition, the reflectivity measurements are performed on a monolayer, which reduces resolution but may also impact interactions as compared to the bilayer membrane of a cell.
In contrast, the Prol–Gly subphase caused some change to the reflectivity profile, especially for the XRR data. When the tail thickness was allowed to vary, it increased to 12.7 Å. The neutron data, however, could be fitted with a tail thickness as low as 11 Å, although the high background in this data makes fitting parameters uncertain.
Given the low (molar) concentrations of DES in the subphase, and the low resolution achievable with a monolayer, definitive conclusions are difficult to draw from this data, other than that both DMSO and Prol–Gly appear to cause a greater disruption to assembled lipid structures than ChCl–Gly or Bet–Gly. This may suggest that cryoprotective efficacy depends on strong lipid interactions. Future studies will use submerged bilayers to obtain better resolution of interactions, but that is beyond the scope of the current study.
4. Conclusions
The research described above utilised advanced nuclear- and synchrotron-based techniques, including reflectivity and macro–ATR–FTIR microspectroscopy, in combination with toxicity assays to investigate why some neoteric solvents are effective cryoprotectants while others are not.
Toxicity assays demonstrated that Prol–Gly was the least toxic of the solvents tested whereas EAA was the most toxic, resulting in extensive cell death with just 1 h exposure. Even DMSO, the current gold standard for cryopreservation, resulted in extensive cell death. Macro–ATR–FTIR showed that those solvents known to act as cryoprotectants (DMSO, Prol–Gly, and EAA) caused biochemical changes to the lipid components of cells; that was not always observed for the other solvents. This was further supported by the reflectivity results which demonstrated that DMSO and Prol–Gly changed the thickness of the lipid monolayer to a greater degree than the other DESs tested. This suggests that they are inserting into, or interacting with, the lipid molecules. In the case of EAA, although similar changes were observed in the lipid region, additional changes were observed in the nucleic acid region which may be related to the higher toxicity of EAA.
Ultimately, the results from this study suggest a strong correlation between lipid interactions and successful cryopreservation (as observed for DMSO, Prol–Gly, and EAA). This is the first study to utilise a combination of cell viability, reflectivity, and macro–ATR–FTIR techniques to explore the interactions underlying the cryoprotective effect of neoteric solvents.
However, there are still many unanswered questions that need to be addressed, such as the complex relationship between cryoprotection, toxicity, and lipid interactions. Future research will expand on the above studies by investigating the components of the DESs separately, utilising reflectivity from submerged bilayers to obtain higher resolution, and systematically varying the solvent components to examine the effect of changing the tail length or ionic species. The effects of changing the composition of the components will also be explored. These new insights will provide even more clarity on what drives lipid (and nucleic acid) interactions and will thus lead to the rational design of neoteric solvents for specific applications, including cryopreservation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biophysica3020021/s1; Figure S1: Top left: microscope image of fixed, untreated HaCat cells prior to the synchrotron macro-ATR-FTIR imaging. Top right: HCA image obtained from the corresponding synchrotron macro-ATR-FTIR dataset of the same HaCat cells. Bottom: five average spectra extracted from each of the five colored clusters. Note: black arrow indicates the spectral cluster selected for subsequent PCA analysis; Figure S2: Regions of interest containing the key biochemical information in the acquired synchrotron macro-ATR-FTIR spectra; Figure S3: (A–C) PCA loadings plots of HaCat cells treated with DMSO compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. A significance cut-off of 0.07 was applied to PC–1 and 3, and of 0.1 was applied to PC–2. (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes; Figure S4: (A–C) PCA loadings plot of HaCat cells treated with ChCl-Gly compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. Significance cut off of 0.07 applied to PC–1 and of 0.1 to PC–2 and PC–3. (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes; Figure S5: (A–C) PCA loadings plots of HaCat cells treated with Bet-Gly compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. Significance cut off of 0.07 applied to PC–1 and of 0.1 to PC–2 and PC–3. (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes; Figure S6: (A–C) PCA loadings plots of HaCat cells treated with Prol-Gly compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. A significance cut-off of 0.07 was applied to PC–1 and of 0.1 was applied to PC–2 and PC–3. (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes; Figure S7: PCA loadings plots of HaCat cells treated with ChCl–Gal compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. Significance cut off of 0.05 was applied to PC–1 and of 0.1 to PC–2 and PC–3. (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes; Figure S8: (A–C) PCA loadings plots of HaCat cells treated with Bet-Gal compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. A significance cut off of 0.07 was applied to PC–1 and PC–2 and of 0.1 to PC–3 (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes; Figure S9: PCA loadings plots of HaCat cells treated with Prol-Gal compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. Significance cut offs of 0.05, 0.07 and 0.1 were applied for PC–1, 2 and 3 respectively. (D) PCA scores plot showing the separation of the cell clusters along PC–1 and PC–3 axes; Figure S10: (A–C) PCA loadings plots of HaCat cells treated with EAA compared to the untreated control. The assignment of key absorption peaks is shown in Table S1. A significance cut-off of 0.07 was applied to PC–1 and PC–2 and of 0.1 was applied to PC–3. (D,E) PCA scores plot showing the separation of the cell clusters along PC–1, PC–2 and PC–3 axes; Figure S11: Comparison of average 2nd derivative spectra of the DMSO-treated and untreated control cells, showing the differences in the key biochemical compositions (i.e., lipids, proteins and nucleic acids); Figure S12: Comparison of average 2nd derivative spectra of treated cells and untreated control cells, showing the differences in the key biochemical compositions (i.e., lipids, proteins and nucleic acids); Figure S13: FTIR spectra of the deep eutectic solvents used in this study; Figure S14: Neutron and X-ray reflectivity data with best fits for a POPC monolayer; Table S1: Band assignment of the loaded peaks observed in the PCA loadings plots for each treatment [,,,,,,,,,,,,,,].
Author Contributions
Conceptualization, S.J.B.; methodology, S.J.B., Z.L.S., L.Z.Y.H., A.E., A.N.A., J.V., S.A.H., T.L.G. and G.B.; formal analysis, S.J.B., Z.L.S., A.E., A.N.A., J.V., S.A.H., T.L.G. and G.B.; investigation, S.J.B., Z.L.S., L.Z.Y.H., A.E., A.N.A., J.V., S.A.H., T.L.G. and G.B.; writing—original draft preparation, S.J.B.; writing—review and editing, S.J.B., Z.L.S., L.Z.Y.H., A.E., A.N.A., J.V., S.A.H., T.L.G. and G.B.; supervision, S.J.B., A.E., T.L.G. and G.B.; project administration, S.J.B., A.E., T.L.G. and G.B.; funding acquisition, S.J.B., A.E. and G.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by an Australian Institute of Nuclear Science and Engineering (AINSE) Early Career Researcher Grant (ECRG) (S.J.B.) and Postgraduate Research Award (PGRA) (Z.L.S. and L.Z.Y.H.). The synchrotron macro–ATR–FTIR experiment was undertaken at the Infrared Microspectroscopy (IRM) beamline at the Australian Synchrotron (Victoria, Australia), part of ANSTO. A.E. is supported by an Australian Research Council (ARC) Discovery Early Career Research Award (DECRA) (DE220100511). G.B. and S.J.B. acknowledge the support of the ARC Research Council grant DP190101010. We acknowledge the support of the Australian Centre for Neutron Scattering, ANSTO, and the Australian Government through the National Collaborative Research Infrastructure Strategy, in supporting the neutron research infrastructure used in this work via ACNS proposal 9771. A.N.A. acknowledges the support of the Australian Research Council (ARC) through its Centre of Excellence for Nanoscale BioPhotonics (CE140100003).
Data Availability Statement
Data is available upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pegg, D.E. Principles of Cryopreservation. In Cryopreservation and Freeze-Drying Protocols; Day, J.G., Stacey, G.N., Eds.; Humana Press: Totowa, NJ, USA, 2007; pp. 39–57. [Google Scholar]
- Fuller, B.; Paynter, S.; Watson, P. Cryopreservation of Human Gametes and Embryos. In Life in the Frozen State; Fuller, B.J., Lane, N., Benson, E.E., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 505–539. [Google Scholar]
- Holley, A.; Marks, D.C.; Johnson, L.; Reade, M.C.; Badloe, J.F.; Noorman, F. Frozen Blood Products: Clinically Effective and Potentially Ideal for Remote Australia. Anaesth. Intensive Care 2013, 41, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Berz, D.; McCormack, E.M.; Winer, E.S.; Colvin, G.A.; Quesenberry, P.J. Cryopreservation of hematopoietic stem cells. Am. J. Hematol. 2007, 82, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.H.; Park, S.C.; Yang, J.H.; Kim, J.Y.; Seok, J.H.; Park, U.S.; Choi, C.W.; Lee, S.R.; Han, J. Cryopreservation and its clinical applications. Integr. Med. Res. 2017, 6, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarczyk, A.; Funnekotter, B.; Turner, S.R.; Bunn, E.; Bryant, G.; Hunt, T.E.; Mancera, R.L. Development of cryopreservation for Loxocarya cinerea—An endemic australian plant species important for post-mining restoration. Cryoletters 2013, 34, 508–519. [Google Scholar] [PubMed]
- Ballou, J.D. Potential contribution of cryopreserved germ plasm to the preservation of genetic diversity and conservation of endangered species in captivity. Cryobiology 1992, 29, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Walters, C.; Wheeler, L.; Stanwood, P.C. Longevity of cryogenically stored seeds. Cryobiology 2004, 48, 229–244. [Google Scholar] [CrossRef]
- Sputtek, A.; Sputtek, R. Cryopreservation in Transfusion Medicine and Hematology. In Life in the Frozen State; Fuller, B.J., Lane, N., Benson, E.E., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 483–504. [Google Scholar]
- Hunt, C.J. Cryopreservation of Human Stem Cells for Clinical Application: A Review. Transfus. Med. Hemother. 2011, 38, 107–123. [Google Scholar] [CrossRef]
- Mazur, P. Cryobiology: The Freezing of Biological Systems. Science 1970, 168, 939–949. [Google Scholar] [CrossRef]
- Manuchehrabadi, N.; Gao, Z.; Zhang, J.; Ring, H.L.; Shao, Q.; Liu, F.; McDermott, M.; Fok, A.; Rabin, Y.; Brockbank, K.G.M.; et al. Improved tissue cryopreservation using inductive heating of magnetic nanoparticles. Sci. Transl. Med. 2017, 9, eaah4586. [Google Scholar] [CrossRef]
- Ardehali, A. 1. While millions and millions of lives have been saved, organ transplantation still faces massive problems after 50 years; organ preservation is a big part of the solution. Cryobiology 2015, 71, 164–165. [Google Scholar] [CrossRef]
- Khush, K.K.; Zaroff, J.G.; Nguyen, J.; Menza, R.; Goldstein, B.A. National Decline in Donor Heart Utilization with Regional Variability: 1995–2010. Am. J. Transplant. 2015, 15, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Smith, A.U.; Parkes, A.S. Revival of Spermatozoa after Vitrification and Dehydration at Low Temperatures. Nature 1949, 164, 666. [Google Scholar] [CrossRef] [PubMed]
- Lovelock, J.E.; Bishop, M.W.H. Prevention of Freezing Damage to Living Cells by Dimethyl Sulphoxide. Nature 1959, 183, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.; Bryant, G. Cellular cryobiology: Thermodynamic and mechanical effects. Int. J. Refrig. 2001, 24, 438–450. [Google Scholar] [CrossRef]
- Fuller, B.J. Cryoprotectants: The essential antifreezes to protect life in the frozen state. Cryoletters 2004, 25, 375–388. [Google Scholar] [PubMed]
- Windrum, P.; Morris, T.C.M. Severe neurotoxicity because of dimethyl sulphoxide following peripheral blood stem cell transplantation. Bone Marrow Transplant. 2003, 31, 315. [Google Scholar] [CrossRef]
- Benekli, M.; Anderson, B.; Wentling, D.; Bernstein, S.; Czuczman, M.; McCarthy, P. Severe respiratory depression after dimethylsulphoxide-containing autologous stem cell infusion in a patient with AL amyloidosis. Bone Marrow Transplant. 2000, 25, 1299–1301. [Google Scholar] [CrossRef]
- Raju, R.; Bryant, S.J.; Wilkinson, B.L.; Bryant, G. The need for novel cryoprotectants and cryopreservation protocols: Insights into the importance of biophysical investigation and cell permeability. Biochim. Biophys. Acta (BBA) Gen. Subj. 2021, 1865, 129749. [Google Scholar] [CrossRef]
- Verheijen, M.; Lienhard, M.; Schrooders, Y.; Clayton, O.; Nudischer, R.; Boerno, S.; Timmermann, B.; Selevsek, N.; Schlapbach, R.; Gmuender, H.; et al. DMSO induces drastic changes in human cellular processes and epigenetic landscape in vitro. Sci. Rep. 2019, 9, 4641. [Google Scholar] [CrossRef]
- Bryant, S.J.; Awad, M.N.; Elbourne, A.; Christofferson, A.J.; Martin, A.V.; Meftahi, N.; Drummond, C.J.; Greaves, T.L.; Bryant, G. Deep eutectic solvents as cryoprotective agents for mammalian cells. J. Mater. Chem. B 2022, 10, 4546–4560. [Google Scholar] [CrossRef]
- Bryant, S.J.; Brown, S.J.; Martin, A.V.; Arunkumar, R.; Raju, R.; Elbourne, A.; Bryant, G.; Drummond, C.J.; Greaves, T.L. Cryopreservation of mammalian cells using protic ionic liquid solutions. J. Colloid Interface Sci. 2021, 603, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Christofferson, A.J.; Greaves, T.L.; McConville, C.F.; Bryant, G.; Elbourne, A. Bulk and interfacial nanostructure and properties in deep eutectic solvents: Current perspectives and future directions. J. Colloid Interface Sci. 2022, 608, 2430–2454. [Google Scholar] [CrossRef] [PubMed]
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Properties and Applications. Chem. Rev. 2008, 108, 206–237. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 1, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Gertrudes, A.; Craveiro, R.; Eltayari, Z.; Reis, R.L.; Paiva, A.; Duarte, A.R.C. How Do Animals Survive Extreme Temperature Amplitudes? The Role of Natural Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2017, 5, 9542–9553. [Google Scholar] [CrossRef]
- Kuroda, K.; Komori, T.; Ishibashi, K.; Uto, T.; Kobayashi, I.; Kadokawa, R.; Kato, Y.; Ninomiya, K.; Takahashi, K.; Hirata, E. Non-aqueous, zwitterionic solvent as an alternative for dimethyl sulfoxide in the life sciences. Commun. Chem. 2020, 3, 163. [Google Scholar] [CrossRef]
- Kato, Y.; Uto, T.; Tanaka, D.; Ishibashi, K.; Kobayashi, A.; Hazawa, M.; Wong, R.W.; Ninomiya, K.; Takahashi, K.; Hirata, E.; et al. Synthetic zwitterions as efficient non-permeable cryoprotectants. Commun. Chem. 2021, 4, 151. [Google Scholar] [CrossRef]
- Jesus, A.R.; Meneses, L.; Duarte, A.R.C.; Paiva, A. Natural deep eutectic systems, an emerging class of cryoprotectant agents. Cryobiology 2021, 101, 95–104. [Google Scholar] [CrossRef]
- Castro, V.I.B.; Craveiro, R.; Silva, J.M.; Reis, R.L.; Paiva, A.; Duarte, A.R.C. Natural deep eutectic systems as alternative nontoxic cryoprotective agents. Cryobiology 2018, 83, 15–26. [Google Scholar] [CrossRef]
- Li, R.; Hornberger, K.; Dutton, J.R.; Hubel, A. Cryopreservation of Human iPS Cell Aggregates in a DMSO-Free Solution-An Optimization and Comparative Study. Front. Bioeng. Biotechnol. 2020, 8, 1. [Google Scholar] [CrossRef]
- Mbous, Y.P.; Hayyan, M.; Wong, W.F.; Hayyan, A.; Looi, C.Y.; Hashim, M.A. Simulation of Deep Eutectic Solvents’ Interaction with Membranes of Cancer Cells Using COSMO-RS. J. Phys. Chem. B 2020, 124, 9086–9094. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.P.; McCluskey, A.; Atkin, R. Activity and thermal stability of lysozyme in alkylammonium formate ionic liquids-influence of cation modification. Green Chem. 2009, 11, 785–792. [Google Scholar] [CrossRef]
- Bryant, S.J.; Garcia, A.; Clarke, R.J.; Warr, G.G. Selective ion transport across a lipid bilayer in a protic ionic liquid. Soft Matter 2021, 17, 2688–2694. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Atkin, R.; Gradzielski, M.; Warr, G.G. Catanionic Surfactant Self-Assembly in Protic Ionic Liquids. J. Phys. Chem. Lett. 2020, 11, 5926–5931. [Google Scholar] [CrossRef]
- Bryant, S.J.; Atkin, R.; Warr, G.G. Effect of Deep Eutectic Solvent Nanostructure on Phospholipid Bilayer Phases. Langmuir 2017, 33, 6878–6884. [Google Scholar] [CrossRef]
- Bryant, S.J.; Atkin, R.; Warr, G.G. Spontaneous vesicle formation in a deep eutectic solvent. Soft Matter 2016, 12, 1645–1648. [Google Scholar] [CrossRef]
- Greaves, T.L.; Weerawardena, A.; Fong, C.; Krodkiewska, I.; Drummond, C.J. Protic Ionic Liquids: Solvents with Tunable Phase Behavior and Physicochemical Properties. J. Phys. Chem. B 2006, 110, 22479–22487. [Google Scholar] [CrossRef]
- Vongsvivut, J.; Pérez-Guaita, D.; Wood, B.R.; Heraud, P.; Khambatta, K.; Hartnell, D.; Hackett, M.J.; Tobin, M.J. Synchrotron macro ATR-FTIR microspectroscopy for high-resolution chemical mapping of single cells. Analyst 2019, 144, 3226–3238. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Kohler, A.; Kirschner, C.; Oust, A.; Martens, H. Extended Multiplicative Signal Correction as a Tool for Separation and Characterization of Physical and Chemical Information in Fourier Transform Infrared Microscopy Images of Cryo-Sections of Beef Loin. Appl. Spectrosc. 2005, 59, 707–716. [Google Scholar] [CrossRef]
- Correa, E.; Sletta, H.; Ellis, D.I.; Hoel, S.; Ertesvåg, H.; Ellingsen, T.E.; Valla, S.; Goodacre, R. Rapid reagentless quantification of alginate biosynthesis in Pseudomonas fluorescens bacteria mutants using FT-IR spectroscopy coupled to multivariate partial least squares regression. Anal. Bioanal. Chem. 2012, 403, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, S.; Shaw, Z.L.; Vongsvivut, J.; Crawford, R.J.; Dupont, M.F.; Boyce, K.J.; Gangadoo, S.; Bryant, S.J.; Bryant, G.; Cozzolino, D.; et al. Analysis of Pathogenic Bacterial and Yeast Biofilms Using the Combination of Synchrotron ATR-FTIR Microspectroscopy and Chemometric Approaches. Molecules 2021, 26, 3890. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.J.; Prescott, S.W. refnx: Neutron and X-ray reflectometry analysis in Python. J. Appl. Crystallogr. 2019, 52, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Höftberger, M.; Althammer, M.; Foissner, I.; Tenhaken, R. Galactose induces formation of cell wall stubs and cell death in Arabidopsis roots. Planta 2022, 256, 26. [Google Scholar] [CrossRef]
- Yu, Z.-W.; Quinn, P.J. Solvation effects of dimethyl sulphoxide on the structure of phospholipid bilayers. Biophys. Chem. 1998, 70, 35–39. [Google Scholar] [CrossRef]
- Yu, Z.-W.; Quinn, P.J. The modulation of membrane structure and stability by dimethyl sulphoxide (Review). Mol. Membr. Biol. 1998, 15, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.M.; Cheng, C.-Y.; Israelachvili, J.N.; Han, S. Communication: Contrasting effects of glycerol and DMSO on lipid membrane surface hydration dynamics and forces. J. Chem. Phys. 2016, 145, 041101. [Google Scholar] [CrossRef]
- Gordeliy, V.I.; Kiselev, M.A.; Lesieur, P.; Pole, A.V.; Teixeira, J. Lipid Membrane Structure and Interactions in Dimethyl Sulfoxide/Water Mixtures. Biophys. J. 1998, 75, 2343–2351. [Google Scholar] [CrossRef]
- Hammond, O.S.; Bowron, D.T.; Edler, K.J. The Effect of Water upon Deep Eutectic Solvent Nanostructure: An Unusual Transition from Ionic Mixture to Aqueous Solution. Angew. Chem. Int. Ed. 2017, 56, 9782–9785. [Google Scholar] [CrossRef]
- Posada, E.; López-Salas, N.; Jiménez Riobóo, R.J.; Ferrer, M.L.; Gutiérrez, M.C.; del Monte, F. Reline aqueous solutions behaving as liquid mixtures of H-bonded co-solvents: Microphase segregation and formation of co-continuous structures as indicated by Brillouin and 1H NMR spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 17103–17110. [Google Scholar] [CrossRef]
- Gutiérrez, M.C.; Ferrer, M.L.; Mateo, C.R.; del Monte, F. Freeze-drying of aqueous solutions of deep eutectic solvents: A suitable approach to deep eutectic suspensions of self-assembled structures. Langmuir 2009, 25, 5509–5515. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Karlsson, J.O.M.; Aksan, A. FTIR Analysis of Molecular Changes Associated with Warming Injury in Cryopreserved Leukocytes. Langmuir 2019, 35, 7552–7559. [Google Scholar] [CrossRef] [PubMed]
- Wolkers, W.F.; Oldenhof, H. Use of FTIR Spectroscopy to Study Cells and Tissues during Cryopreservation Processing and Thermal Therapies. In Multiscale Technologies for Cryomedicine; He, X., Bischof, J.C., Eds.; World Scientific: Singapore, 2016; pp. 353–370. [Google Scholar]
- Dabkowska, A.P.; Collins, L.E.; Barlow, D.J.; Barker, R.; McLain, S.E.; Lawrence, M.J.; Lorenz, C.D. Modulation of Dipalmitoylphosphatidylcholine Monolayers by Dimethyl Sulfoxide. Langmuir 2014, 30, 8803–8811. [Google Scholar] [CrossRef]
- Movasaghi, Z.; Rehman, S.; ur Rehman, D.I. Raman Spectroscopy of Biological Tissues. Appl. Spectrosc. Rev. 2008, 43, 134–179. [Google Scholar] [CrossRef]
- Huleihel, M.; Salman, A.; Erukhimovitch, V.; Ramesh, J.; Hammody, Z.; Mordechai, S. Novel spectral method for the study of viral carcinogenesis in vitro. J. Biochem. Biophys. Methods 2002, 50, 111–121. [Google Scholar] [CrossRef]
- Dovbeshko, G.I.; Gridina, N.Y.; Kruglova, E.B.; Pashchuk, O.P. FTIR spectroscopy studies of nucleic acid damage. Talanta 2000, 53, 233–246. [Google Scholar] [CrossRef]
- Fujioka, N.; Morimoto, Y.; Arai, T.; Kikuchi, M. Discrimination between normal and malignant human gastric tissues by Fourier transform infrared spectroscopy. Cancer Detect. Prev. 2004, 28, 32–36. [Google Scholar] [CrossRef]
- Paluszkiewicz, C.; Kwiatek, W.M. Analysis of human cancer prostate tissues using FTIR microspectroscopy and SRIXE techniques. J. Mol. Struct. 2001, 565–566, 329–334. [Google Scholar] [CrossRef]
- Chiriboga, L.; Xie, P.; Yee, H.; Vigorita, V.; Zarou, D.; Zakim, D.; Diem, M. Infrared spectroscopy of human tissue. I. Differentiation and maturation of epithelial cells in the human cervix. Biospectroscopy 1998, 4, 47–53. [Google Scholar] [CrossRef]
- Yang, Y.; Sulé-Suso, J.; Sockalingum, G.D.; Kegelaer, G.; Manfait, M.; El Haj, A.J. Study of tumor cell invasion by Fourier transform infrared microspectroscopy. Biopolymers 2005, 78, 311–317. [Google Scholar] [CrossRef]
- Eckel, R.; Huo, H.; Guan, H.-W.; Hu, X.; Che, X.; Huang, W.-D. Characteristic infrared spectroscopic patterns in the protein bands of human breast cancer tissue. Vib. Spectrosc. 2001, 27, 165–173. [Google Scholar] [CrossRef]
- Dovbeshko, G.I.; Chegel, V.I.; Gridina, N.Y.; Repnytska, O.P.; Shirshov, Y.M.; Tryndiak, V.P.; Todor, I.M.; Solyanik, G.I. Surface enhanced IR absorption of nucleic acids from tumor cells: FTIR reflectance study. Biopolymers 2002, 67, 470–486. [Google Scholar] [CrossRef] [PubMed]
- Gazi, E.; Dwyer, J.; Gardner, P.; Ghanbari-Siahkali, A.; Wade, A.P.; Miyan, J.; Lockyer, N.P.; Vickerman, J.C.; Clarke, N.W.; Shanks, J.H.; et al. Applications of Fourier transform infrared microspectroscopy in studies of benign prostate and prostate cancer. J. Pathol. 2003, 201, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Andrus, P.G.; Strickland, R.D. Cancer grading by Fourier transform infrared spectroscopy. Biospectroscopy 1998, 4, 37–46. [Google Scholar] [CrossRef]
- Yoshida, S.; Miyazaki, M.; Sakai, K.; Takeshita, M.; Yuasa, S.; Sato, A.; Kobayashi, T.; Watanabe, S.; Okuyama, H. Fourier transform infrared spectroscopic analysis of rat brain microsomal membranes modified by dietary fatty acids: Possible correlation with altered learning behavior. Biospectroscopy 1997, 3, 281–290. [Google Scholar] [CrossRef]
- Fung, M.F.K.; Senterman, M.K.; Mikhael, N.Z.; Lacelle, S.; Wong, P.T.T. Pressure-tuning fourier transform infrared spectroscopic study of carcinogenesis in human endometrium. Biospectroscopy 1996, 2, 155–165. [Google Scholar] [CrossRef]
- Fabian, H.; Jackson, M.; Murphy, L.; Watson, P.H.; Fichtner, I.; Mantsch, H.H. A comparative infrared spectroscopic study of human breast tumors and breast tumor cell xenografts. Biospectroscopy 1995, 1, 37–45. [Google Scholar] [CrossRef]
- Wood, B.R.; Quinn, M.A.; Tait, B.; Ashdown, M.; Hislop, T.; Romeo, M.; McNaughton, D. FTIR microspectroscopic study of cell types and potential confounding variables in screening for cervical malignancies. Biospectroscopy 1998, 4, 75–91. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).