
Mechanistic Insights into the Inhibition of SARS-CoV-2 Main Protease by Clovamide and Its Derivatives: In Silico Studies




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
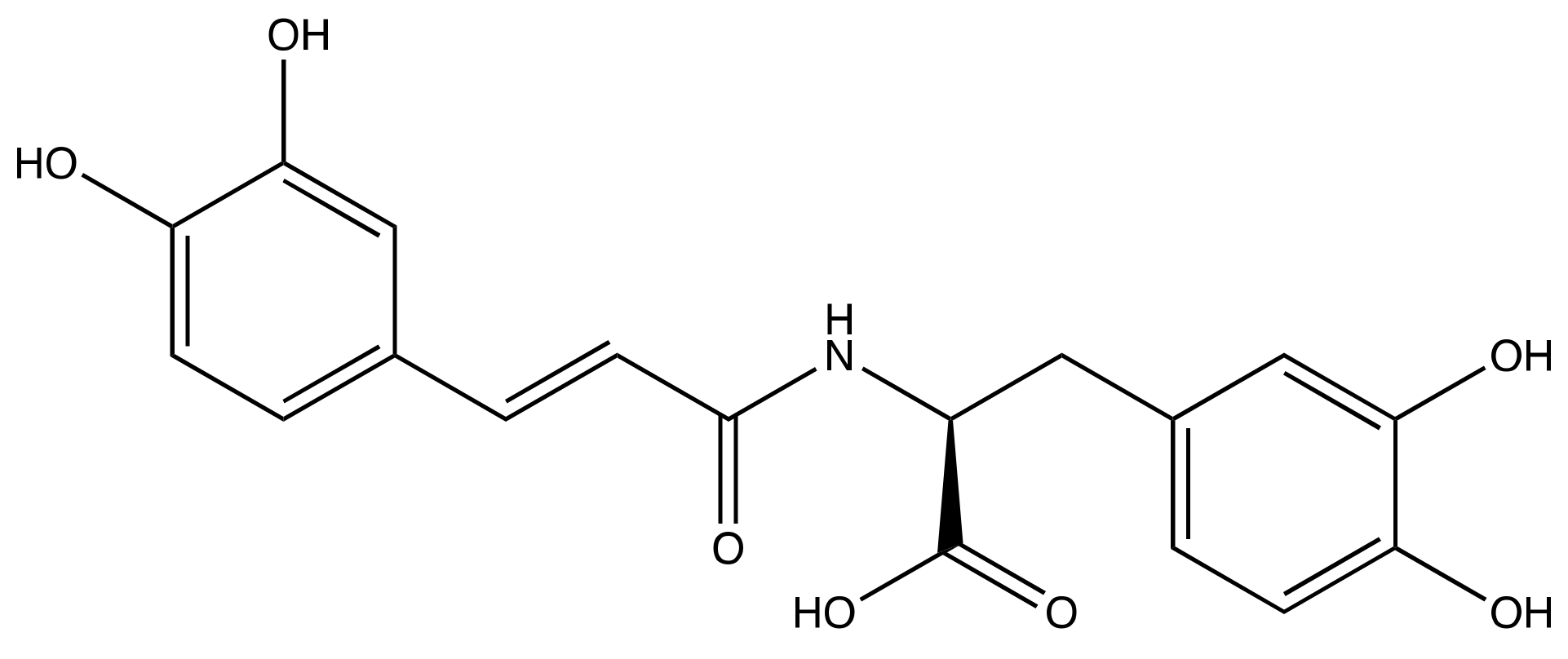
:1. Introduction
2. Materials and Methods
2.1. Density Functional Theory
2.2. Molecular Docking and Dynamics
3. Results
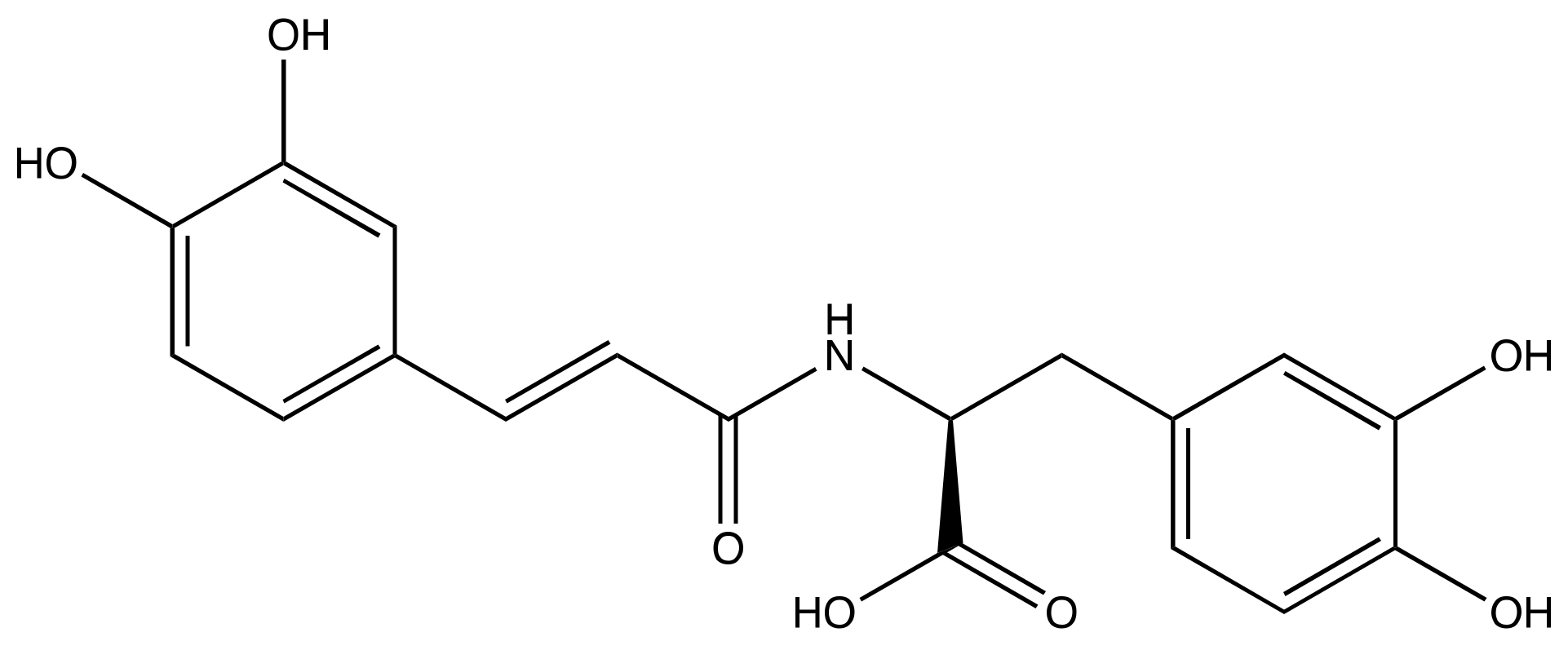
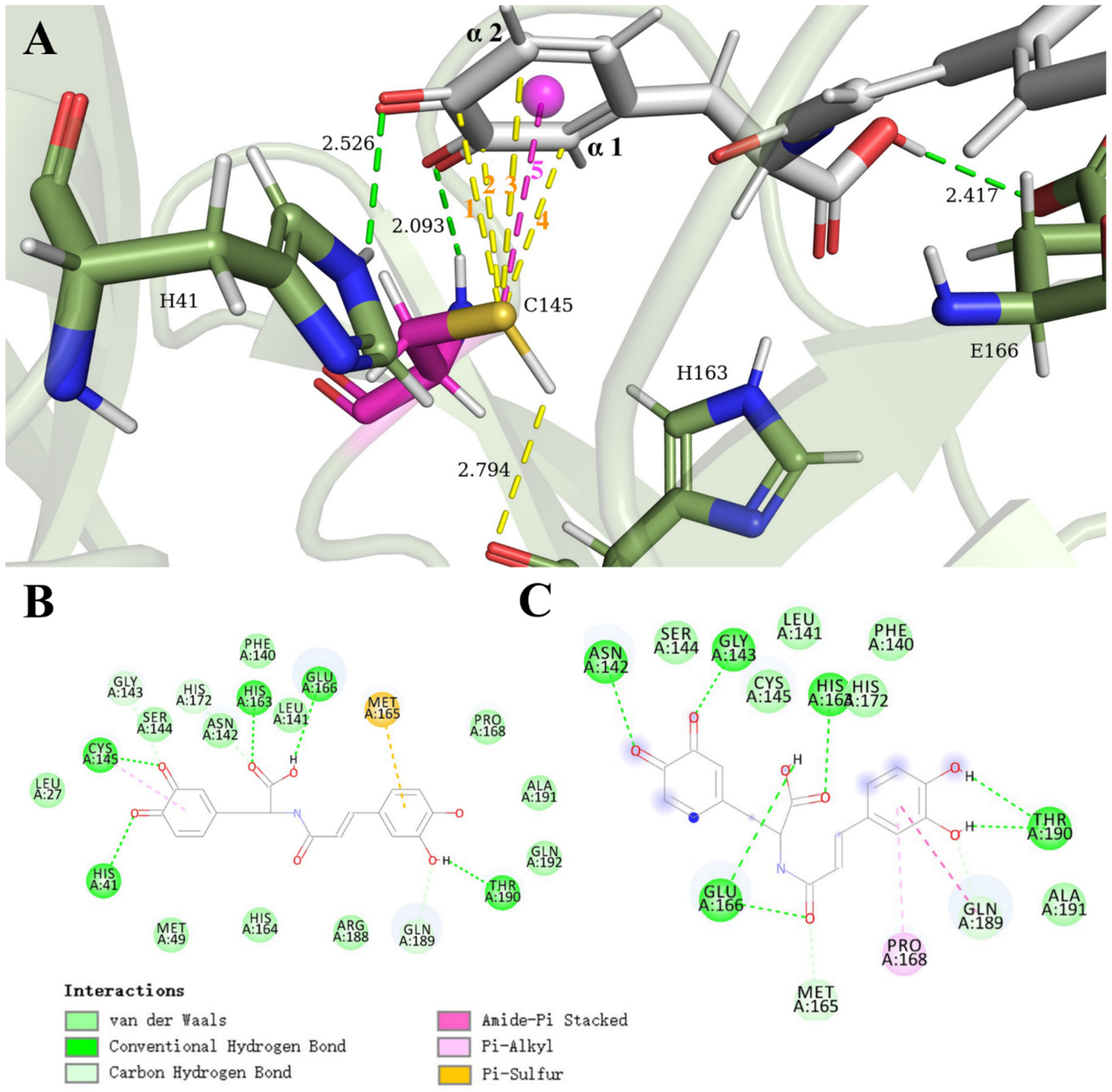
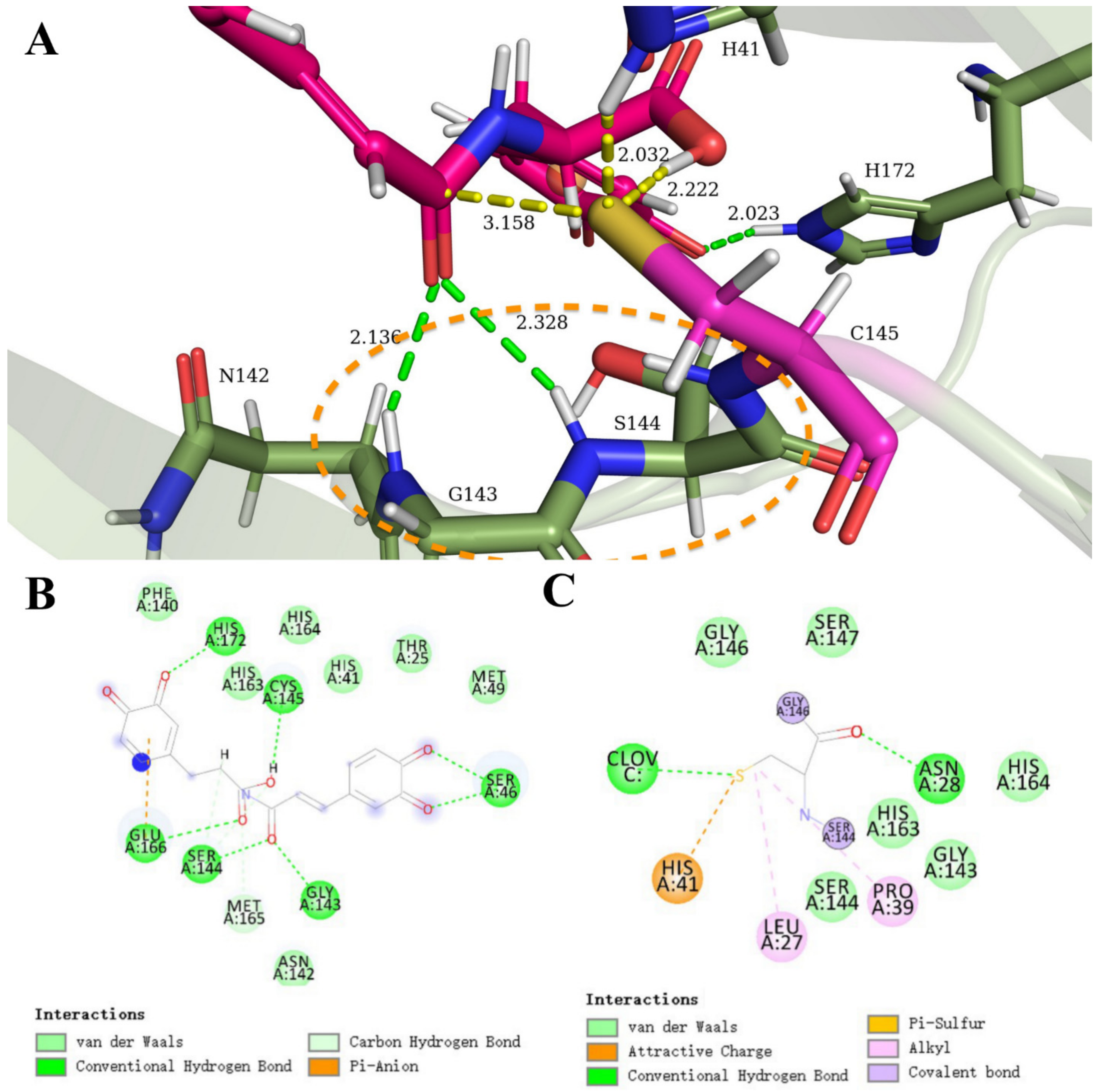
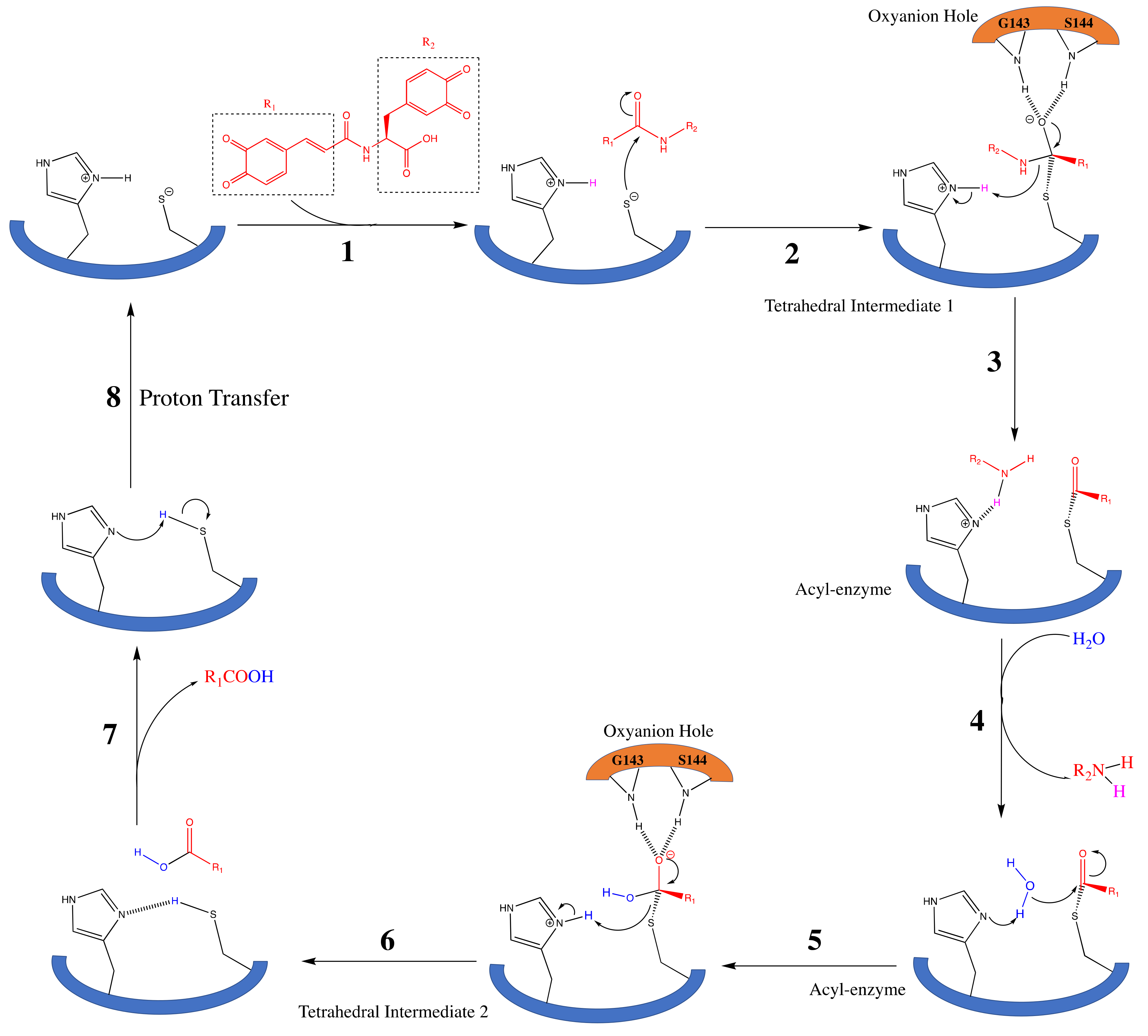
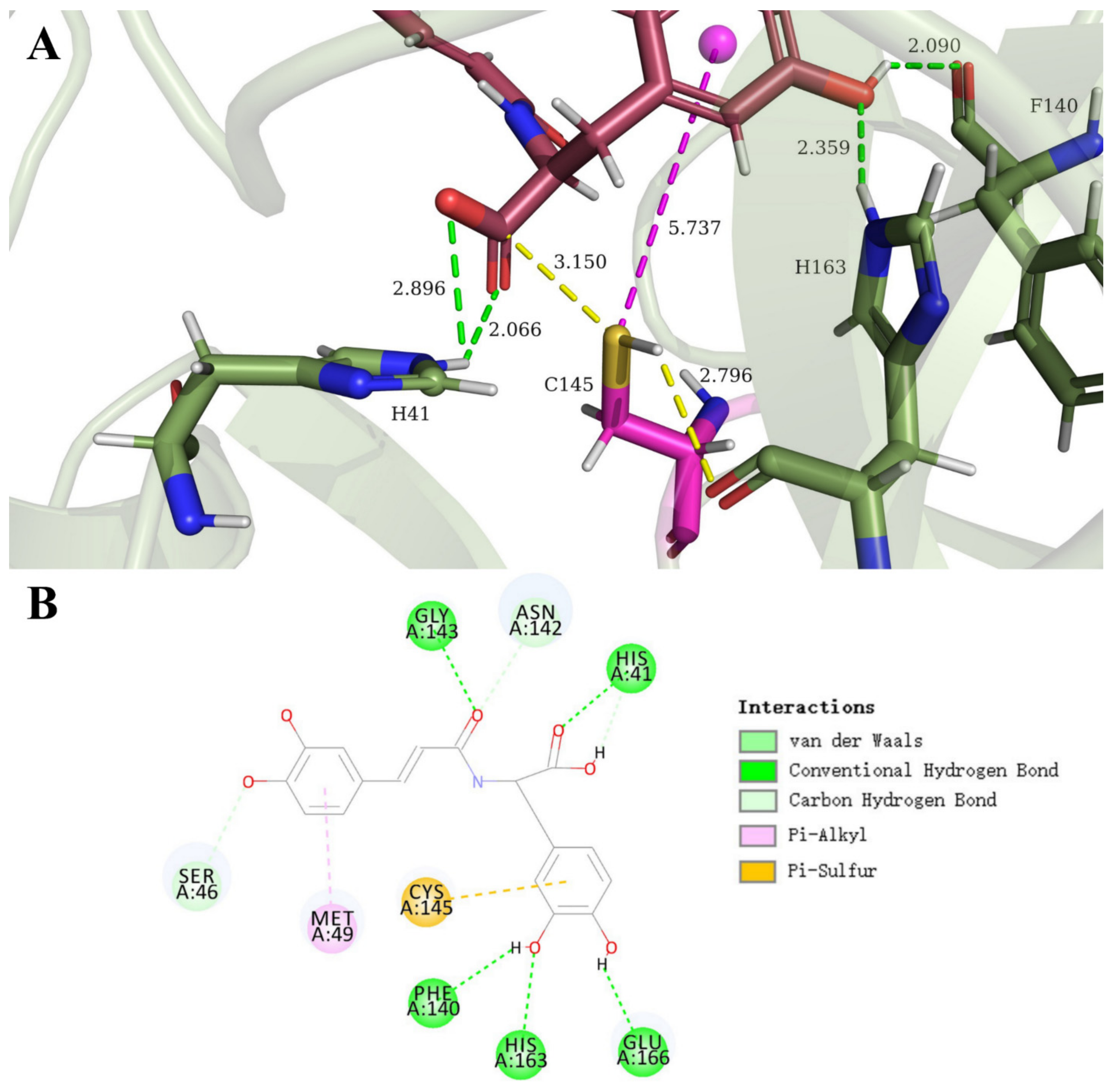
3.1. SARS-CoV-2 Mpro and Clovamide Molecule
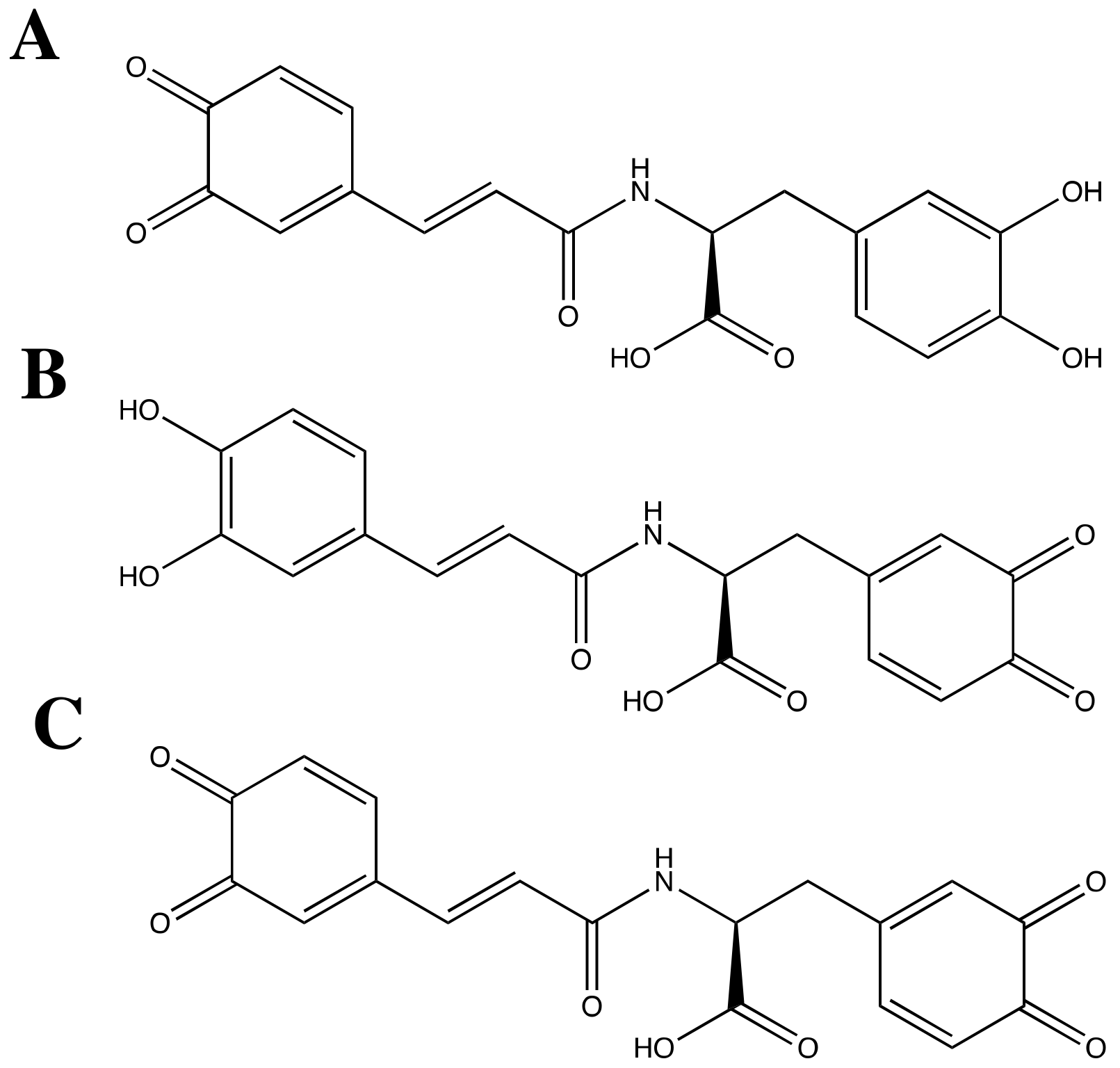
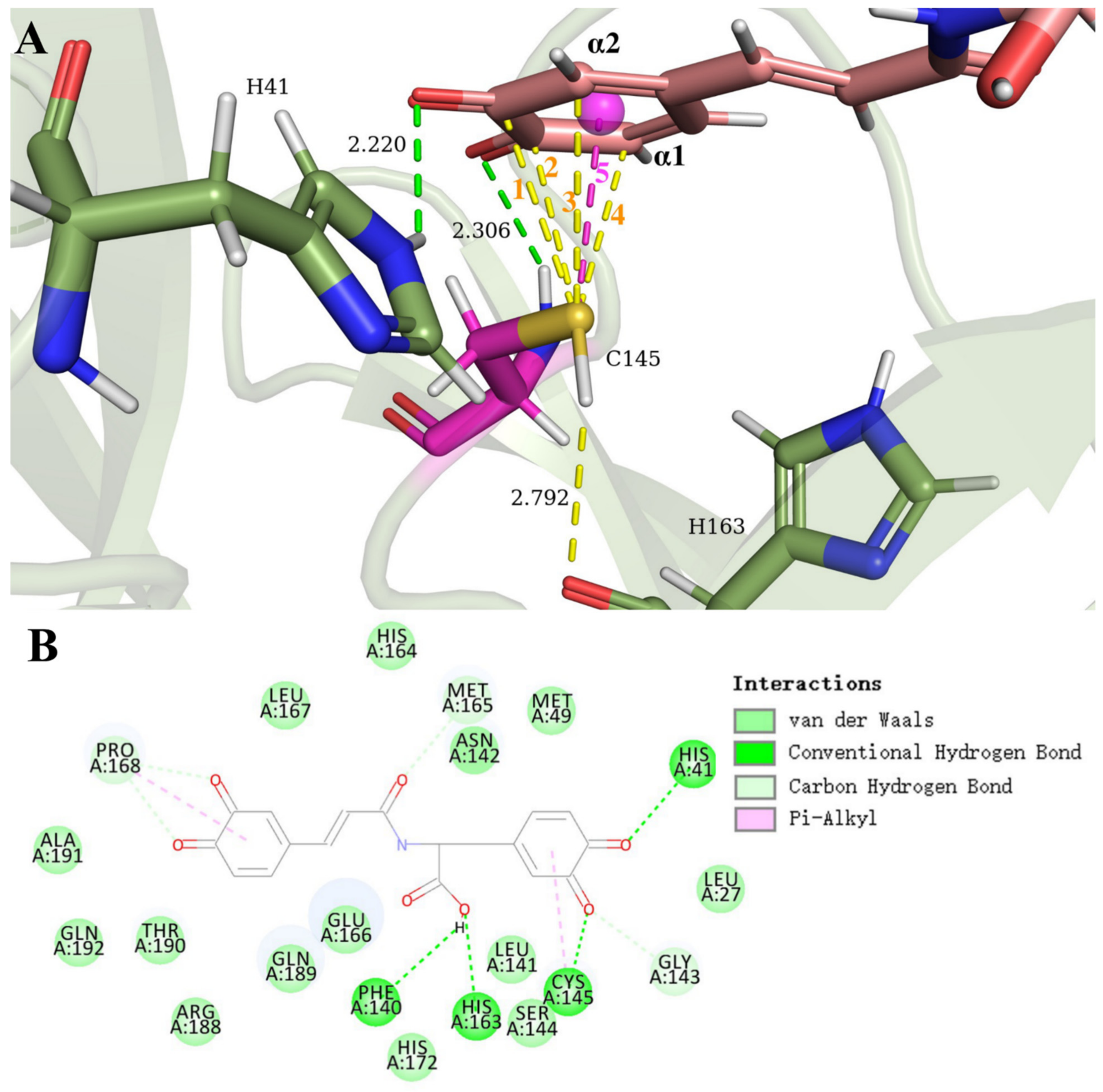
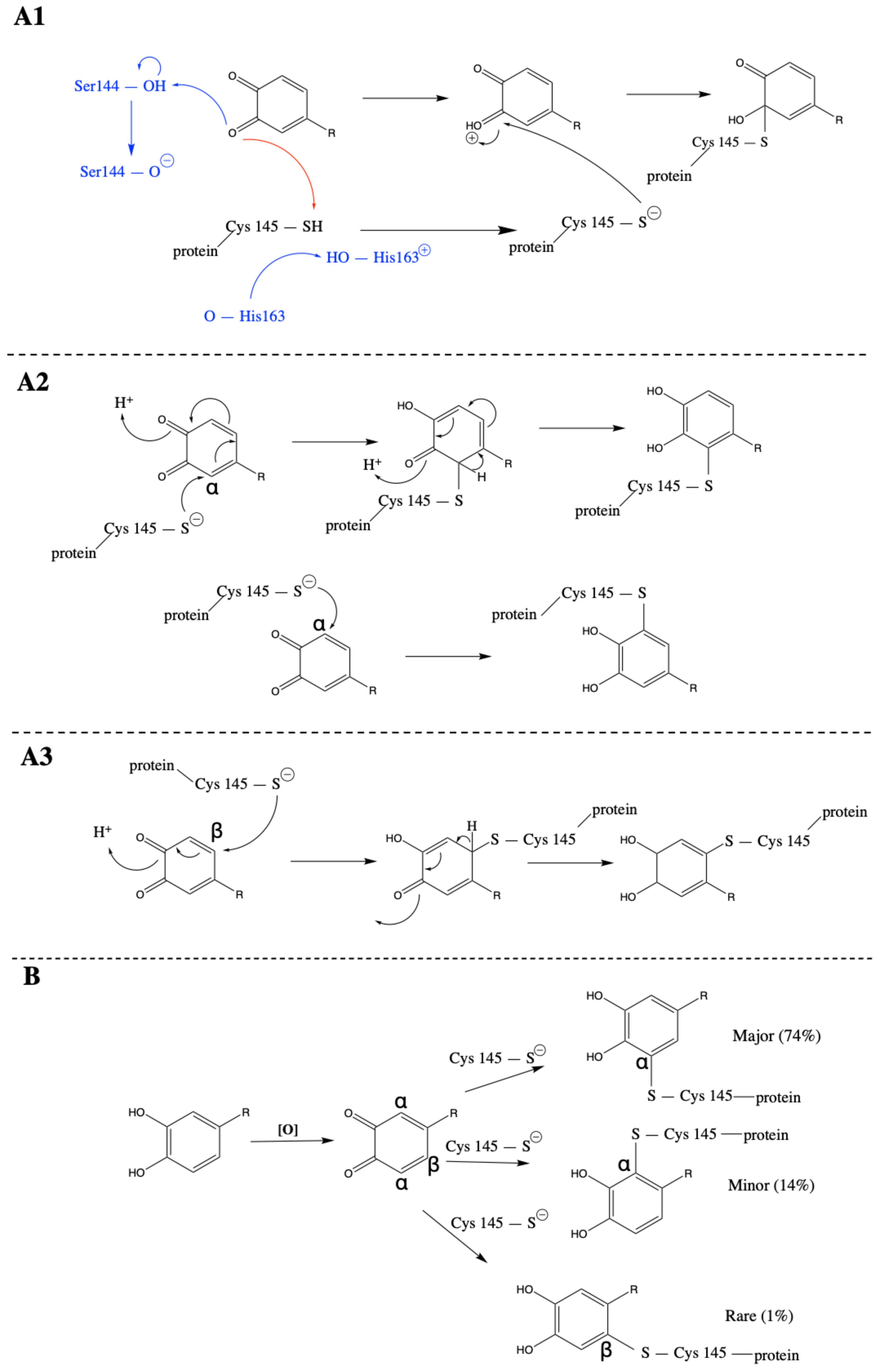
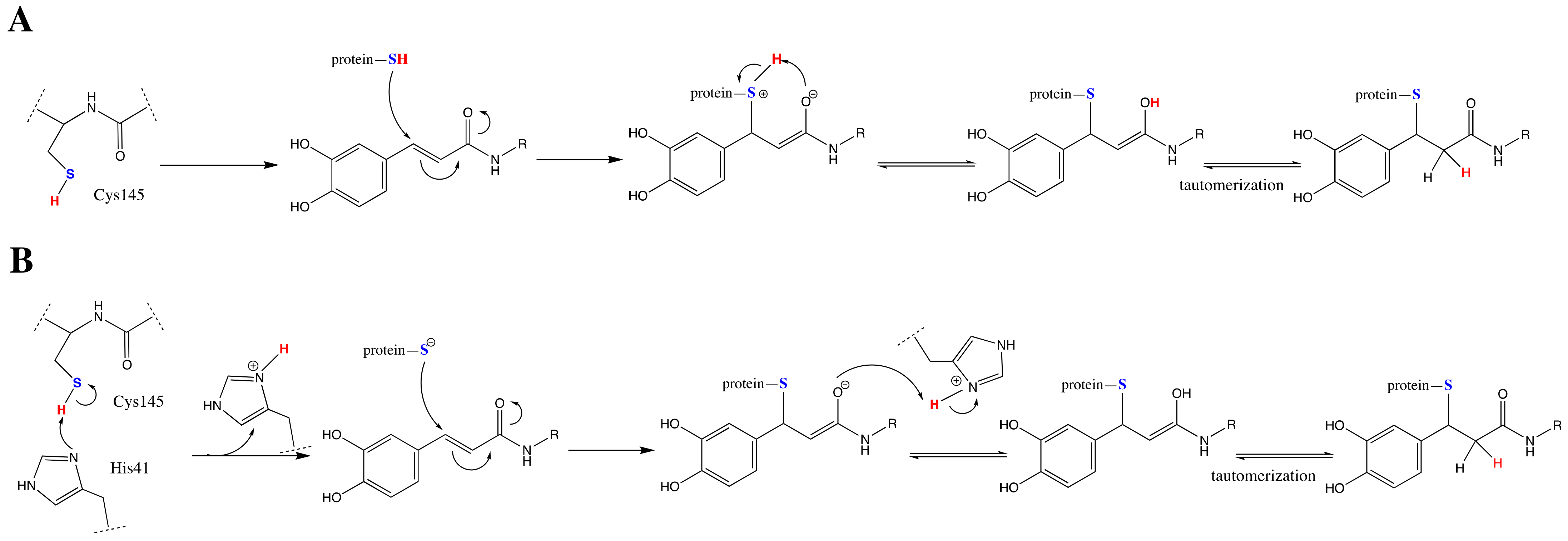
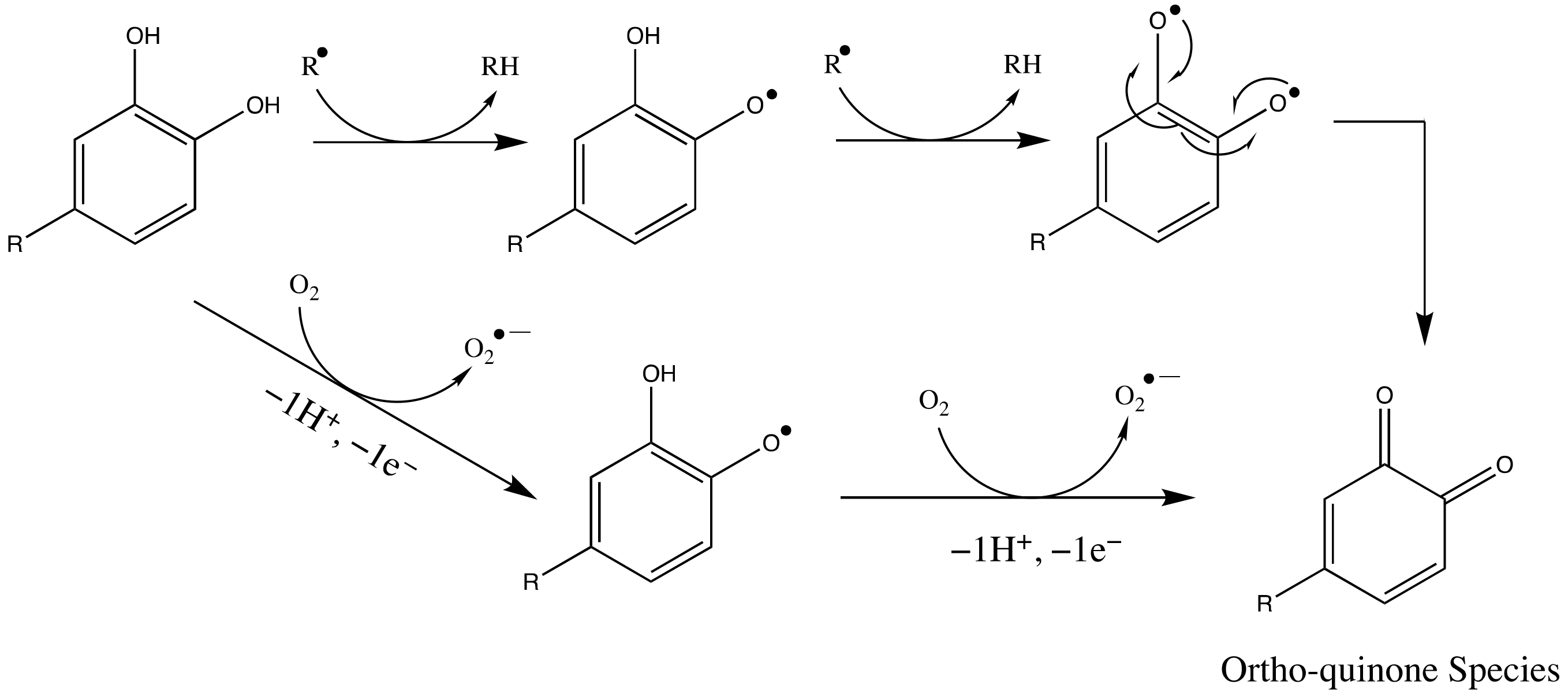
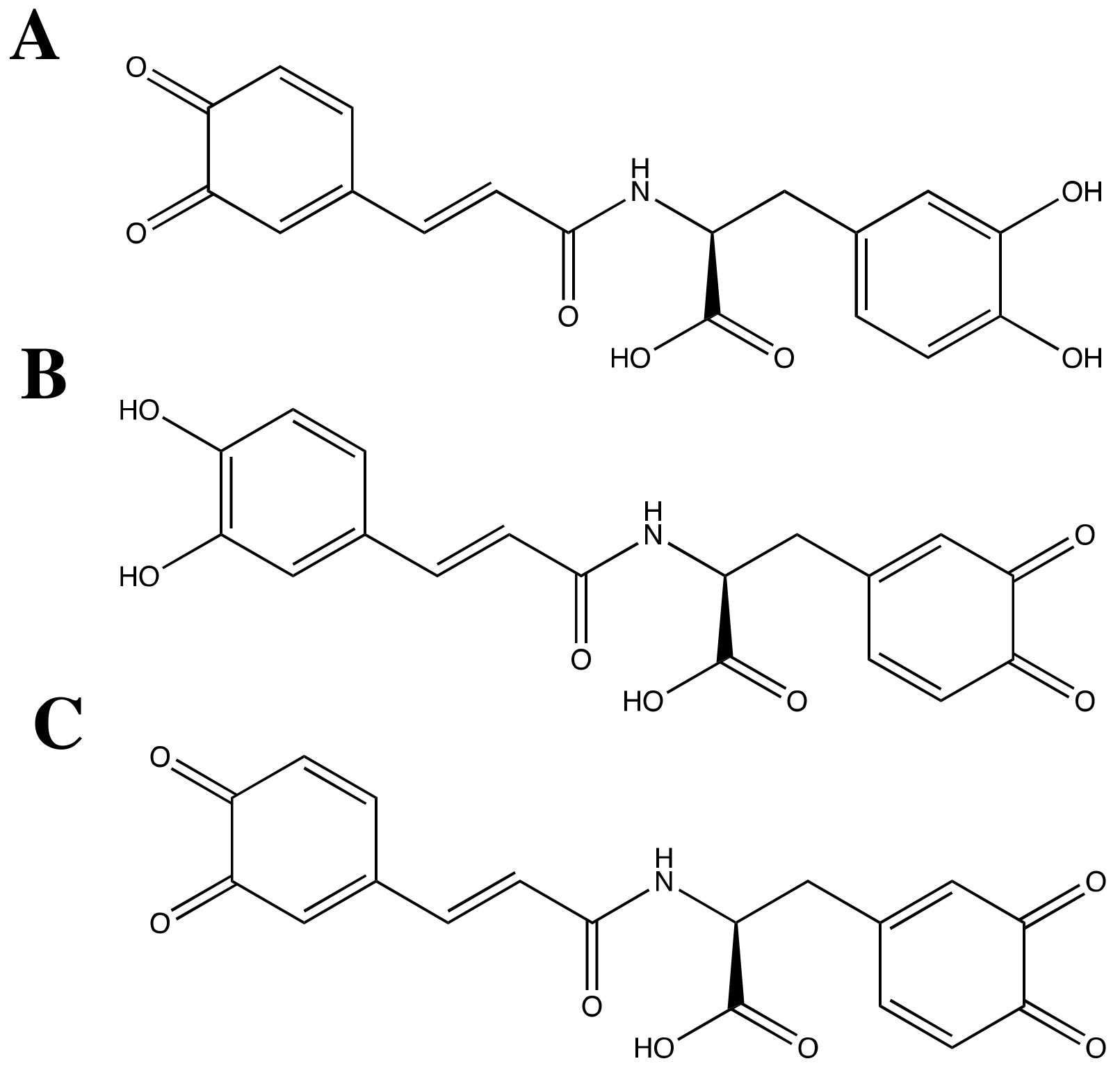
3.2. SARS-CoV-2 Mpro and Ortho-Quinone Derivatives of Clovamide
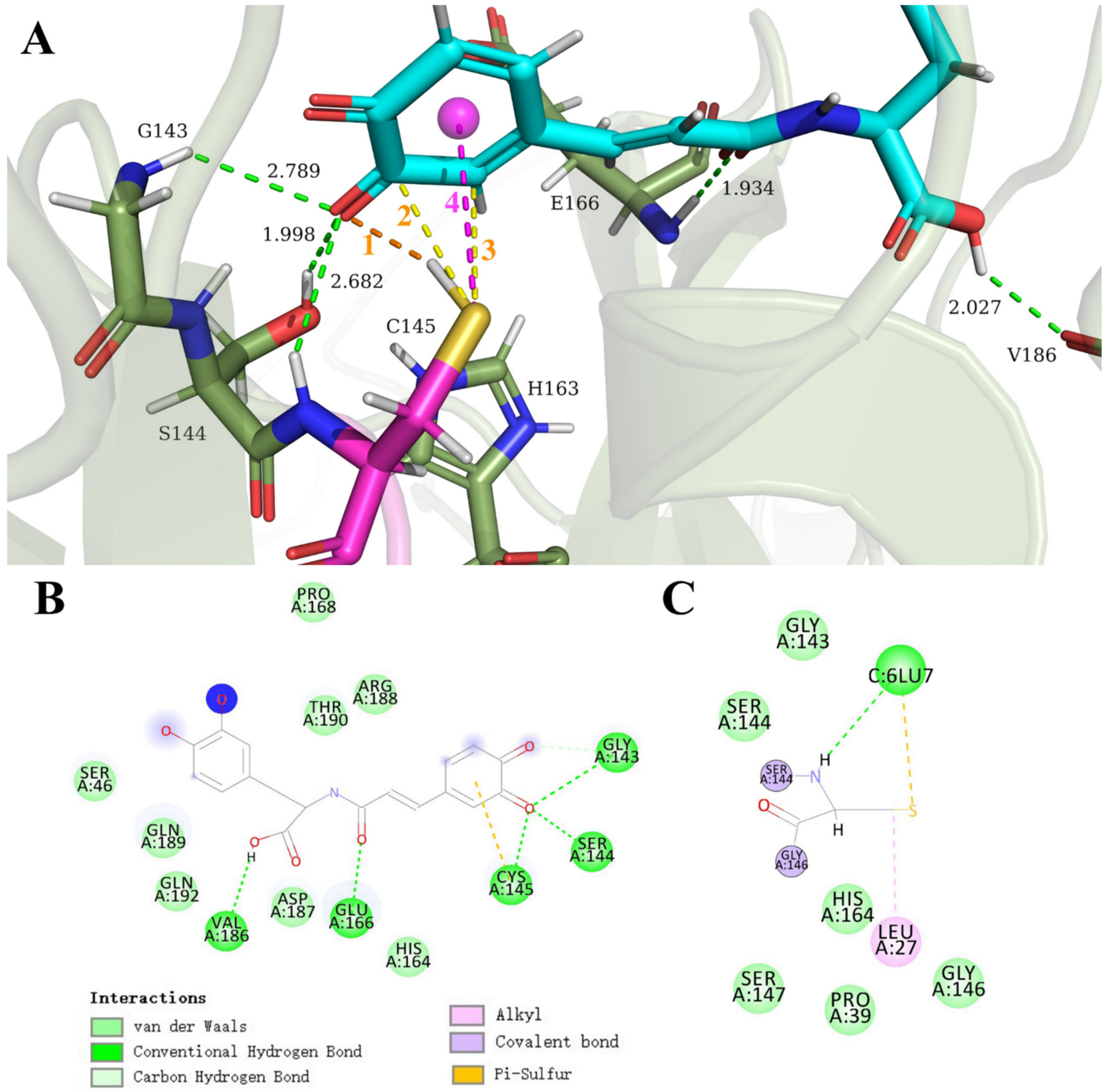
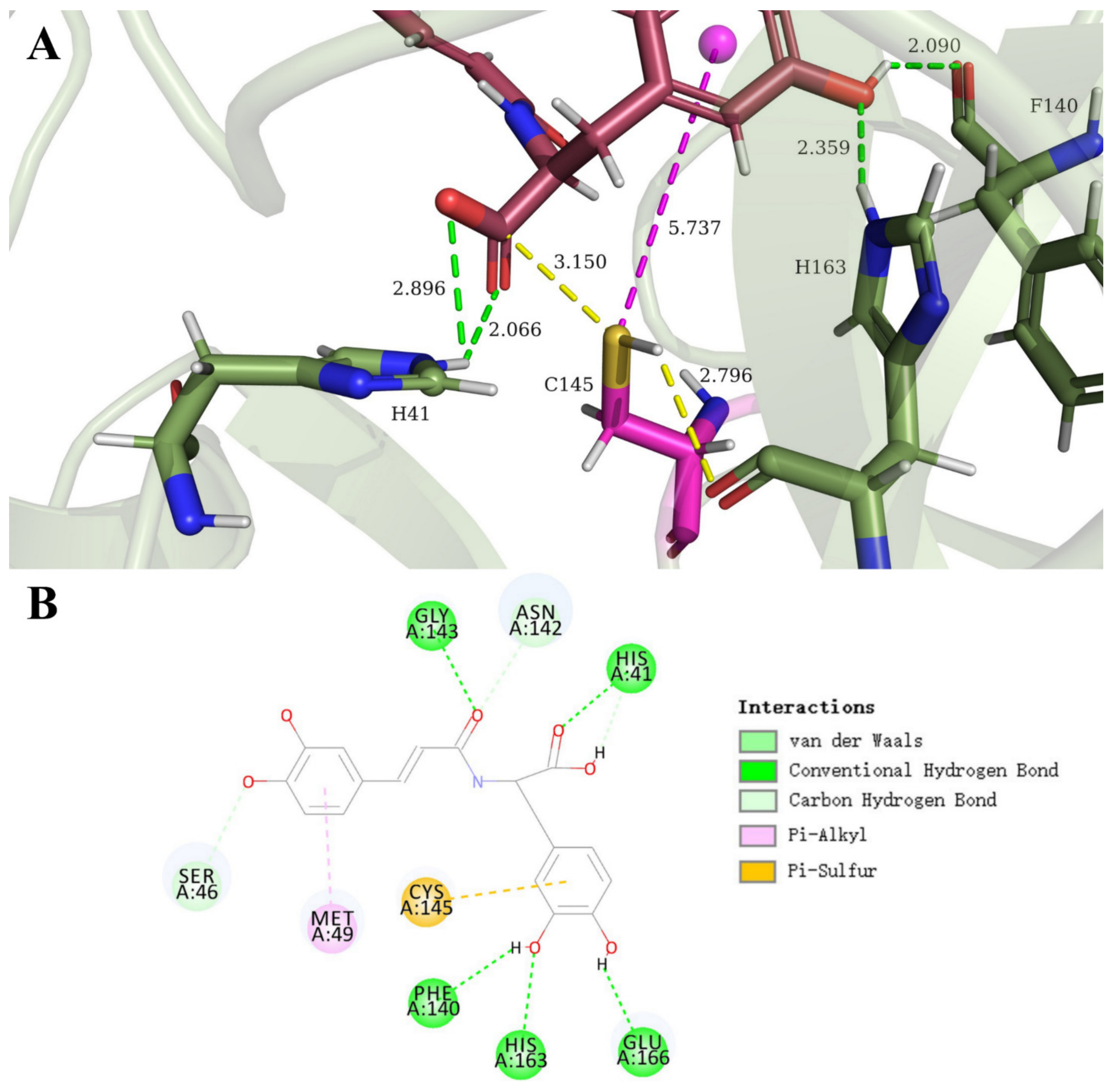
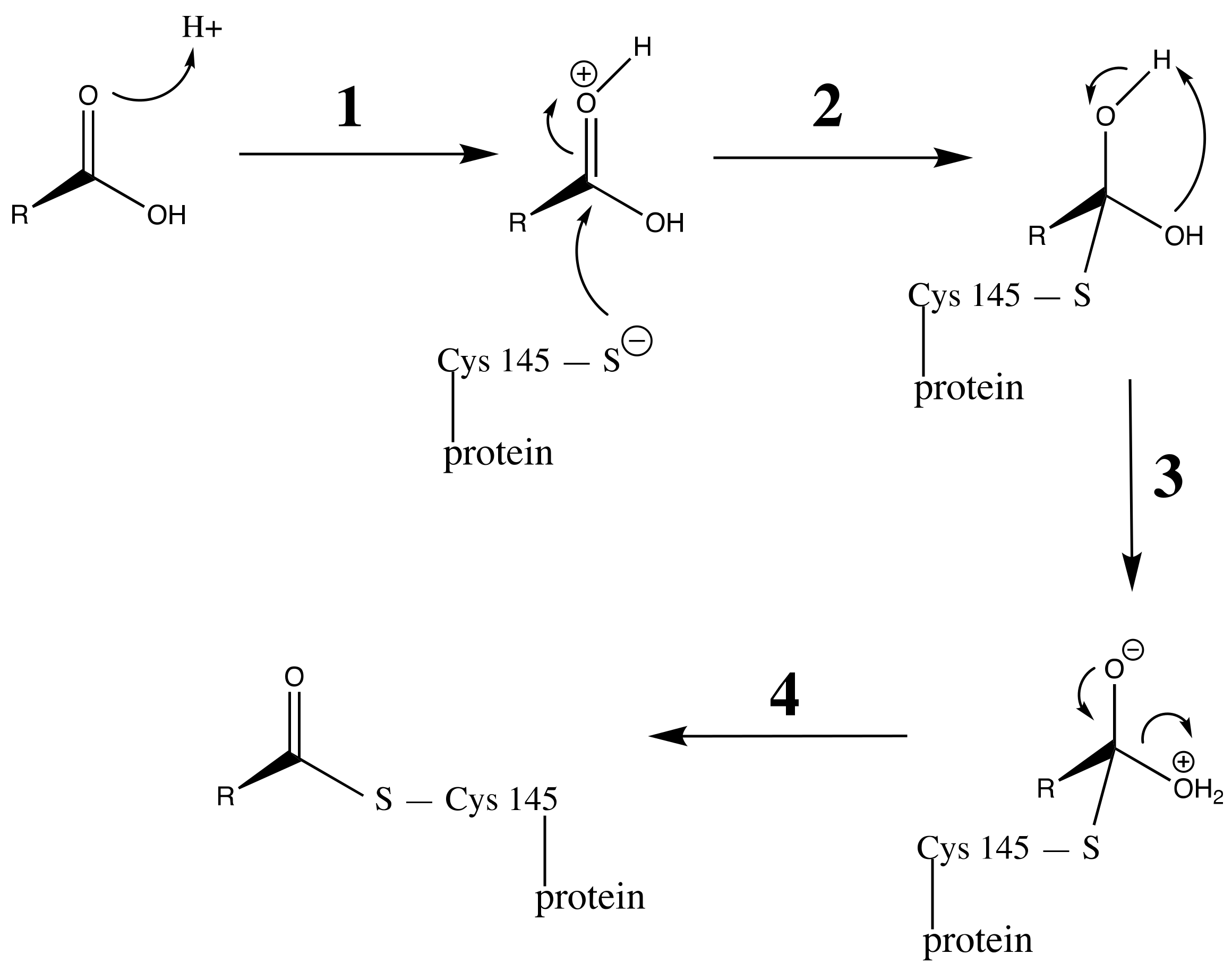
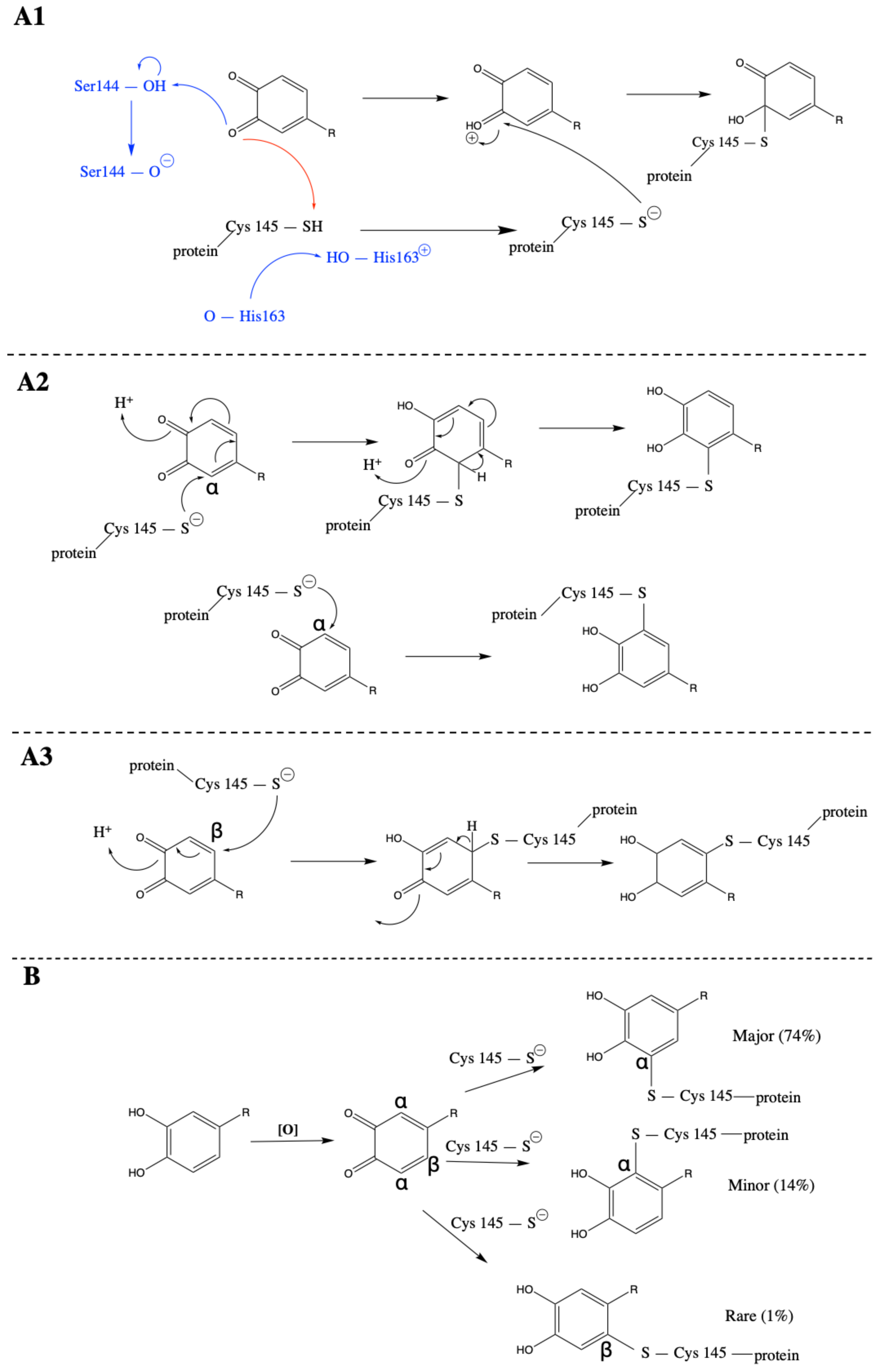
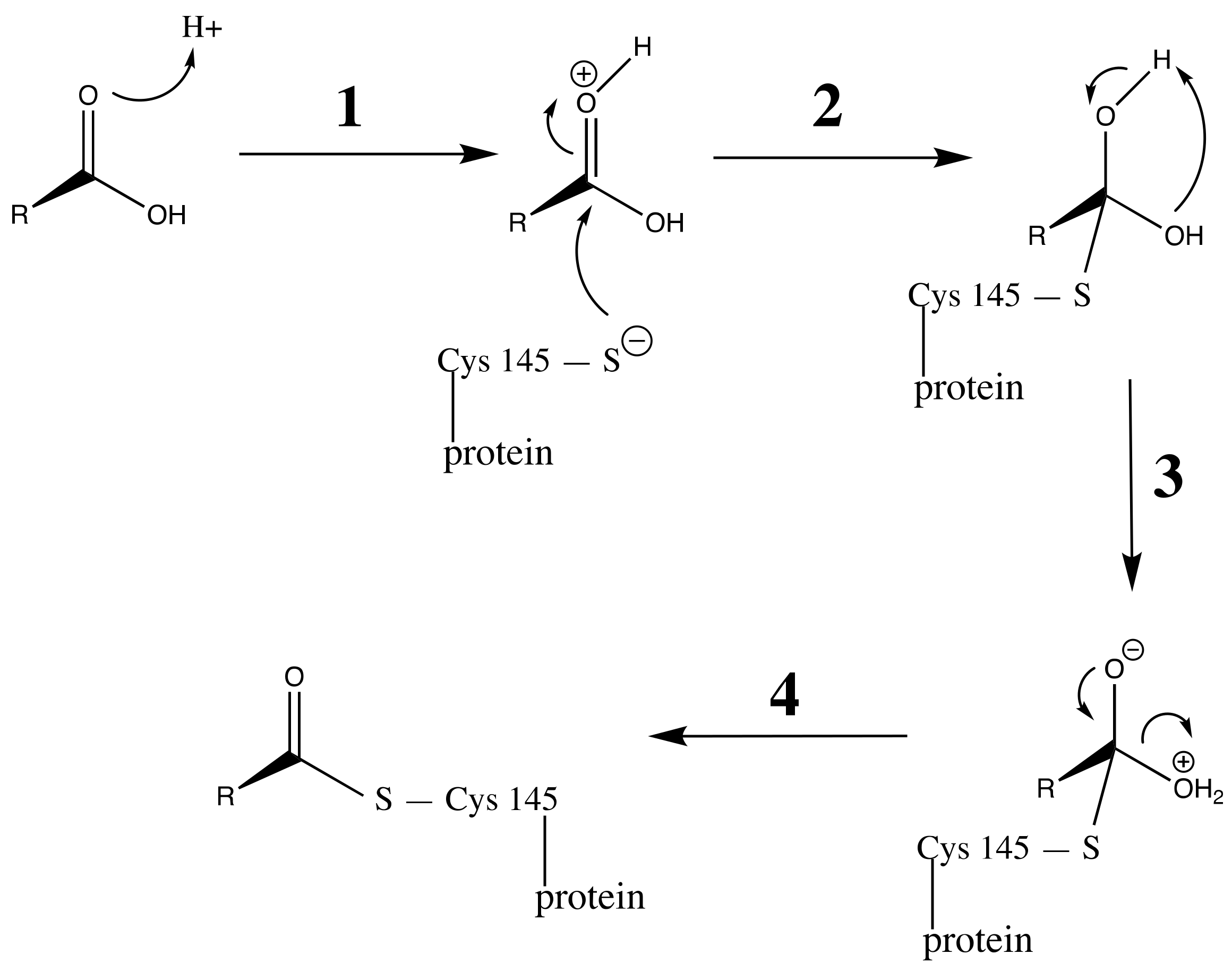
3.3. Carboxylic Acid Is the Key Substrate Functional Group for C145 Thiolate Attacks
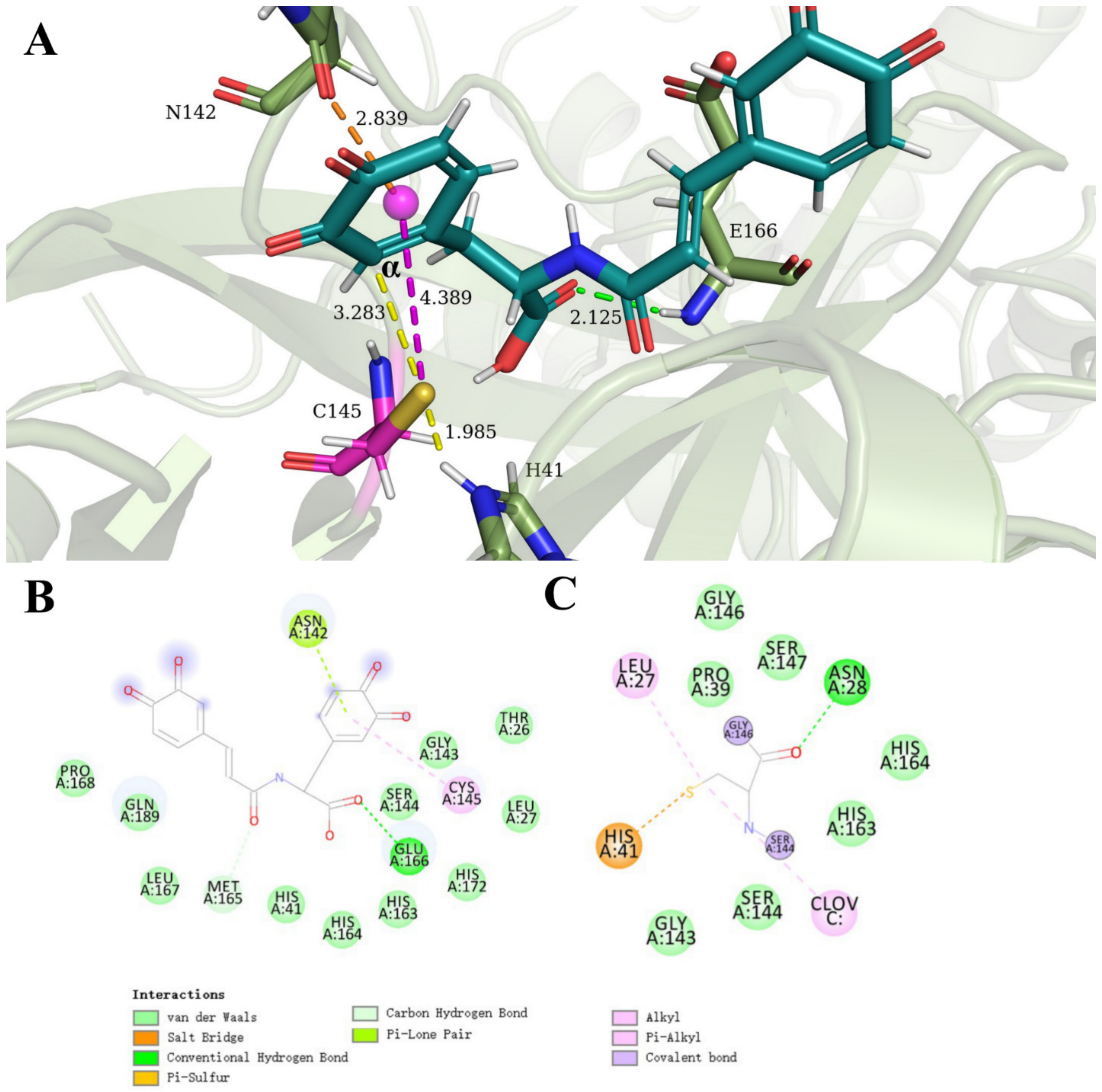
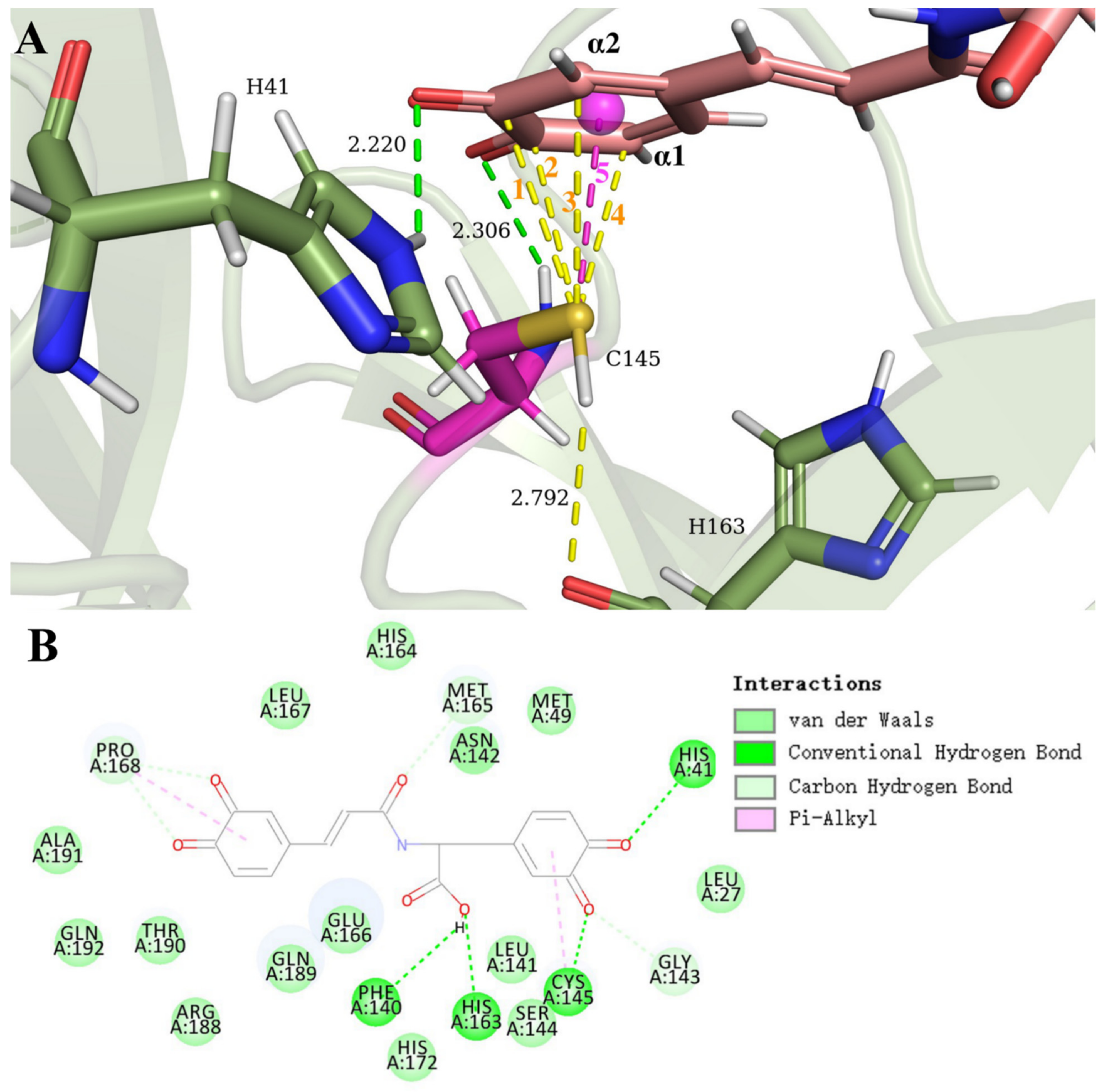
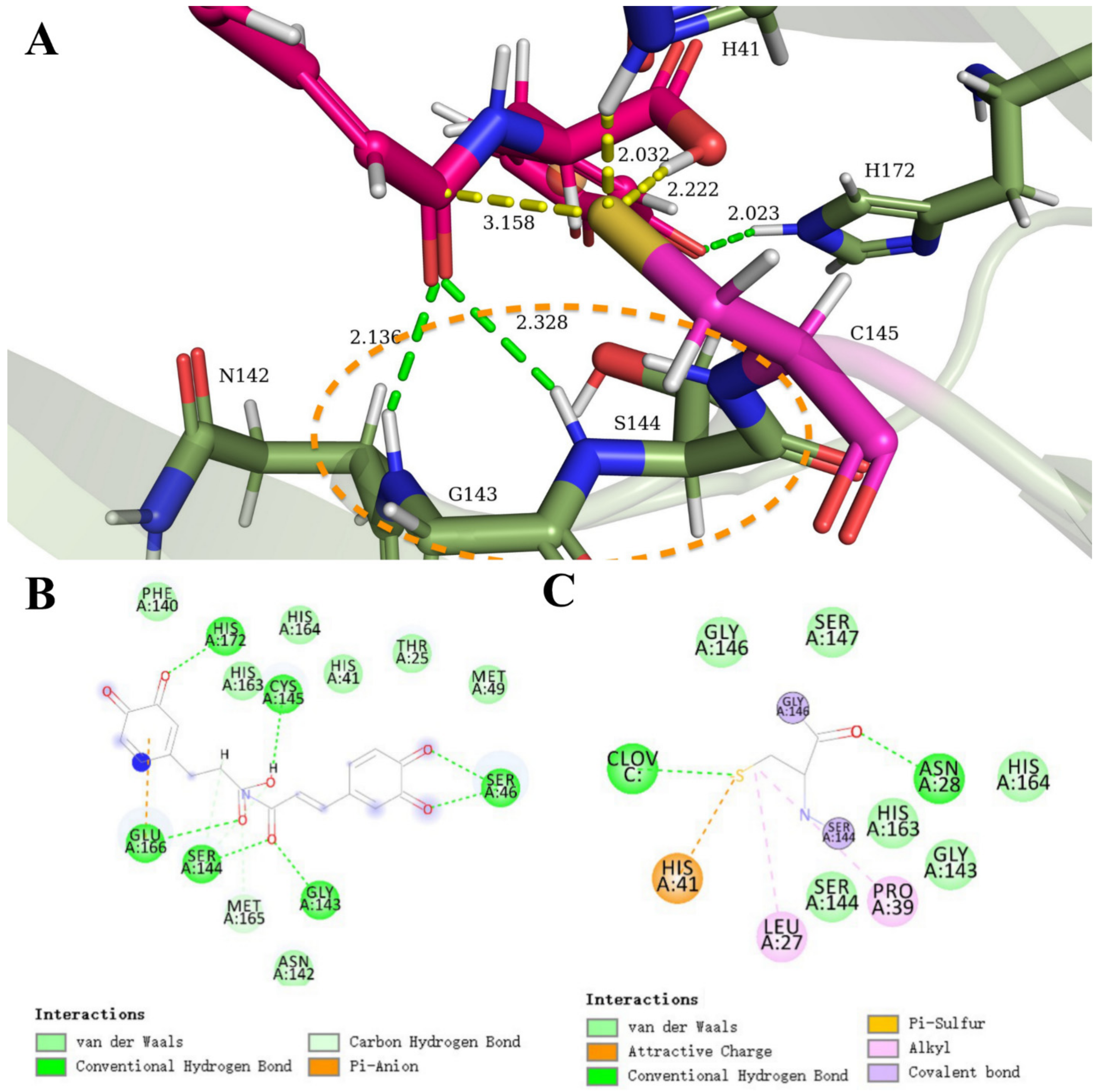
3.4. Clovamide and Analogs in the Zwitterionic Cys–His Mpro Catalytic Site
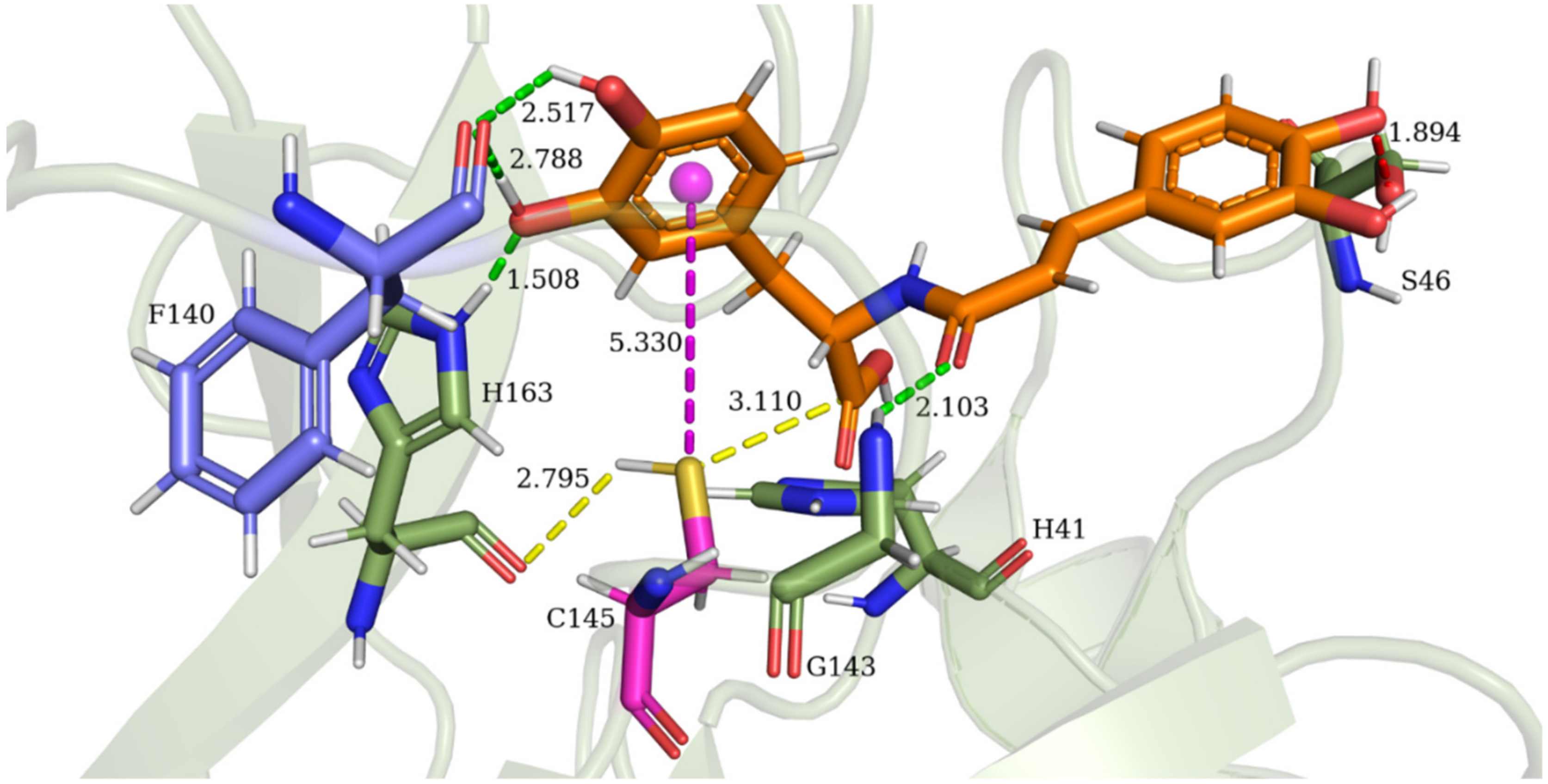
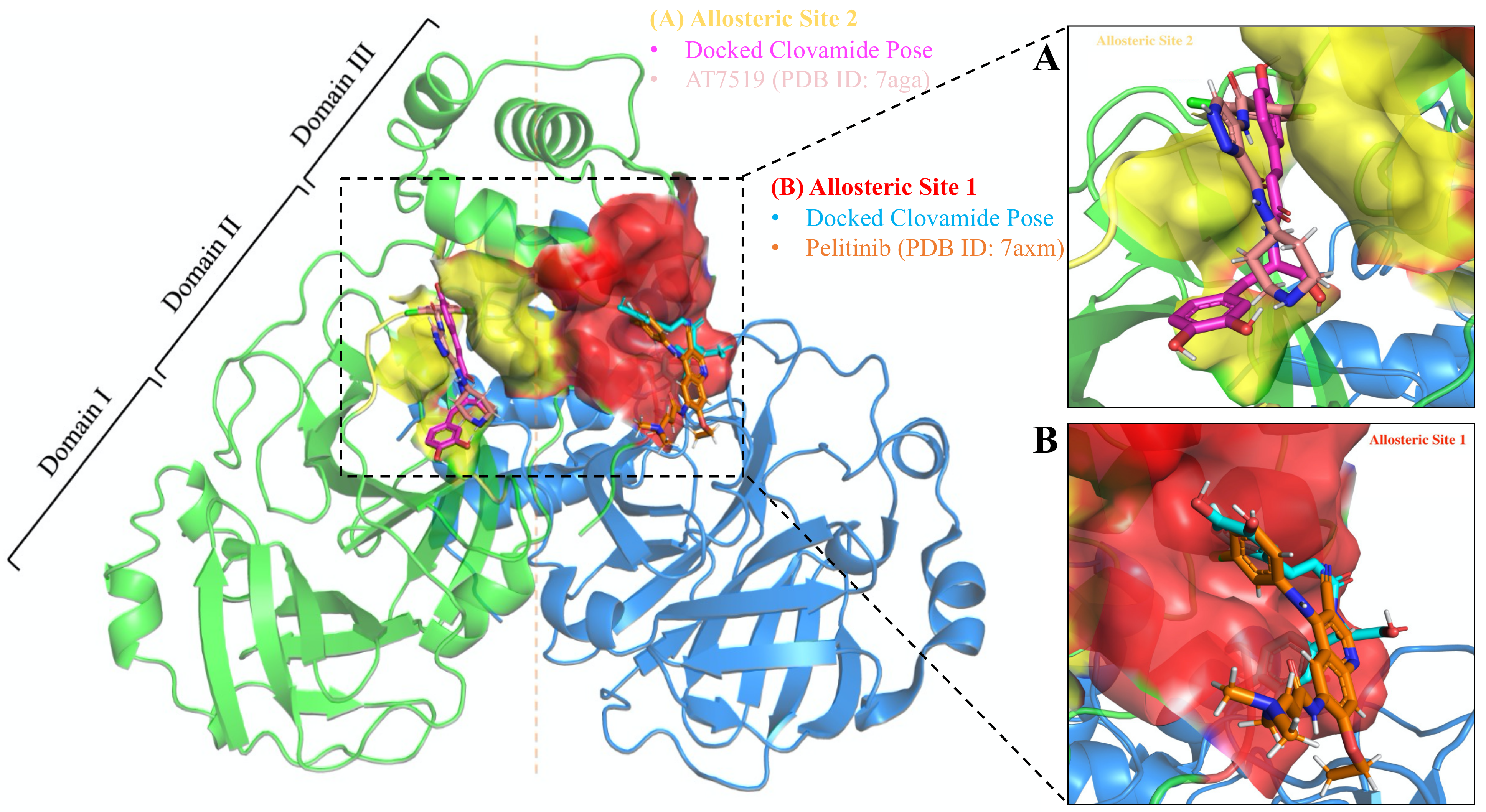
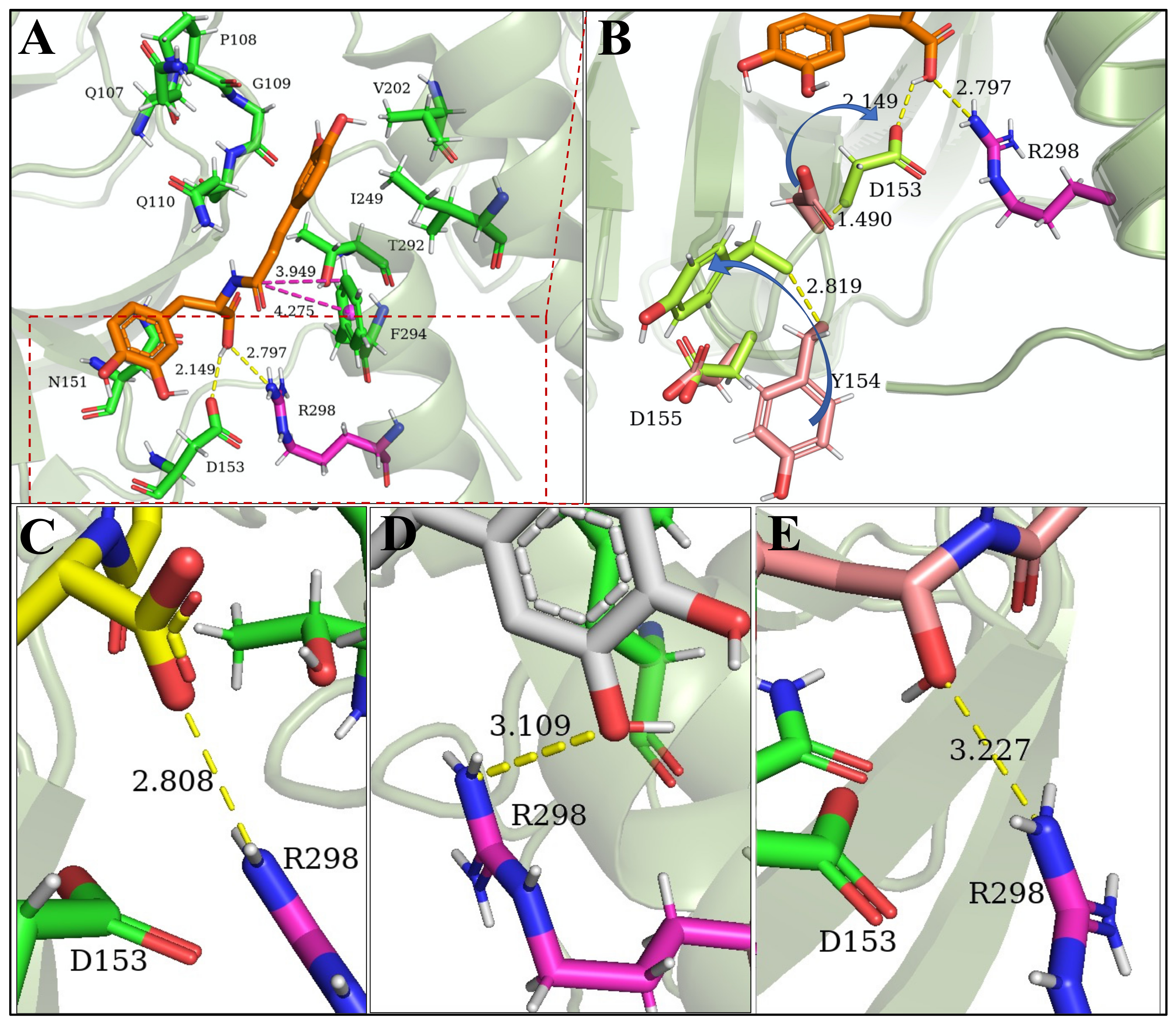
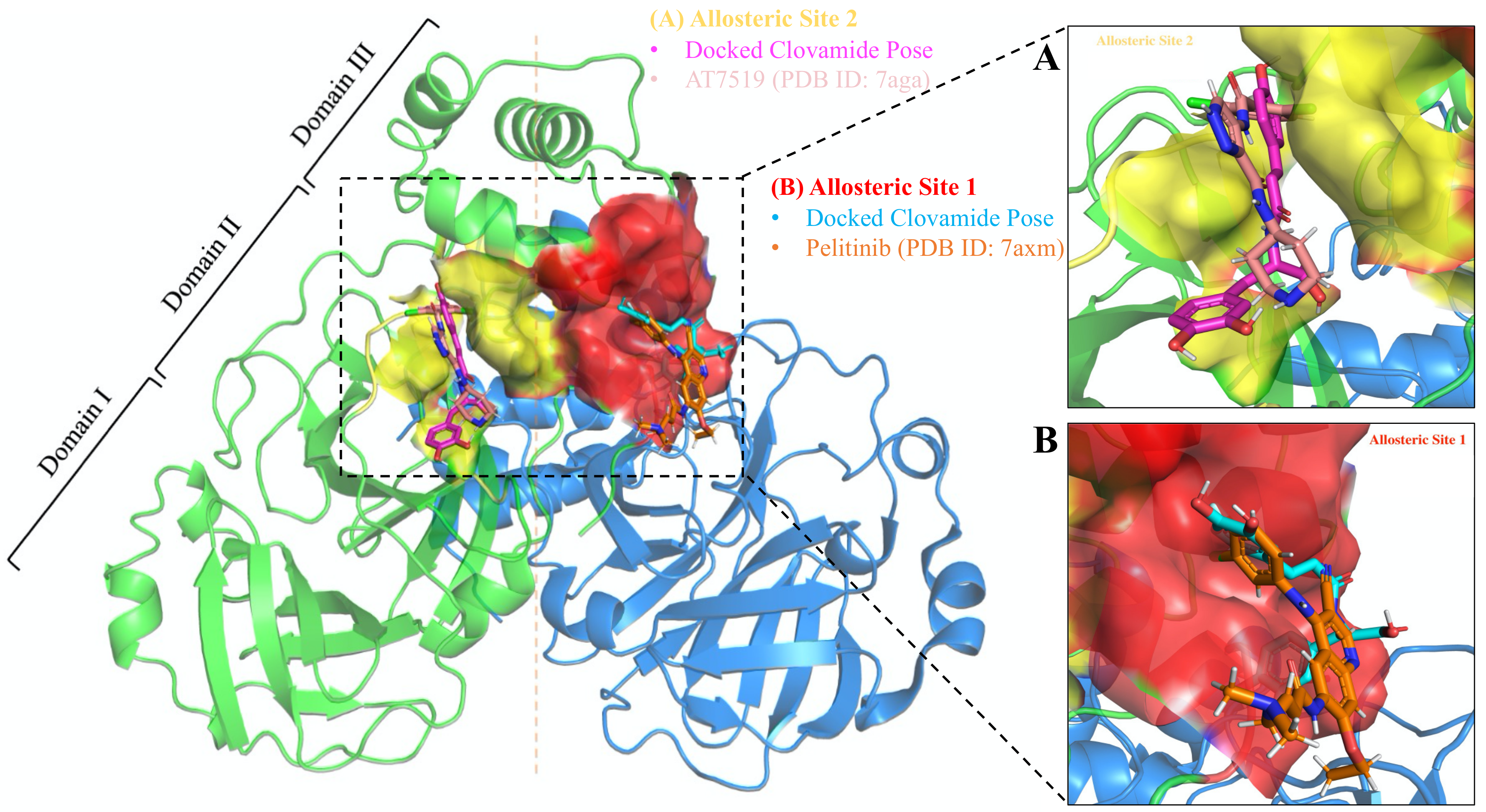
3.5. Allosteric Inhibitions: Clovamide Acts as SARS-CoV-2 Mpro Non-Competitive/Mixed-Type Inhibitor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Goyal, B.; Goyal, D. Targeting the dimerization of the main protease of coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: State of the art and future opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lubin, J.H.; Zardecki, C.; Dolan, E.M.; Lu, C.; Shen, Z.; Dutta, S.; Westbrook, J.D.; Hudson, B.P.; Goodsell, D.S.; Williams, J.K.; et al. Evolution of the SARS-CoV-2 proteome in three dimensions (3D) during the first six months of the COVID-19 pandemic. bioRxiv 2020, in press. [Google Scholar]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Notka, F.; Meier, G.; Wagner, R. Concerted inhibitory activities of Phyllanthus amarus on HIV replication in vitro and ex vivo. Antivir. Res. 2004, 64, 93–102. [Google Scholar] [CrossRef]
- Rehman, S.; Ashfaq, U.A.; Riaz, S.; Javed, T.; Riazuddin, S. Antiviral activity of Acacia nilotica against hepatitis C virus in liver infected cells. Virol. J. 2011, 8, 220. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 3449–3458. [Google Scholar] [CrossRef]
- Orhan, I.E.; Senol Deniz, F.S. Natural products as potential leads against coronaviruses: Could they be encouraging structural models against SARS-CoV-2? Nat. Prod. Bioprospect. 2020, 10, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Twilley, D.; Esmear, T.; Oosthuizen, C.B.; Reid, A.-M.; Nel, M.; Lall, N. Anti-SARS-CoV natural products with the potential to inhibit SARS-CoV-2 (COVID-19). Front. Pharmacol. 2020, 11, 1663–9812. [Google Scholar] [CrossRef]
- Annunziata, G.; Zamparelli, M.S.; Santoro, C.; Ciampaglia, R.; Stornaiuolo, M.; Tenore, G.C.; Sanduzzi, A.; Novellino, E. May polyphenols have a role against coronavirus infection? An overview of in vitro evidence. Front. Med. 2020, 7, 240. [Google Scholar] [CrossRef] [PubMed]
- El-Mordy, F.M.A.; El-Hamouly, M.M.; Ibrahim, M.T.; El-Rheem, G.A.; Aly, O.M.; El-Kader, A.M.A.; Youssif, K.A.; Abdelmohsen, U.R. Inhibition of SARS-CoV-2 main protease by phenolic compounds from Manilkara hexandra (Roxb.) dubard assisted by metabolite profiling and in silico virtual screening. RSC Adv. 2020, 10, 32148–32155. [Google Scholar] [CrossRef]
- Mishara, A.; Pathak, Y.; Tripathi, V. Natural compounds as potential inhibitors of novel coronavirus (COVID-19) main protease: An in silico study. Res. Square 2020, in press. [Google Scholar]
- Narkhede, R.R.; Pise, A.V.; Cheke, R.S.; Shinde, S.D. Recognition of natural products as potential inhibitors of COVID-19 main protease (Mpro): In-silico evidences. Nat. Prod. Bioprospect. 2020, 10, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, I.L.; Revel, J.S.; Stevens, J.F. Potential use of polyphenols in the battle against COVID-19. Curr. Opin. Food Sci. 2020, 32, 149–155. [Google Scholar] [CrossRef]
- Zhu, Y.; Xie, D.-Y. Docking characterization and in vitro inhibitory activity of flavan-3-ols and dimeric proanthocyanidins against the main protease activity of SARS-Cov-2. Front. Plant Sci. 2020, 11, 601316. [Google Scholar] [CrossRef]
- Yoshihara, T.; Yoshikawa, H.; Sakamura, S.; Sakuma, T. Clovamides; l-dopa conjugated with trans- and cis-caffeic acids in red clover (Trifolium pratense). Agric. Biol. Chem. 1974, 38, 1107–1109. [Google Scholar] [CrossRef]
- Locatelli, M.; Travaglia, F.; Giovannelli, L.; Coisson, J.D.; Bordiga, M.; Pattarino, F.; Arlorio, M. Clovamide and phenolics from cocoa beans (Theobroma cacao L.) inhibit lipid peroxidation in liposomal systems. Food Res. Int. 2013, 50, 129–134. [Google Scholar] [CrossRef]
- Dong, J.; Cui, Y.; Li, S.; Le, W. Current pharmaceutical treatments and alternative therapies of Parkinson’s disease. Curr. Neuropharmacol. 2016, 14, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Belli, S.; Caruso, F.; Roy, G.; Rossi, M. Antioxidant studies by hydrodynamic voltammetry and DFT, quantitative analyses by HPLC-DAD of clovamide, a natural phenolic compound found in Theobroma Cacao L. beans. Food Chem. 2021, 341, 128260. [Google Scholar] [CrossRef]
- Sullivan, M.L.; Zeller, W.E. Efficacy of various naturally occurring caffeic acid derivatives in preventing post-harvest protein losses in forages. J. Sci. Food Agric. 2013, 93, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.E.; Madureira, D.; Capani, F.; Aon-Bertolino, L.; Saraceno, E.; Alvarez-Giraldez, L.D. The role of catechols and free radicals in benzene toxicity: An oxidative DNA damage pathway. Environ. Mol. Mutagen. 2009, 50, 771–780. [Google Scholar] [CrossRef]
- Montine, T.J.; Picklo, M.J.; Amarnath, V.; Whetsell, W.O.; Graham, D.G. Neurotoxicity of endogenous cysteinylcatechols. Exp. Neurol. 1997, 148, 26–33. [Google Scholar] [CrossRef]
- Knollenberg, B.J.; Li, G.-X.; Lambert, J.D.; Maximova, S.N.; Guiltinan, M.J. Clovamide, a hydroxycinnamic acid amide, is a resistance factor against Phytophthora spp. in Theobroma cacao. Front. Plant Sci. 2020, 11, 617520. [Google Scholar] [CrossRef] [PubMed]
- Yamane, H.; Konno, K.; Sabelis, M.; Takabayashi, J.; Sassa, T.; Oikawa, H. Chemical defence and toxins of plants. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 339–385. [Google Scholar]
- Bittner, S. When quinones meet amino acids: Chemical, physical and biological consequences. Amino Acids 2006, 30, 205–224. [Google Scholar] [CrossRef]
- Ito, S.; Sugumaran, M.; Wakamatsu, K. Chemical reactivities of ortho-quinones produced in living organisms: Fate of quinonoid products formed by tyrosinase and phenoloxidase action on phenols and catechols. Int. J. Mol. Sci. 2020, 21, 6080. [Google Scholar] [CrossRef] [PubMed]
- Shitrit, A.; Zaidman, D.; Kalid, O.; Bloch, I.; Doron, D.; Yarnizky, T.; Buch, I.; Segev, I.; Ben-Zeev, E.; Segev, E.; et al. Conserved interactions required for inhibition of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Sci. Rep. 2020, 10, 20808. [Google Scholar] [CrossRef]
- El-Sharawy, R.T.; Elkhateeb, A.; Marzouk, M.M.; El-Latif, R.R.A.; Abdelrazig, S.E.; El-Ansari, M.A. Antiviral and antiparasitic activities of clovamide: The major constituent of Dichrostachys cinerea (L.) Wight et Arn. J. Appl. Pharm. Sci. 2017, 7, 219–223. [Google Scholar]
- Mandal, S.; Moudgil, M.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular docking in modern drug discovery: Principles and recent applications. In Drug Descovery and Development: New Advances; Gaitonde, V., Karmakar, P., Trivedi, A., Eds.; IntechOpen: London, UK, 2019. [Google Scholar]
- Ahmadi, S.; Herrera, L.B.; Chehelamirani, M.; Hostas, J.; Jalife, S.; Salahub, D.R. Multiscale modeling of enzymes: QM-cluster, QM/MM, and QM/MM/MD: A tutorial review. Int. J. Quantum Chem. 2018, 118, e25558. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Riley, K.E.; Vondrášek, J.; Hobza, P. Performance of the DFT-D method, paired with the PCM implicit solvation model, for the computation of interaction energies of solvated complexes of biological interest. Phys. Chem. Chem. Phys. 2007, 9, 5555–5560. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M.; Himo, F. The quantum chemical cluster approach for modeling enzyme reactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 323–356. [Google Scholar] [CrossRef]
- Bigler, D.J.; Peterson, L.W.; Cafiero, M. Effects of implicit solvent and relaxed amino acid side chains on the MP2 and DFT calculations of ligand–protein structure and electronic interaction energies of dopaminergic ligands in the SULT1A3 enzyme active site. Comput. Theor. Chem. 2015, 1051, 79–92. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Nikolantonaki, M.; Jourdes, M.; Shinoda, K.; Teissedre, P.-L.; Quideau, S.; Darriet, P. Identification of adducts between an odoriferous volatile thiol and oxidized grape phenolic compounds: Kinetic study of adduct formation under chemical and enzymatic oxidation conditions. J. Agric. Food Chem. 2012, 60, 2647–2656. [Google Scholar] [CrossRef]
- Fujimoto, A.; Masuda, T. Chemical interaction between polyphenols and a cysteinyl thiol under radical oxidation conditions. J. Agric. Food Chem. 2012, 60, 5142–5151. [Google Scholar] [CrossRef]
- Watson, R.R.; Preedy, V.R.; Zibadi, S. Polyphenols: Mechanisms of Action in Human Health and Disease; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Caruso, F.; Rossi, M.; Pedersen, J.Z.; Incerpi, S. Computational studies reveal mechanism by which quinone derivatives can inhibit SARS-CoV-2. Study of embelin and two therapeutic compounds of interest, methyl prednisolone and dexamethasone. J. Infect. Public Health 2020, 13, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Caruso, F.; Singh, M.; Belli, S.; Berinato, M.; Rossi, M. Interrelated mechanism by which the methide quinone celastrol, obtained from the roots of tripterygium wilfordii, inhibits main protease 3CLpro of COVID-19 and acts as superoxide radical scavenger. Int. J. Mol. Sci. 2020, 21, 9266. [Google Scholar] [CrossRef] [PubMed]
- Strauss, E. Coenzyme A biosynthesis and enzymology. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 351–410. [Google Scholar]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Tan, K.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Room-temperature X-ray crystallography reveals the oxidation and reactivity of cysteine residues in SARS-CoV-2 3CL Mpro: Insights into enzyme mechanism and drug design. IUCrJ 2020, 7, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Pant, S.; Zhang, Q.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020, 295, 17365–17373. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Phyical Data, 72th ed.; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Schirmeister, T. (S)-Thiirancarboxylic acid as a reactive building block for a new class of cysteine protease inhibitors. Bioorganic Med. Chem. Lett. 2000, 10, 2647–2651. [Google Scholar] [CrossRef]
- Paasche, A.; Zipper, A.; Schäfer, S.; Ziebuhr, J.; Schirmeister, T.; Engels, B. Evidence for substrate binding-induced zwitterion formation in the catalytic cys-his dyad of the SARS-CoV main protease. Biochemistry 2014, 53, 5930–5946. [Google Scholar] [CrossRef]
- Novotný, J.; Bazzi, S.; Marek, R.; Kozelka, J. Lone-pair-π interactions: Analysis of the physical origin and biological implications. Phys. Chem. Chem. Phys. 2016, 18, 19472–19481. [Google Scholar] [CrossRef] [Green Version]
- Mooibroek, T.J.; Gamez, P.; Reedijk, J. Lone pair-π interactions: A new supramolecular bond? CrystEngComm 2008, 10, 1501–1515. [Google Scholar] [CrossRef]
- Blow, D.M. Structure and mechanism of chymotrypsin. Accounts Chem. Res. 1976, 9, 145–152. [Google Scholar] [CrossRef]
- Kozelka, J. Lone pair-π interactions in biological systems: Occurrence, function, and physical origin. Eur. Biophys. J. 2017, 46, 729–737. [Google Scholar] [CrossRef]
- El-Baba, T.J.; Lutomski, C.A.; Kantsadi, A.L.; Malla, T.R.; John, T.; Mikhailov, V.; Bolla, J.R.; Schofield, C.J.; Zitzmann, N.; Vakonakis, I.; et al. Allosteric inhibition of the SARS-CoV-2 main protease: Insights from mass spectrometry based assays. Angew. Chem. Int. Ed. 2020, 59, 23544–23548. [Google Scholar] [CrossRef]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Verschueren, K.H.; Anand, K.; Shen, J.; Yang, M.; Xu, Y.; Rao, Z.; Bigalke, J.; Heisen, B.; Mesters, J.R.; et al. pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: Molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005, 354, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Krone, M.W.; Travis, C.R.; Lee, G.Y.; Eckvahl, H.J.; Houk, K.N.; Waters, M.L. More than π-π-π stacking: Contribution of amide-π and CH-π interactions to crotonyllysine binding by the AF9 YEATS domain. J. Am. Chem. Soc. 2020, 142, 17048–17056. [Google Scholar] [CrossRef]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- WHO. Therapeutics and COVID-19: Living Guideline. 2021. Available online: https://www.who.int/publications-detail-redirect/WHO-2019-nCoV-therapeutics-2021.2 (accessed on 7 August 2021).
- Jorge, A. Hydroxychloroquine in the prevention of COVID-19 mortality. Lancet Rheumatol. 2021, 3, e2–e3. [Google Scholar] [CrossRef]
- Rochwerg, B.; Siemieniuk, R.A.; Agoritsas, T.; Lamontagne, F.; Askie, L.; Lytvyn, L.; Agarwal, A.; Leo, Y.-S.; Macdonald, H.; Zeng, L.; et al. A living WHO guideline on drugs for COVID-19. BMJ 2020, 370, m3379. [Google Scholar] [CrossRef]
- Tummino, T.A.; Rezelj, V.V.; Fischer, B.; Fischer, A.; O’Meara, M.J.; Monel, B.; Vallet, T.; White, K.M.; Zhang, Z.; Alon, A.; et al. Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2. Science 2021, 373, 541–547. [Google Scholar] [CrossRef]
- Feng, J.-H.; Hu, X.-L.; Lv, X.-Y.; Wang, B.; Lin, J.; Zhang, X.-Q.; Ye, W.-C.; Xiong, F.; Wang, H. Synthesis and biological evaluation of clovamide analogues with catechol functionality as potent Parkinson’s disease agents in vitro and in vivo. Bioorganic Med. Chem. Lett. 2019, 29, 302–312. [Google Scholar] [CrossRef]
- Hu, X.-L.; Lin, J.; Lv, X.-Y.; Feng, J.-H.; Zhang, X.-Q.; Wang, H.; Ye, W.-C. Synthesis and biological evaluation of clovamide analogues as potent anti-neuroinflammatory agents in vitro and in vivo. Eur. J. Med. Chem. 2018, 151, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Tipton, K.F. Assessment of enzyme inhibition: A review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules 2017, 22, 1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, N.; Caruso, F.; Rossi, M. Mechanistic Insights into the Inhibition of SARS-CoV-2 Main Protease by Clovamide and Its Derivatives: In Silico Studies. Biophysica 2021, 1, 377-404. https://doi.org/10.3390/biophysica1040028
Ye N, Caruso F, Rossi M. Mechanistic Insights into the Inhibition of SARS-CoV-2 Main Protease by Clovamide and Its Derivatives: In Silico Studies. Biophysica. 2021; 1(4):377-404. https://doi.org/10.3390/biophysica1040028
Chicago/Turabian StyleYe, Naike, Francesco Caruso, and Miriam Rossi. 2021. "Mechanistic Insights into the Inhibition of SARS-CoV-2 Main Protease by Clovamide and Its Derivatives: In Silico Studies" Biophysica 1, no. 4: 377-404. https://doi.org/10.3390/biophysica1040028
APA StyleYe, N., Caruso, F., & Rossi, M. (2021). Mechanistic Insights into the Inhibition of SARS-CoV-2 Main Protease by Clovamide and Its Derivatives: In Silico Studies. Biophysica, 1(4), 377-404. https://doi.org/10.3390/biophysica1040028