
The Link between Aggrecan and Familial Osteochondritis Dissecans

 , ,
, , 
{kind=link}
Abstract
:1. Introduction
2. Aggrecanopathies and OCD
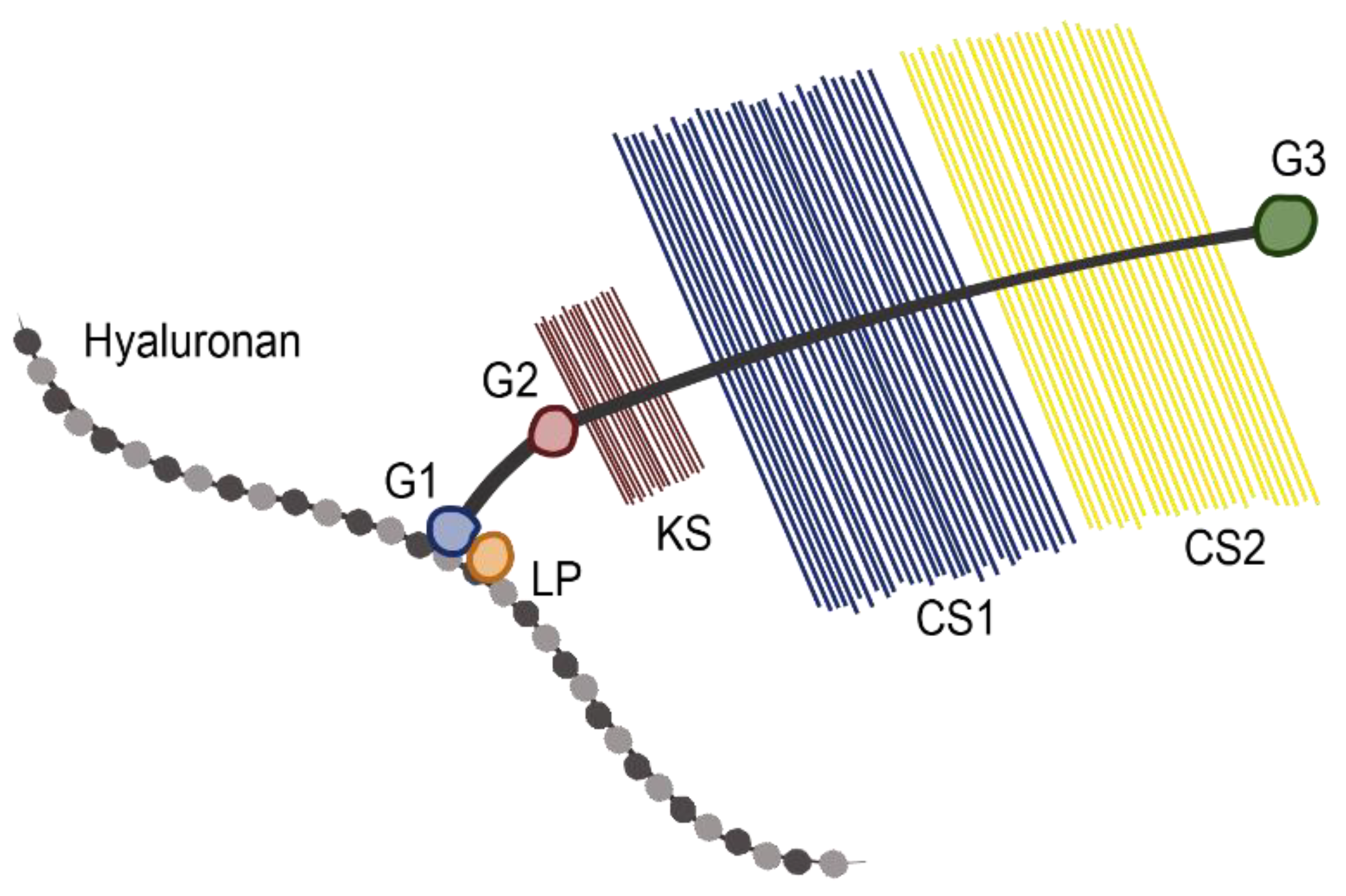
2.1. Role of Aggrecan in Normal Physiology
2.2. Diseases Associated with ACAN Mutations
2.3. Genetic Link to OCD
3. Current Research on Aggrecan Mutations
3.1. Natural Aggrecan Mutations in Animals
3.2. In Vitro Studies
3.3. Case Reports and Treatment
3.3.1. Pharmacological Treatments
3.3.2. Conservative Treatments
3.3.3. Surgical Treatments
4. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Handbook of Histology Methods for Bone and Cartilage; Humana Press: Totowa, NJ, USA, 2003.
- Dzaja, I.; Khalid, S. Hip and knee osteoarthritis. In Osteoarthritis: Pathogenesis, Diagnosis, Available Treatments, Drug Safety, Regenerative and Precision Medicine; Springer: Cham, Switzerland, 2015; pp. 29–42. ISBN 9783319195605. [Google Scholar]
- Krishnan, Y.; Grodzinsky, A.J. Cartilage diseases. Matrix Biol. 2018, 71–72, 51–69. [Google Scholar] [CrossRef]
- Roughley, P.J.; Mort, J.S. The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 2014, 1, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, A.; Keret, D.; Macewen, G. Familial Osteochondritis Dissecans. Orthopedics 1990, 13, 1259–1262. [Google Scholar]
- Grässel, S.; Aszódi, A. Cartilage: Pathophysiology; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; Volume 2, ISBN 9783319458038. [Google Scholar]
- Cole, B.J.; Malek, M.M. Articular Cartilage Lesions; Springer: New York, NY, USA, 2004. [Google Scholar]
- Andriolo, L.; Crawford, D.C.; Reale, D.; Zaffagnini, S.; Candrian, C.; Cavicchioli, A.; Filardo, G. Osteochondritis Dissecans of the Knee: Etiology and Pathogenetic Mechanisms. A Systematic Review. Cartilage 2020, 11, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, K.; Middleton, R. Familial osteochondritis dissecans: A dysplasia of articular cartilage? Skelet. Radiol. 1985, 13, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Bruns, J.; Werner, M.; Habermann, C. Osteochondritis Dissecans: Etiology, Pathology, and Imaging with a Special Focus on the Knee Joint. Cartilage 2018, 9, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jordan, J.M. Epidemiology of osteoarthritis. Clin. Geriatr. Med. 2010, 26, 355–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldring, M.B.; Culley, K.L.; Otero, M. Pathogenesis of osteoarthritis in general. In Cartilage: Pathophysiology; Springer International Publishing: Cham, Switzerland, 2017; Volume 2, pp. 1–25. ISBN 9783319458038. [Google Scholar]
- Grässel, S.; Aszódi, A. Cartilage: Physiology and Development; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; Volume 1, ISBN 9783319295688. [Google Scholar]
- Krakow, D. Heritable Diseases of Connective Tissue. In Kelley and Firestein’s Textbook of Rheumatology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1797–1815. [Google Scholar]
- Laor, T.; Zbojniewicz, A.M.; Eismann, E.A.; Wall, E.J. Juvenile osteochondritis dissecans: Is it a growth disturbance of the secondary physis of the epiphysis? Am. J. Roentgenol. 2012, 199, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Olstad, K.; Shea, K.G.; Cannamela, P.C.; Polousky, J.D.; Ekman, S.; Ytrehus, B.; Carlson, C.S. Juvenile osteochondritis dissecans of the knee is a result of failure of the blood supply to growth cartilage and osteochondrosis. Osteoarthr. Cartil. 2018, 26, 1691–1698. [Google Scholar] [CrossRef] [Green Version]
- Gornitzky, A.L.; Mistovich, R.J.; Atuahuene, B.; Storey, E.P.; Ganley, T.J. Osteochondritis Dissecans Lesions in Family Members: Does a Positive Family History Impact Phenotypic Potency? Clin. Orthop. Relat. Res. 2017, 475, 1573–1580. [Google Scholar] [CrossRef] [Green Version]
- Polousky, J.D. Juvenile osteochondritis dissecans. Sports Med. Arthrosc. 2011, 19, 56–63. [Google Scholar] [CrossRef] [PubMed]
- van Weeren, P.R.; Olstad, K. Pathogenesis of osteochondrosis dissecans: How does this translate to management of the clinical case? Equine Vet. Educ. 2016, 28, 155–166. [Google Scholar] [CrossRef]
- Gans, I.; Grant, S.F.A.; Ganley, T.J. The Genetic Nature of Osteochondritis Dissecans: A Systematic Review and Call for Improved Studies. Univ. Pa. Orthop. J. 2013, 23, 14–16. [Google Scholar]
- Stattin, E.L.; Tegner, Y.; Domellöf, M.; Dahl, N. Familial osteochondritis dissecans associated with early osteoarthritis and disproportionate short stature. Osteoarthr. Cartil. 2008, 16, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentchordi-Montane, L.; Aza-Carmona, M.; Benito-Sanz, S.; Barreda-Bonis, A.C.; Sanchez-Garre, C.; Prieto-Matos, P.; Ruiz-Ocana, P.; Lechuga-Sancho, A.; Carcavilla-Urqui, A.; Mulero-Collantes, I.; et al. Heterozygous aggrecan variants are associated with short stature and brachydactyly: Description of 16 probands and a review of the literature. Clin. Endocrinol. (Oxf.) 2018, 88, 820–829. [Google Scholar] [CrossRef]
- Nilsson, O.; Guo, M.H.; Dunbar, N.; Popovic, J.; Flynn, D.; Jacobsen, C.; Lui, J.C.; Hirschhorn, J.N.; Baron, J.; Dauber, A. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J. Clin. Endocrinol. Metab. 2014, 99, E1510–E1518. [Google Scholar] [CrossRef]
- Sevane, N.; Dunner, S.; Boado, A.; Cañon, J. Candidate gene analysis of osteochondrosis in Spanish Purebred horses. Anim. Genet. 2016, 47, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Laverty, S.; Ionescu, M.; Marcoux, M.; Boure, L.; Doize, B.; Poole, A.R. Alterations in cartilage type-II procollagen and aggrecan contents in synovial fluid in equine osteochondrosis. J. Orthop. Res. 2000, 18, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Kiani, C.; Chen, L.; Wu, Y.J.; Yee, A.J.; Yang, B.B. Structure and function of aggrecan. Cell Res. 2002, 12, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Yanagishita, M. Function of proteoglycans in the extracellular matrix. Pathol. Int. 1993, 43, 283–293. [Google Scholar] [CrossRef]
- Gibson, B.G.; Briggs, M.D. The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J. Rare Dis. 2016, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cooke, M.E.; Lawless, B.M.; Jones, S.W.; Grover, L.M. Matrix degradation in osteoarthritis primes the superficial region of cartilage for mechanical damage. Acta Biomater. 2018, 78, 320–328. [Google Scholar] [CrossRef]
- Baker, N.; Sharpe, P.; Culley, K.; Otero, M.; Bevan, D.; Newham, P.; Barker, W.; Clements, K.M.; Langham, C.J.; Goldring, M.B.; et al. Dual regulation of metalloproteinase expression in chondrocytes by Wnt-1-inducible signaling pathway protein 3/CCN6. Arthritis Rheum 2012, 64, 2289–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caterson, B.; Flannery, C.R.; Hughes, C.E.; Little, C.B. Mechanisms involved in cartilage proteoglycan catabolism. Matrix Biol. 2000, 19, 333–344. [Google Scholar] [CrossRef]
- Watanabe, H.; Yamada, Y.; Kimata, K. Roles of aggrecan, a large chondroitin sulfate proteoglycan, in cartilage structure and function. J. Biochem. 1998, 124, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.B.; Zhang, Y.; Cao, L.; Yang, B.L. Aggrecan and link protein affect cell adhesion to culture plates and to type II collagen. Matrix Biol. 1998, 16, 541–561. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Y.; Yang, B.B. Expression of the G1 domain of aggrecan interferes with chondrocyte attachment and adhesion. Matrix Biol. 1998, 17, 379–392. [Google Scholar] [CrossRef]
- Stavber, L.; Hovnik, T.; Kotnik, P.; Lovrečić, L.; Kovač, J.; Tesovnik, T.; Bertok, S.; Dovč, K.; Debeljak, M.; Battelino, T.; et al. High frequency of pathogenic ACAN variants including an intragenic deletion in selected individuals with short stature. Eur. J. Endocrinol. 2020, 182, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Tatsi, C.; Gkourogianni, A.; Mohnike, K.; DeArment, D.; Witchel, S.; Andrade, A.C.; Markello, T.C.; Baron, J.; Nilsson, O.; Jee, Y.H. Aggrecan mutations in nonfamilial short stature and short stature without accelerated skeletal maturation. J. Endocr. Soc. 2017, 1, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Gkourogianni, A.; Andrew, M.; Tyzinski, L.; Crocker, M.; Douglas, J.; Dunbar, N.; Fairchild, J.; Funari, M.F.A.A.; Heath, K.E.; Jorge, A.A.L.L.; et al. Clinical Characterization of Patients With Autosomal Dominant Short Stature due to Aggrecan Mutations. J. Clin. Endocrinol. Metab. 2017, 102, 460–469. [Google Scholar] [CrossRef]
- Petrie, P. Aetiology of Osteochondritis Dissecans. Failure to Establish a Familial Background. J Bone Jt. Surg Br. 1977, 59, 366–367. [Google Scholar] [CrossRef]
- Mubarak, S.J.; Carroll, N.C. Familial Osteochondritis Dissecans of the Knee. Clin. Orthop. Relat. Res. 1979, 140, 131–136. [Google Scholar] [CrossRef]
- Mei-Dan, O.; Mann, G.; Steinbacher, G.; Cugat, R.B.; Alvarez, P.D. Bilateral Osteochondritis Dissecans of the Knees in Monozygotic Twins: The Genetic Factor and Review of the Etiology. Am. J. Orthop. 2009, 38, E152–E155. [Google Scholar] [PubMed]
- Richie, L.B.; Sytsma, M.J. Matching osteochondritis dissecans lesions in identical twin brothers. Orthopedics 2013, 36. [Google Scholar] [CrossRef] [Green Version]
- Stattin, E.L.; Wiklund, F.; Lindblom, K.; Önnerfjord, P.; Jonsson, B.A.; Tegner, Y.; Sasaki, T.; Struglics, A.; Lohmander, S.; Dahl, N.; et al. A Missense Mutation in the Aggrecan C-type Lectin Domain Disrupts Extracellular Matrix Interactions and Causes Dominant Familial Osteochondritis Dissecans. Am. J. Hum. Genet. 2010, 86, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Yamada, Y. Chondrodysplasia of gene knockout mice for aggrecan and link protein. Glycoconj. J. 2002, 19, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Domowicz, M.S.; Cortes, M.; Henry, J.G.; Schwartz, N.B. Aggrecan modulation of growth plate morphogenesis. Dev. Biol. 2009, 329, 242–257. [Google Scholar] [CrossRef] [Green Version]
- Metzger, J.; Gast, A.C.; Schrimpf, R.; Rau, J.; Eikelberg, D.; Beineke, A.; Hellige, M.; Distl, O. Whole-genome sequencing reveals a potential causal mutation for dwarfism in the Miniature Shetland pony. Mamm. Genome 2017, 28, 143–151. [Google Scholar] [CrossRef]
- Xu, M.; Stattin, E.-L.; Shaw, G.; Heinegard, D.; Sullivan, G.; Wilmut, I.; Colman, A.; Onnerfjord, P.; Khabut, A.; Aspberg, A.; et al. Chondrocytes Derived From Mesenchymal Stromal Cells and Induced Pluripotent Cells of Patients with Familial Osteochondritis Dissecans Exhibit an Endoplasmic Reticulum Stress Response and Defective Matrix Assembly. Stem. Cells Transl. Med. 2016, 5, 1171–1181. [Google Scholar] [CrossRef]
- Crippa, M.; Giangiobbe, S.; Villa, R.; Bestetti, I.; De Filippis, T.; Fatti, L.; Taurino, J.; Larizza, L.; Persani, L.; Bellini, F.; et al. A balanced reciprocal translocation t(10;15)(q22.3;q26.1) interrupting ACAN gene in a family with proportionate short stature. J. Endocrinol. Invest. 2018, 41, 929–936. [Google Scholar] [CrossRef]
- Andrew, T.A.; Spivey, J.; Lindebaum, R.H. Familial osteochondritis dissecans and dwarfism. Acta Orthop. 1981, 52, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Moy, O.J.; Peimer, C.A.; Nakamura, T.; Howard, C.; Ko, S.H.; Lee, T.C.; Nishiwaki, Y. An experimental study on costal osteochondral graft. Osteoarthr. Cartil. 2012, 20, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Steen, M.; Pfundt, R.; Maas, S.J.W.H.; Bakker-van Waarde, W.M.; Odink, R.J.; Hokken-Koelega, A.C.S. ACAN gene mutations in short children born SGA and response to growth hormone treatment. J. Clin. Endocrinol. Metab. 2017, 102, 1458–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, W.E.; Neufurth, M.; Wang, S.; Tolba, E.; Schroder, H.C.; Wang, X. Morphogenetically active scaffold for osteochondral repair (polyphosphate/alginate/N,O-carboxymethyl chitosan). Eur. Cell. Mater. 2016, 31, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Yellin, J.L.; Trocle, A.; Grant, S.F.; Hakonarson, H.; Shea, K.G.; Ganley, T.J. Candidate Loci are Revealed by an Initial Genome-wide Association Study of Juvenile Osteochondritis Dissecans. J. Pediatr. Orthop. 2017, 37, e32–e36. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.; Delgado, A.; Sanchez, E.; Fernandez, A.; Hernandez, A.; Evora, C. Repair of an osteochondral defect by sustained delivery of BMP-2 or TGFbeta1 from a bilayered alginate-PLGA scaffold. J. Tissue Eng. Regen. Med. 2014, 8, 521–533. [Google Scholar] [CrossRef]
- Mei-Dan, O.; Maoz, G.; Swartzon, M.; Onel, E.; Kish, B.; Nyska, M.; Mann, G. Treatment of Osteochondritis Dissecans of the Ankle with Hyaluronic Acid Injections: A Prospective Study. Foot Ankle Int. 2008, 29, 1171–1178. [Google Scholar] [CrossRef]
- Beck, J.J.; Sugimoto, D.; Micheli, L. Sustained Results in Long-Term Follow-Up of Autologous Chondrocyte Implantation (ACI) for Distal Femur Juvenile Osteochondritis Dissecans (JOCD). Adv. Orthop. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Growth Hormone Treatment in Patients With Aggrecan (ACAN) Deficiency—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03288103?term=acan%2C+aggrecan&draw=2&rank=1 (accessed on 9 March 2021).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozere, S.; Chergui, S.; Cooke, M.E.; Pauyo, T.; Rosenzweig, D.H. The Link between Aggrecan and Familial Osteochondritis Dissecans. Surgeries 2021, 2, 128-138. https://doi.org/10.3390/surgeries2020012
Ozere S, Chergui S, Cooke ME, Pauyo T, Rosenzweig DH. The Link between Aggrecan and Familial Osteochondritis Dissecans. Surgeries. 2021; 2(2):128-138. https://doi.org/10.3390/surgeries2020012
Chicago/Turabian StyleOzere, Samantha, Sami Chergui, Megan E. Cooke, Thierry Pauyo, and Derek H. Rosenzweig. 2021. "The Link between Aggrecan and Familial Osteochondritis Dissecans" Surgeries 2, no. 2: 128-138. https://doi.org/10.3390/surgeries2020012
APA StyleOzere, S., Chergui, S., Cooke, M. E., Pauyo, T., & Rosenzweig, D. H. (2021). The Link between Aggrecan and Familial Osteochondritis Dissecans. Surgeries, 2(2), 128-138. https://doi.org/10.3390/surgeries2020012