Abstract
In recent years, the upgrading of lignocellulose bio-oils from fast-pyrolysis by means of ketonization has emerged as a frontier research domain to produce a new generation of biofuels. Propionic acid (PA) ketonization is extensively investigated as a model reaction over metal oxides, but the activity of other materials, such as metal phosphates, is mostly unknown. Therefore, PA ketonization was preliminarily investigated in the gas phase over both phosphates and oxides of Al, Zr, and La. Their catalytic activity was correlated to the physicochemical properties of the materials characterized by means of XRD, XRF, BET N2 porosimetry, and CO2- and NH3-TPD. Noteworthy, monoclinic ZrO2 proved to be the most promising candidate for the target reaction, leading to a 3-pentanone productivity as high as 5.6 h−1 in the optimized conditions. This value is higher than most of those reported for the same reaction in both the academic and patent literature.
1. Introduction
The progressive shift from traditional fossil fuels towards renewable biofuels for energy production and transports is considered an attractive strategy to reduce greenhouse gas emissions and contribute to climate change mitigation [1]. However, to achieve these goals without adversely affecting the environment or social sustainability, biofuels must be produced in a sustainable way, i.e., their production must not rely on edible crops and should not result in either direct or indirect land use change [2,3].
Keeping this in mind, the flash-pyrolysis of lignocellulosic biomasses (e.g., non-edible crops that do not create an additional demand for land such as agricultural and forestry residues and industrial wastes from food or the pulp and paper industry) [1], stands out among other synthetic strategies aimed at biofuel production thanks to the large availability of raw materials and process robustness, which permits to treat almost any type of biomass, thus allowing to valorize raw materials otherwise considered as a waste [4].
State-of-art fast pyrolysis technologies can convert biomass into a liquid oil with yields as high as 70–80 wt.% [5]. However, this product is a complex mixture of oxygenated compounds (e.g., carboxylic acids, phenolics, furanics, and other small, oxygenated molecules) [4] and cannot be used as fuel due to its very high oxygen content (up to 50 wt.%, including water) and its high acidity [6]. Moreover, its hydrophilicity prevents its use in blends with traditional fuels [4]. Consequently, the use of bio-oil as fuel requires its upgrading via chemical processes capable of reducing its oxygen (and water) content and removing its acidity, resulting in an enhanced liquid fuel with increased calorific power, higher hydrophobicity, and higher miscibility with traditional fuels.
The most investigated upgrading processes involve the removal of oxygen from crude bio-oil by means of hydrodeoxygenation reactions, which are catalyzed by noble metals in the presence of molecular hydrogen (H2) at high pressure and coproduce water [7]. This leads to the recurring formation of two phases at the end of the treatment: a hydrophilic oxygen-rich phase and an upgraded, hydrophobic organic phase. As a result of the hydrogenation of low molecular weight oxygenates, a gaseous stream consisting of light alkanes and olefins is also obtained. As an example, in analogy with the well-established hydrodesulfurization (HDS) process of fossil oils, the hydrodeoxygenation (HDO) of bio-oils can be carried out in a continuous-flow system in the pressure range of 80–300 bar and temperature range of 300–400 °C [8], with HDS catalysts (Co-MoS2/Al2O3, Ni-MoS2/Al2O3) or supported noble metals catalysts (Pd/C, Ru/C) [9]. These processes, however, are extremely hydrogen-demanding due to the high oxygen content in crude bio-oils, and, on top of that, the low molecular weight oxygenates that represent a significant fraction of bio-oils are lost as gases upon hydrogenation [10]. The sustainability of HDO of bio-oils could be greatly improved by developing a gas-phase catalytic transfer hydrogenation (CHT) process at atmospheric pressure, to be carried out using light renewables alcohols (e.g., MeOH and EtOH) instead of H2 [11]. Despite that, an intermediate upgrading process capable of reducing the alcohol or H2-consumption of the subsequent HDO process, to be carried out during (e.g., catalytic fast-pyrolysis with zeolites) [12] or right after the fast-pyrolysis (e.g., direct vapor-phase upgrading with metal oxides) [13], is highly desirable and has inspired the industrial and academic research over the past two decades.
In this context, since bio-oil is characterized by the presence of light carboxylic acids such as acetic acid (AA) and propionic acid (PA) up to 10 mol % [14], their continuous gas-phase ketonization is considered a promising upgrading alternative, when carried out right after the pyrolysis and before the condensation of the resulting bio-oil, which also avoids unnecessary expense for re-vaporization. As depicted in Scheme 1, the ketonization reaction results in the removal of the acid moieties from crude bio-oils, while at the same time the oxygen content in the liquid is reduced, and the coupling of light acids produces heavier ketones that are less likely to be degraded into light gases during the subsequent HDO step [10]. Moreover, ketones can be further coupled with each other via aldol condensation, leading to the formation of longer chain molecules, further enhancing the calorimetric properties of the final mixture [15,16]. It is worth noticing that, during such upgrading process, several reactions in addition to ketonization can lead to the formation of CO2 (e.g., direct decarboxylation of carboxylic acids or aquathermolysis reactions occurring on the heavier components of the bio-oil [17,18,19]).
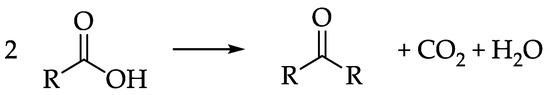
Scheme 1.
Generic stoichiometry of carboxylic acid ketonization.
The gas-phase ketonization of acids and/or esters [20] may occur following two distinct mechanisms:
- “Bulk” mechanism [21]: metal oxides with low lattice energy and/or very high basicity such as alkali earth oxides (e.g., MgO, CaO, BaO) [22] and rare earth oxides (e.g., La2O3, Pr6O11, Nd2O3) [23] react with carboxylic acids vapors producing carboxylate salts and water. When the reaction temperature is high enough to trigger the thermal decomposition of carboxylate salts, the recombination of the resulting radical fragments produces a ketone and a metal carbonate (or an oxycarbonate depending on the nature of the metal cation). Finally, a catalytic cycle is established when the reaction temperature is high enough to decompose the most stable metal carbonate (or oxycarbonate), producing CO2 and regenerating the pristine metal oxide.
- “Surface” mechanism: metal oxides possessing high lattice energy (e.g., ZrO2, TiO2, MnO2, CeO2) and zeolites do not react with carboxylic acids vapors to produce bulk carboxylate salts. Nonetheless, acids are strongly chemisorbed, and the reaction remains confined over the catalyst surface. Unlike the bulk mechanism, the surface mechanism is not of the radical type, and to occur, it strictly requires the presence of an acidic hydrogen in α-position with respect to the carboxylic group in at least one of the two acids that take part in the reaction [10].
Despite the large number of studies focusing on surface ketonization published over the last three decades, both experimentally [21,22,24,25] and computationally [26,27], a general agreement on how the α-hydrogen is involved is still the subject of debate, and several mechanisms involving the participation of different intermediates have been proposed (e.g., via the direct concerted coupling of carboxylates [28], via ketene [29], via β-ketoacid [30], via carboxylic anhydride [31], or via a dianion interaction with a catalyst surface with a negatively charged α-carbon and a bidentate carboxylate group [21]). Nonetheless, several experimental evidence, carefully reviewed by Pham and co-workers [10] and by Sels and co-workers [32], suggest that the mechanism involving the formation of a β-ketoacid is the most probable one over the transition metal oxides.
The ketonization of AA [33] and PA [34] have been investigated as model reactions over a variety of metal oxides supported on SiO2, Al2O3 and TiO2 by Glinski and co-workers. Despite AA and PA, ketonization occurred to a certain extent over all the catalysts investigated, and the most active oxides were found to be those of U, Th, Mn, Ce, La, and Zr. Following these two systematic screenings, an increasing number of authors and inventors focused their attention on catalytic materials consisting of amphoteric and/or reducible metal oxides such as TiO2 [35], ZrO2 [36,37,38,39], CeO2 and MnO2 [24], La2O3, and other rare-earth oxides [40]. Mixed metal oxides, such as Ce/Zr/O [41], Ce/Zr/Mn/O [42], Mn/Ce/O [24,43], Ce/Fe/O [44], Mn/Zn-chromite [45], and Mg/Al/Ce/O [46] and supported catalysts such as MnO2/Al2O3 [37], TiO2/C [47] and Ru/TiO2 [25] have been claimed to be active as well.
It has been demonstrated that both basic and acidic sites are involved in ketonization [48], and it is largely accepted that the most active sites consist of strong, adjacent Lewis acid-base pairs [10,32]. Lewis acidic sites (e.g., coordinatively unsaturated metal cations) can interact with the oxygens of the carboxylic group as shown in Scheme 2a, enhancing the acidity of both the carboxylic proton and the α-proton, thus favoring their abstraction by adjacent basic sites (e.g., oxygen anions). On the other hand, once the carboxylic proton and the α-proton are abstracted by vicinal oxygen atoms (Scheme 2, reactions b and c), acidic sites are needed to coordinate and stabilize the resulting nucleophilic species (e.g., dianions or 1-hydroxy-enolates, as shown in Scheme 2, reactions c and d, respectively) long enough to allow them to react with another carboxylate anion producing the β-ketoacid intermediate (Scheme 2, reaction e). At the temperatures at which ketonization usually occurs, β-ketoacids are unstable and immediately undergo a decarboxylation via a six-membered cyclic transition state, leading to an enol that readily tautomerize to the corresponding ketone (Scheme 2, reaction f).
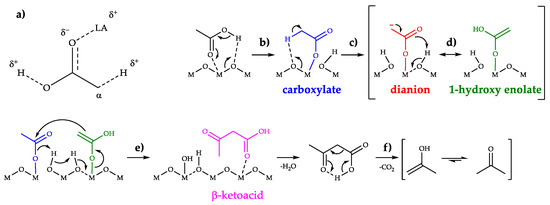
Scheme 2.
(a) AA carbonyl group interaction with surface Lewis Acid (LA) leading to the weakening of both the carboxyl O-H bond and the α-carbon C-H bond, thus enhancing the acidity of both the protons; (b) AA chemisorption over metal oxides to form a monodentate carboxylate anion (bidentate and bridging configuration are also possible [49]); (c) α-proton abstraction from a carboxylate anion to form a dianion or (d) enolization of the carboxylate anion to form a 1-hydroxylate enolate anion; € coupling between an electrophilic carboxylate anion and a nucleophilic 1-hydroxy enolate anion to form a β-ketoacid and water; (f) β-ketoacid decarboxylation to yield the ketone.
In addition to a strong amphoteric character, the presence of Lewis acid-base pairs has been linked to the presence of surface defects such as oxygen vacancies and coordinatively unsaturated metal cations [10,25,50]. Such defects can be formed by the partial reduction of the surface of the metal oxide upon interaction with the reactant and, not surprisingly, readily reducible metal oxides that often display high ketonization activity.
For these reasons, catalyst reduction with molecular hydrogen prior or during ketonization has been investigated as a mean to enhance catalyst activity. As an example, using H2 instead of N2 as a carrier significantly increases the activity of ZrO2, CeO2, and Ce/Zr/O mixed oxides for valeric acid ketonization [51]. This behavior was explained by the formation of surface oxygen vacancies and Zr3+ and Ce3+ coordinatively unsaturated cations.
Despite a wide literature regarding ketonization over metal oxides being available, the activity of metal phosphates and how their properties (e.g., nature of the cation Mn+, stoichiometry between P and M, polymorph, acid-base, morphological properties, and so on) influence their catalytic activity are mostly unknown. Moreover, even if excellent catalyst screenings are available for metal oxides [33,34], these works’ lack an equally thorough characterization of catalyst acid-base properties, redox capacity, and structure activity relationships.
Therefore, here we present an in-depth investigation of the continuous-flow, gas-phase ketonization reaction of PA to produce 3-pentanone (3-P) over a series of metal phosphates (Al/P/O, Zr/P/O and La/P/O) and a comprehensive comparison, through both catalytic tests and characterization, with the corresponding metal oxides (γ-Al2O3, ZrO2, and La2O3).
These phosphates and oxides have been carefully chosen with the aim to compare materials with a wide range of different acid-based properties (e.g., La2O3 is a strong base, ZrO2 is amphoteric, and γ-Al2O3 is a strong Lewis acid) and assess structure–activity relationships. SiO2 was also included in the study to complete the range of acid-base properties of oxides with a mild Brønsted acidity and because it is often used as support for ketonization catalysts.
After the first screening, the best catalyst (zirconium oxide) was further investigated by varying the synthetic method: one sample of zirconia with tetragonal crystal structure and one sample with monoclinic structure were obtained by means of precipitation techniques; a third sample of zirconia possessing the monoclinic structure but higher specific surface area, low crystallinity, high defectivity, and enhanced basicity was obtained by means of a hydrothermal method.
After testing in industrially relevant reaction conditions (e.g., 30 mol % PA in the feed, 350 °C, time factor = W/F = 0.1 s g/mL), the hydrothermally synthetized zirconia was found to be the best catalyst and afforded a 3-P productivity (calculated as the mass of product obtained in 1 h divided by the mass of the catalyst used) equal to 5.6 h−1, which is, at the best of our knowledge, better than all the others reported in the literature so far.
2. Materials and Methods
2.1. Catalyst Preparation
Catalysts will be referred to as follows: Al/P/O, Zr/P/O, and La/P/O for Al, Zr, and La phosphate, respectively; Al2O3 and La2O3 for Al and La oxide, respectively; m-ZrO2-PR, t-ZrO2-PR, and m-ZrO2-HT for Zr oxides (in this case, the first letter stands for the crystal structure (m = monoclinic, t = tetragonal), while the last two letters indicate the preparation method (PR = precipitation, HT = hydrothermal)).
Al/P/O, Zr/P/O, and La/P/O were synthesized by means of co-precipitation, using a procedure adapted from the literature [52]. Briefly, the metal precursor AlCl3 (Alfa Aesar, 99%, Haverhill, MA, USA), La(NO3)3∙6H2O (Alfa Aesar, 99.9%), or ZrOCl2∙8H2O (Alfa Aesar, 98%) was dissolved in 300 mL of deionized water to obtain a 1 mol/L solution. Then, 300 mL of aqueous H3PO4 (1 mol/L) was added to the first solution under continuous stirring, and finally, the pH was risen from 1–1.5 to 7.0 adding aqueous NH3 (Sigma Aldrich, 28–30 wt.%, St. Louis, MO, USA) to promote the precipitation of a metal phosphate. The suspensions were aged for 3–4 h under stirring, filtered over a Buchner funnel, and washed with 2 L of distilled water to remove adsorbed chloride, nitrate, and ammonium ions. Finally, the resulting wet solid was dried overnight at 120 °C and calcined at 550 °C for 3 h with a heating rate of 5 °C/min.
Al2O3 was a commercial reference material (SASOL Puralox SCFa140, 98%, Sandton, South Africa) as well as SiO2 (GRACE 360, 98%, Columbia, Maryland, USA); before use, they were calcined for 3 h with a heating rate of 5 °C/min up to 600 °C and 650 °C, respectively.
La2O3 and m-ZrO2-PR were synthesized modifying the precipitation technique used for the preparation of the respective phosphates. In this case, La(NO3)3∙6H2O or ZrOCl2∙8H2O was dissolved in deionized water to obtain a 0.5 mol/L solution (60 mL for La and 70 mL for Zr); such solutions were slowly added dropwise to aqueous NH3 (2 mol/L, 300 mL for La and 400 mL for Zr) under vigorous stirring. The pH of the basic solution was maintained constant and equal to 11.5 during the addition by adding concentrated NH3 (28–30 wt.%). Precipitates were aged for 1–2 h under stirring, filtered over a Buchner funnel, and washed with 2 L of distilled water for each 3 g of material to remove adsorbed chloride, nitrate, and ammonium ions. Finally, the resulting wet solid was dried at 120 °C overnight and calcined at 550 °C (Zr) or 750 °C (La) for 3 h with a heating rate of 5 °C/min.
t-ZrO2-PR [53,54] and m-ZrO2-HT [55] were prepared according to methods described elsewhere.
Prior to use, catalysts were formed in pellets by compressing the powder into a self-sustaining disk (≈1 mm in height and 3 cm in diameter); then, the disk was crushed using appropriate sieves in order to obtain pellets with a granulometry between 30 and 60 mesh.
2.2. Catalyst Characterization
The XRD powder patterns of all materials were acquired using a Philips X’Pert diffractometer with Bragg-Brentano geometry, equipped with a pulse height analyzer and a secondary curved graphite-crystal monochromator. The X-ray source that was used emits the characteristic Cu Kα radiation (λ = 1.54178 Å) filtered by a Ni foil to suppress the Kβ lines.
The phosphorus/metal (P/M) atomic ratios of the different phosphates were determined by means of wavelength dispersive X-ray fluorescence, which was carried out with a PANalytical Axios Advanced WD-XRF spectrometer equipped with a Rh-anode X-ray tube (maximum power = 4 kW). The analysis was carried out in vacuum on samples shaped into a 13 mm diameter disk.
The specific surface area (SSA) of catalysts were calculated with the BET method after acquiring the N2 adsorption/desorption isotherms at −196 °C (77 K) using a Micromeritics ASAP 2020 instrument.
Temperature programmed desorption (TPD) of NH3 and CO2 were carried out with a Micromeritics Autochem II 2920 instrument coupled with a Cirrus 2 quadrupole mass spectrometer to measure the total acidity and total basicity of all materials. The quantification of effluents was carried out by recording the intensity of the following ion current signals: NH3 (m/z = 17), CO2 (m/z = 44), and H2O (m/z = 18). In a typical experiment, 0.2 g of material was put in a quartz tube and heated up to calcination temperature at a rate of 10 °C/min in 30 mL/min of pure He flow to clean the catalyst surface from adsorbed water and carbonates. The final temperature was kept for 60 min. After cooling, NH3 chemisorption was conducted at 100 °C for 20 min by flowing 30 mL/min of 10% NH3/He, while CO2 chemisorption was performed at 40 °C for 60 min by flowing 30 mL/min of 10% CO2/He. After chemisorption, all samples were flown with 30 mL/min of He for 60 min to remove the weakly physisorbed probe molecules. Finally, the temperature programmed desorption (TPD) was carried out by heating the sample up to calcination temperature at the rate of 10 °C/min and keeping the final temperature for 60 min.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of adsorbed pyridine, followed by desorption at increasing temperature, was carried out to characterize the distribution of Lewis and Brønsted acidic sites of the most active catalyst (m-ZrO2-HT). DRIFTS spectra were acquired with a Bruker Vertex 70 instrument equipped with a Pike DiffusIR cell attachment and a MCT detector. Spectra were recorded carrying out 128 scans in the spectral region of 4000–600 cm−1 with a resolution of 4 cm−1. The spectra were recorded as follows: first, KBr was loaded in the DRIFTS cell and pre-treated at 450 °C under a flow of He (10 mL/min) for 45 min in order to remove adsorbed molecules. Then, the cell was cooled down to 50 °C, and the background spectra were acquired at different temperatures (with steps of 50 °C up to 450 °C). Then, m-ZrO2-HT was loaded in the DRIFTS cell and was subjected to the same procedure carried out over KBr (e.g., pre-treatment at 450 °C in He flow for 45 min, cooling to 50 °C, and recording of spectra at 50, 100, 150, 200, 250, 300, 350, 400, and 450 °C). Next, the adsorption of pyridine was carried out after cooling down to 50 °C by injecting a pulse (2 μL) of this molecular probe in the He flow. The adsorption process was monitored by recording a spectrum every minute until the intensity of the bands of pyridine in the spectra remained constant. Finally, spectra of pyridine adsorbed over m-ZrO2-HT were recorded at increasing temperatures up to 450 °C, with steps of 50 °C.
2.3. Catalytic Tests
All catalytic tests were carried out using the gas-phase rig shown in the electronic supporting information (ESI, Figure S1) and performed at atmospheric pressure using a conventional fixed bed down-flow quartz reactor; all catalysts were charged in the form of pellets with particle size between 30 and 60 mesh. The liquid reactant PA (Alfa Aesar, 99%) was injected with a KD Scientific Legacy 100 volumetric pump in a stainless-steel line (1/16 inches) and directly driven ≈ 5 cm above the catalytic bed with 12 mL/min of N2. Another stainless-steel line (1/8 inches) drove a second flux of pre-heated N2 (210 °C) to the top of the reactor to obtain the desired total N2 flux.
Usually, the catalyst was heated up to reaction temperature at 10 °C/min under nitrogen flow (the same flow that will be used to carry out the catalytic test) and then was kept at this temperature for 30 min before starting to feed the reactant.
The effluent from the reactor was bubbled through two cold traps in series filled with acetonitrile (AcCN, Sigma-Aldrich, 99.8%) and kept at 0 °C by means of an ice bath to absorb the condensable products. The so-obtained reaction mixtures were transferred into a 100 mL flask from the cold traps at regular intervals of time, and 1 g of dodecane (Sigma-Aldrich, 99%) solution (4 × 10−5 mol/g) was added as the internal standard.
Gaseous products exiting the cold trap were driven to an online GC equipped with two sample/injection loops in series. Both the liquid reaction mixtures and gases exiting from the cold trap were analyzed by means of gas chromatography with a Hewlett Packard 5890 Series II GC instrument, equipped with FID and TCD detectors. The quantification of the condensed products was carried out offline with an Agilent J and W DB-1701 capillary column (25 m × 530 μm × 1.05 μm) connected to the FID detector. The quantification of the gaseous products was carried out online using an Agilent CP-Molsieve 5A capillary column (25 m × 530 μm × 50 μm, to elute H2, O2, and N2) and an Agilent CP-SilicaPLOT capillary column (30 m × 530 μm × 6 μm, to elute CO, CO2, CH4, and ethylene).
An Agilent Technologies 6890 gas-chromatographer equipped with an Agilent HP-5 capillary column (30 m × 250 μm × 1.05 μm) and coupled with an Agilent Technologies 5973 mass analyzer (GC-MS) were used to identify unknown products (Agilent Technologies, Santa Clara, CA, USA); moreover, the retention times of unknown products were compared with those of pure reference standards.
The following equations were used to calculate PA conversion (XPA), yields (Yi), selectivity (Si), sum of yields (Yield Sum), and molar balance (Yield Sum/XMP).
XPA = [(molPA IN − molPA OUT)/(molPA IN)] ∙ 100,
Yi = νι ∙ (moli out/molPA IN) ∙ 100,
ν: stoichiometric factor (e.g., ν3-P = 2)
Si = (Yi/XPA) ∙ 100
Yield Sum = Σi Yi
Molar Balance = Yield Sum/XPA = (Σi Yi)/XMP ∙ 100
3. Results and Discussion
3.1. Catalyst Characterization
The main physicochemical features of the catalysts (specific surface area, P/M atomic ratio, density of acidic sites, and density of basic sites) are summarized in Table 1. Powder X-ray diffraction analyses, as well as NH3 and CO2-TPD profiles, are reported in the electronic supporting information (ESI, Figures S2–S10). The densities of acidic and basic sites reported in Table 1 are expressed as μmol/m2 to allow an easier comparison of the intrinsic acidity and basicity of materials in spite of their very different measured SSA.

Table 1.
Physicochemical features of fresh catalysts.
As expected, all the phosphates displayed higher acidity than their relative oxides in the trend Al > Zr > La without any appreciable basicity, as proved by the absence of any desorption peak of CO2 (a similar profile was observed also in the case of SiO2). On the other hand, the density of acidic sites of these materials (19.3, 13.0, and 10.4 μmol/m2 for Al/P/O, Zr/P/O, and La/P/O, respectively) was two orders of magnitude larger than the one of SiO2 and ≥2-fold the one of Al2O3. It seems that the overall density of acidic sites of metal phosphates increased with an increase of the electronegativity of the metal cation (e.g., Al > Zr > La), but their strength does not follow the same trend. In fact, the material possessing the strongest acid sites (e.g., NH3 desorption temperature > 400 °C) was Zr/P/O and not Al/P/O. The desorption of NH3 at such a high temperature could be related to the structure of Zr/P/O; in fact, the P/Zr atomic ratio = 1.46 measured by means of XRF (in agreement with the one measured by Sushkevich et al. [52]) was intermediate between the value of 1.34 expected from Zr3(PO4)4 and the value of 2 expected from the layered hydrogen phosphate phases α-Zr(HPO4)2·H2O, γ-Zr(H2PO4)(PO4)∙2H2O, and from ZrP2O7. This fact suggests that Zr/P/O was probably a mixture of different polymorphs, possibly containing P as PO43−, as HPO42− and as H2PO43−. These layered hydrogen phosphate phases (e.g., α-Zr(HPO4)2∙H2O) are known for their capacity to bond NH3 stronger in their interlayers than on their surface [56].
On the other hand, among oxides, m-ZrO2 showed the best balance between acidic and basic sites while Al2O3 and La2O3 showed higher acidity and basicity, respectively. Al2O3 is a well-known Lewis acid, as demonstrated by the DRIFTS characterization of adsorbed pyridine reported in the literature [57], while La2O3, according to NH3-TPD results, possesses negligible acidity.
The analysis of the TPD profiles of t-ZrO2-PR, m-ZrO2-PR, and m-ZrO2-HT (shown in Figures S5b, S6b, and S7b, respectively) showed that the density of basic sites for these materials follows the order m-ZrO2-HT (5.0 μmol/m2) > m-ZrO2-PR (3.5 μmol/m2) > t-ZrO2-PR (1.2 μmol/m2), and the CO2 adsorption capacity of the monoclinic phase is higher than the one of tetragonal phase, in agreement with Pokrovski et al. [58].
The ratio between Lewis and Brønsted acidic sites over monoclinic zirconia (m-ZrO2-HT sample) was assessed by means of DRIFTS characterization of adsorbed pyridine in the temperature range 50–450 °C. The results of this characterization are shown in Figure S11; all spectra are characterized by the presence of three strong bands centered at 1603 cm−1, 1575 cm−1, and 1443 cm−1, respectively, which are attributable to the presence of pyridine bonded to Lewis acidic sites [59]. On the other hand, the band attributable to pyridine bonded as pyridinium ion to strong Brønsted acidic sites (1637 cm−1) is absent, and the one of pyridine bonded to weak Bronsted sites (1558 cm−1) is extremely weak. Therefore, it can be concluded that monoclinic zirconia possesses mainly Lewis acidity. Analogous results are reported in the literature for tetragonal zirconia [53].
The DRIFTS spectra of pyridine desorption at increasing temperature, available in the literature for Al/P/O [60], Zr/P/O [61], and La/P/O [62], show that, after thermal pre-treatment, these materials mostly bind pyridine with their Lewis acidic sites, and Brønsted sites, even if present, are a minority. The amount of Brønsted sites increase in the order Al > Zr > La. A more detailed and extensive discussion about the characterization of catalysts can be found in the ESI (Chapter S1). To sum up, the TPD characterization of metal oxides and phosphates, in agreement with the literature, showed that the total density of acidic sites follows the order Al/P/O > Zr/P/O > La/P/O > Al2O3 > ZrO2 > SiO2 > La2O3, while the total density of basic sites follows the order La2O3 > ZrO2 > Al2O3 > SiO2 ≈ Al/P/O ≈ Zr/P/O ≈ La/P/O. In respect to metal oxides, the presence of phosphorus in metal phosphates reduced the basicity to zero regardless of the cation; on the other hand, both the density and the strength of acidic sites increased greatly.
These trends in the acid/base properties of oxides and phosphates can be rationalized to a certain extent considering the percentage of ionic character of the M-O bond involved, which can be estimated from the difference in electronegativity (χ) between oxygen (χO) and the metal (χM), according to the following equation:
Metal oxides are ordered three-dimensional arrays of O2− anions and Mn+ cations; the former act as strong Lewis bases, while the latter act as Lewis acids. O2− anions share more electron density with electronegative metal cations; therefore, the more electronegative the metal cations are, the weaker the nucleophilic/basic character of O2− anions bound to them will be.
The Si-O bond in SiO2 is characterized by a low ionic character (χSi = 1.9, ionic character = 44.7%), and, therefore, O2− anions do not behave as Lewis bases. Moreover, Si4+ do not possess Lewis acidity, and this is why SiO2 displays only a weak Brønsted acidity. On the other hand, the slightly amphoteric character displayed by Al2O3 depends on the higher ionic character of Al-O bond (χAl = 1.61, ionic character = 56.7%) that makes its O2− anions weakly basic and on the strong Lewis acidity of aluminum. The even higher basicity displayed by ZrO2 and La2O3 can be explained as well by the increasing ionic character of the Zr-O bond (χZr = 1.33, ionic character = 67.1%) and of the La-O bond (χLa = 1.1, ionic character = 74.6%), and by the Lewis acidity displayed by these two elements.
In metal phosphates, oxygens are present in the form of PO43− (a well-known weak base) and share their electrons with the highly electronegative P5+ cations (χP = 2.19, ionic character = 32.3%), which possess a Lewis acidity much stronger than Al3+. As a result, the introduction of phosphorus totally suppresses the nucleophilicity/basicity of oxygens in respect to metal oxides. On the other hand, acidity is enhanced because pending PO43− tetrahedra saturate their excess negative charge with H+ ions, therefore the presence of Brønsted acidic sites (P-OH) on metal phosphates surface should be expected. Moreover, in metal phosphates, the same metal cation receives less electronic density from its counter ion (the electron poor oxygens of PO43−) than in metal oxides (where counterions are more electron rich O2− anions). For these reasons, the Lewis acidity of metal cations in phosphates is likely to be larger than the one of the same metal cations in oxides.
3.2. Catalytic Tests
3.2.1. Catalyst Screening: Oxides and Phosphates
As a preliminary study, the effect of the time on stream on the short-term stability (e.g., 6 h) was investigated using Al/P/O, the phosphate with the higher density of acidic sites. Figure 1a shows the outcome of the ketonization of propionic acid (PA) to 3-pentanone (3-P), carried out in the gas phase at 300 °C, with a feed molar percentage of PA in N2 equal to 6%, and a time factor (W/F, calculated by dividing the mass of the catalyst by the total volumetric flow at reaction temperature) equal to 0.8 s·g/mL. Results of this test are expressed in terms of conversion of propionic acid (X PA), products yield (Y), and molar balance (YS/X = sum of yields divided by conversion).
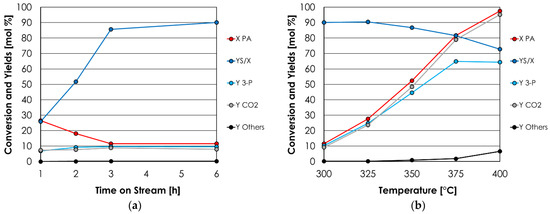
Figure 1.
(a) Catalytic activity of Al/P/O for the ketonization of PA to 3-P as a function of the time on stream. Reaction conditions: temperature = 300 °C, PA = 6 mol % in N2, time factor = W/F = 0.8 s·g/mL. (b) Catalytic activity of Al/P/O for the ketonization of PA to 3-P as a function of reaction temperature. Reaction conditions: temperature = variable, PA = 6 mol % in N2, time factor = W/F = 0.8 s·g/mL. Symbols: propionic acid conversion (X PA, red), sum of yields (YS, blue), 3-pentanone yield (Y 3-P, light blue); CO2 yield (Y CO2, gray); by-products sum of yields (Y others, black).
In these conditions, 3-P and CO2 were the main products, and their yields (light blue line and gray line in Figure 1a) correlated quite well. Al/P/O required about 3 h on stream to reach a steady-state performance, which was maintained during the following 3 h.
Anyway, a similar delay in reaching a steady-state conversion of PA was observed during a blank run (e.g., without catalyst) carried out in the same conditions (Figure S12); therefore, a deactivation of Al/P/O during the first 3 h of reaction was ruled out.
A series of catalytic tests were then carried out in order to determine the optimal reaction temperature for PA ketonization over Al/P/O. Figure 1b shows the results of this temperature screening.
Despite that PA conversion increased with the temperature and was almost complete at 400 °C, the yield of 3-P reached a plateau at 375 °C already and did not exceed 65% with a further increase of the reaction temperature. This trend was accompanied by a progressive worsening of the molar balance, which dropped from 90% (300 °C) to 73% (400 °C). On the other hand, the yield of CO2 was roughly equal to PA conversion in all the temperature ranges investigated and correlated well with the yield of 3-P up to 350 °C. These results suggest that ketonization was the only reaction consuming PA in the temperature range 300–350 °C. However, starting from 375 °C, the resulting 3-P underwent consecutive reactions (e.g., aldol condensations, polymerizations, coking) that reduced its yield and the molar balance, without affecting the yield in CO2. It is worth noticing that the CO2 yield was calculated assuming that it forms only via ketonization (stoichiometric factor ν = 2) and not via the direct decarboxylation of PA (ν = 1); therefore, if the direct decarboxylation of PA had occurred extensively, the calculated yield of CO2 would have exceeded PA conversion. Starting from these results, it was decided to carry out a catalytic screening of the metal oxides and the metal phosphates at 350 °C, which was selected as the optimal temperature to promote ketonization without compromising 3-P selectivity.
The results of this catalyst screening (T = 350 °C, W/F = 0.8 s·g/mL, PA/N2 = 6/94 mol %) are reported as histograms in Figure 2a. However, since PA conversion was complete (with similar yields of 3-P) over both m-ZrO2-PR and La2O3, it was necessary to carry out a different test by charging less catalysts in the reactor, thus reducing the time factor W/F, to avoid a complete conversion of PA that allows to determine which material was the most active. The results of these tests (carried out in the same condition but reducing the W/F from 0.8 to 0.2 s·g/mL) are reported in Figure 2b. The 3-P was the main product of the reaction over all the catalytic systems investigated, and its yield correlated well with the one of the CO2 co-produced during the process (Scheme 1).
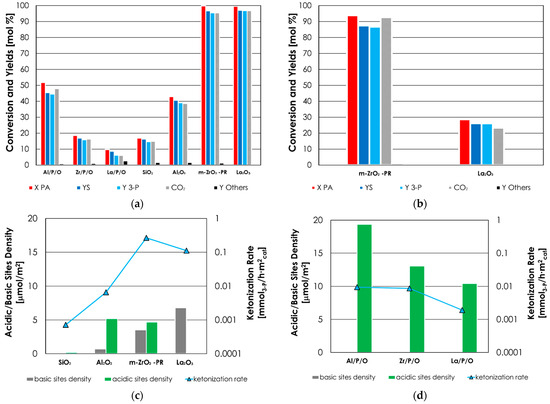
Figure 2.
(a) Catalytic activity of metal oxides and phosphates for the ketonization of PA to 3-P. Reaction conditions: temperature = 350 °C, PA = 6 mol % in N2, time factor = W/F = 0.8 s·g/mL. (b) Catalytic activity of m-ZrO2-PR and La2O3 for the ketonization of PA to 3-P. Reaction conditions: same as (a) but with time factor = W/F = 0.2 s·g/mL. Symbols: propionic acid conversion (X PA, red), sum of yields (YS, blue), 3-pentanone yield (Y 3-P, light blue); CO2 yield (Y CO2, gray); by-products sum of yields (Y Others, black). (c,d) Correlation between the desorbed NH3 (green) and CO2 (gray) and the intrinsic rate of PA ketonization of metal oxides (c) and metal phosphates (d).
The sum of the yields of unwanted by-products (ethylene, propionic anhydride (PAN), and unknown compounds) were always very low (<3%) and represented a significant fraction of the reaction mixture only in the case of La/P/O and SiO2 due to the low PA conversion obtained over these two materials.
The order of activity of metal oxides (inferred from the intrinsic ketonization rates listed in Table 2) was m-ZrO2-PR > La2O3 > Al2O3 > SiO2. This outcome matched quite well the activity orders reported previously in the literature [33,34] and was correlated to the surface acid-base properties of metal oxides (determined by means of CO2- and NH3-TPD), as shown in Figure 2c.
Table 2.
Catalytic activity of metal oxides and metal phosphates for the ketonization of PA to 3-P in the reaction condition of Figure 2a.
m-ZrO2-PR was the best catalyst because of its well-balanced amphoteric character; in fact, it outperformed both Al2O3 and La2O3, although it possesses a lower density of acidic sites in respect to the former and a lower density of basic sites in respect to the latter.
These results represent another strong evidence that the activation of PA via α-proton abstraction, according to the mechanism discussed in Section 1 (see Scheme 2), occurs to a faster rate over catalyst-possessing well-balanced Lewis acid-base pairs due to a cooperative effect between basic and acidic sites.
On the other hand, the comparison between La2O3 and Al2O3 suggests that basicity may play a more important role than Lewis acidity. Finally, SiO2, lacking both Lewis acidity and basicity, was the least active catalyst among oxides.
The correlation between the intrinsic ketonization rates and the acid-base properties of metal phosphates is shown in Figure 2d; in this case, the activity for ketonization increased with an increase in catalyst acid sites density (e.g., La/P/O < Zr/P/O < Al/P/O).
As discussed in Section 3.1, one of the consequences of the presence of phosphorus in a metal phosphate was that the basicity measured by CO2-TPD became negligible in respect to the corresponding oxide. The intrinsic ketonization rates over Zr/P/O and La/P/O were found to be lower than the ones of the corresponding oxides m-ZrO2-PR and La2O3 by more than an order of magnitude, and it is reasonable to ascribe this result to their reduced basicity. This outcome indicates that basicity plays a major role in determining the activity of both m-ZrO2-PR and La2O3.
On the other hand, the same did not apply to the intrinsic rate of ketonization of Al/P/O, which, surprisingly, was higher than the one of Al2O3 in spite of its lower basicity. As mentioned earlier, Al3+ cations in Al/P/O are expected to be more electron-poor than in Al2O3 and to possess, as a consequence, higher Lewis acidity. In fact, oxygen in PO43− anions, being bonded to the highly electronegative P5+ cation, are less prone to delocalize electronic density towards Al3+ in respect to the more electron-rich O2− anions of the corresponding oxide. Since Al2O3 possesses strong Lewis acidity but weak basicity, one can assume that acidity plays a major role with respect to basicity in determining its activity. If that is true, the higher ketonization rate of Al/P/O with respect to Al2O3 can be explained supposing that the positive effect of higher Lewis acidity overcomes the negative effect of lower basicity.
In conclusion, PA ketonization occurred over all the metal phosphates investigated, demonstrating that the presence of basic sites (or more precisely oxygen atoms possessing nucleophilicity/basicity) over catalyst surface, although generally leading to an enhanced ketonization activity, is not absolutely mandatory for the activation of PA via α-proton abstraction. The catalytic activity of metal phosphates was found to increase with increasing acidity (most probably of the Lewis type), and the intrinsic ketonization rate of Al/P/O and Zr/P/O was found to be higher than the one of Al2O3, which is a quite active ketonization catalyst, even if not the best. On the other hand, even the most active and selective metal phosphate (Al/P/O) did not perform as well as ZrO2 and La2O3. In fact, while these two oxides yielded 3-P with >95% selectivity at complete PA conversion already at 350 °C (W/F = 0.8 s·g/mL), Al/P/O converted only 52% of PA with a 3-P selectivity of 87% in the same reaction conditions. Almost complete conversion over Al/P/O was achieved at 400 °C, but in these conditions, the selectivity was as low as 65% due to the occurrence of consecutive reactions that consumed 3-P (Figure 1b).
3.2.2. Zirconium Oxides Screening: Effect of the Synthetic Procedure and Polymorph
At the end of the catalyst screening shown in the previous Section, m-ZrO2-PR was found to be by far the most active and selective catalyst for PA ketonization compared to the oxides and phosphates of aluminum and lanthanum.
On the other hand, zirconium oxide is a versatile material, which may crystallize also with a tetragonal crystal habit, and the sample of monoclinic zirconia used in the screening (e.g., m-ZrO2-PR) possessed a relatively low SSA (36 m2/g), which may be increased by changing the synthetic procedure. Therefore, two other zirconium oxides were synthesized according to different synthetic procedures to obtain materials with different crystal structure (i.e., tetragonal, t-ZrO2-PR, SSA: 123 m2/g) and/or enhanced SSA (i.e., m-ZrO2-HT, SSA: 117 m2/g).
The catalytic activity in PA ketonization of these materials was investigated by feeding an increased concentration of PA in the feed (30 mol % of PA in N2), with a W/F = 0.1 s·g/mL at 350 °C, which are conditions close to those used for industrial operations during 3-P preparation. These catalytic tests were carried out for at least 10 h, and the one with m-ZrO2-HT is shown as a function of the time on stream in ESI, Figure S13, by way of example. Briefly, despite the high concentration of PA in feed, m-ZrO2-HT was stable up to 10 h on stream, the molar balance was remarkably good (always higher than 90%), and 3-P was obtained with ≈95% selectivity at ≈35% PA conversion; the behavior of the other two catalysts (e.g., t-ZrO2-PR and m-ZrO2-HT) was similar in terms of stability and selectivity towards 3-P, but PA conversions were lower.
The comparison between the results of the catalytic tests over t-ZrO2-PR, m-ZrO2-PR, and m-ZrO2-HT (average values over 10 h) is reported in the histogram in Figure 3a. Both m-ZrO2-PR and m-ZrO2-HT were found to be more active than t-ZrO2-PR, despite that the latter was the material possessing the highest SSA; this fact indicates that the monoclinic phase is more active for PA ketonization with respect to the tetragonal phase. Moreover, XRD characterization carried out after the reaction showed that m-ZrO2-PR and m-ZrO2-HT were stable under the reaction condition, while t-ZrO2-PR partially transformed into the monoclinic phase (Figure S14).
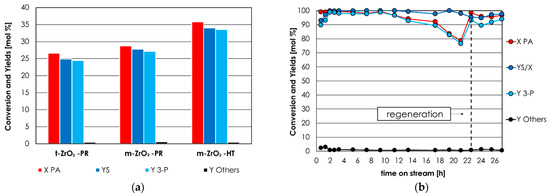
Figure 3.
(a) Propionic acid ketonization over m-ZrO2-HT, m-ZrO2-PR, and t-ZrO2-PR. Reaction conditions: temperature = 350 °C, PA = 30 mol % in N2, time factor = W/F = 0.1 s·g/mL. (b) Propionic acid ketonization over m-ZrO2-HT as a function of the time-on-stream. Reaction conditions: temperature = 400 °C, molar % of propionic acid = 20% in N2, time factor = W/F = 0.1 s·g/mL. Symbols: propionic acid conversion (X PA, red), sum of yields (a) or molar balance (b) (YS and YS/X, respectively, blue), 3-pentanone yield (Y 3-P, light blue); by-products sum of yields (Y Others, black).
The comparison between the activity of m-ZrO2 and t-ZrO2 has been reported by Foraita et al. [63], which, in agreement with our results, claimed that m-ZrO2 is more active than t-ZrO2 because of its superior adsorption capacity for PA and because it forms oxygen vacancies more easily. Moreover, given that ketonization occurs to a faster rate over well-balanced Lewis acid-base pairs, m-ZrO2 is expected to be more active than t-ZrO2 because of CO2- and NH3-TPD characterization, as shown in Section 3.1 (see Table 1).
Figure 3b shows the results of PA ketonization over m-ZrO2-HT as a function of time on stream, obtained by feeding 20 mol % PA with W/F = 0.1 s·g/mL and at 400 °C. These reaction conditions were found to be the optimal compromise between the different needs of an industrial process: (i) a complete conversion of PA is required to minimize reactant recycle; (ii) an excellent selectivity towards 3-P is highly desired to reduce separation and purification costs; (iii) a relatively high concentration of PA in the feed and a contact time as low as possible to increase productivity.
Complete PA conversion and selectivity towards 3-P > 95% were maintained for roughly 11 h, then PA conversion slowly decreased, reaching ≈ 80% after 21 h on stream. Therefore, it should be noted that m-ZrO2-HT lacks the long-term stability required by industrial operations.
This outcome is not surprising because it is a known fact that alkaline metal doping is required to prolong the lifetime of the ZrO2 catalyst in continuous reactors [64], and such doping usually results in a further increase of activity and selectivity [65]. Such a kind of treatment to enhance m-ZrO2-HT long term stability and activity will be investigated in the future.
The opportunity to regenerate m-ZrO2-HT in situ was investigated after 21 h of reaction by interrupting the flow of reactant and by feeding air for 3 h at 450 °C.
As a result of this treatment, m-ZrO2-HT recovered the same activity and selectivity it had at the beginning of the reaction. The results of the catalytic test in Figure 3b (X PA, Y 3-P, and productivity expressed both in terms of mass and in terms of volume) as well as the main reaction conditions (contact time τ, time factor W/F, reaction temperature, and % of PA in the feed) are listed in detail in Table 3 and are compared to the ones of all the catalytic materials reported in the literature, for which we were able to calculate the productivity.
Table 3.
Comparison between m-ZrO2-HT-f (=fresh), m-ZrO2-HT-r (=regenerated), and other catalytic systems reported in patents and academic literature. Symbols: τ = contact time, W/F = time factor, T = reaction temperature, % PA feed = mol % of propionic acid in the feed, X PA = conversion of propionic acid, Y 3-P = yield of 3-pentanone, PM = mass productivity, PV = volume productivity.
To the best of our knowledge, the productivities of 3-P achieved with our m-ZrO2-HT catalyst are the highest reported (5.6 h−1).
4. Conclusions
For the first time, PA ketonization was investigated in the gas phase over metal phosphates of Al, Zr, and La, and their activity were compared to the one shown by related oxides. These materials possess a wide range of different acid-based features, and the correlation between their activity and their physicochemical properties (characterized by means of XRD, XRF, BET N2 porosimetry, and CO2- and NH3-TPD) allowed us to draw a structure–activity relationship and substantially confirm the previous claims of other authors about the superior activity of catalysts possessing well-balanced Lewis acid-base pairs.
In fact, the experimental order of activity inferred from intrinsic ketonization rate was ZrO2 > La2O3 > Al/P/O > Zr/P/O > Al2O3 > La/P/O > SiO2, and ketonization occurred at a faster rate over the catalyst possessing well-balanced acidic and basic sites (ZrO2). The DRIFTS characterization of pyridine adsorbed on m-ZrO2-HT carried out in this study, together with the literature cited, strongly suggested that the most active sites are Lewis acid-base pairs, such as coordinatively unsaturated cations neighboring oxygen vacancies. For non-amphoteric materials, basic sites (La2O3) were more active than acidic sites (Al2O3) in promoting ketonization. SiO2 was the least active catalyst due to its lack of Lewis acidity and basicity.
On the other hand, despite the absence of basic sites, metal phosphates showed a peculiar catalytic activity, which increased with the increasing of acid sites density. In particular, Al/P/O and was found to be more active than γ-Al2O3. The results obtained over metal phosphates indicated that the absence of basic sites was not essential for ketonization to occur.
The best catalyst, zirconium oxide, was further investigated to evaluate the effect of different polymorphs (e.g., tetragonal and monoclinic) and higher surface areas. It was found that both m-ZrO2-PR (36 m2/g) and m-ZrO2-HT (117 m2/g) were more active than t-ZrO2-PR (123 m2/g); therefore, it was concluded that the monoclinic phase is intrinsically more active than the tetragonal, as suggested by the TPD characterization. Moreover, the tetragonal phase showed an intrinsic instability, partially transforming into the monoclinic phase during the reaction.
The higher activity and basicity of m-ZrO2-HT in respect to m-ZrO2-PR was likely to be related to the presence of a higher number of surface defects on the former material, as suggested by the coordination number of the surficial ions and its higher SSA.
The most active zirconium oxide catalyst (m-ZrO2-HT) was investigated for PA ketonization in industrially relevant conditions (W/F = 0.1 s·g/mL, 400 °C, and feeding 20 mol % PA in N2).
Despite that PA complete conversion and 3-P selectivity >95% were achieved, this catalyst was not capable of maintaining these performances for more than 11 h, and its activity decreased significantly after 21 h of reaction. However, after regeneration in situ with air at 450 °C for 3 h, the catalyst recovered most of its initial activity and selectivity. The calculated values of productivity for m-ZrO2-HT in these optimized conditions (superior to all the other reported previously in the literature) and the possibility to regenerate the catalyst with a minimum loss in activity and selectivity, make this material a promising candidate for the preliminary upgrading of bio-oils by means of ketonization, although further development will be needed to improve its long-term stability.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/suschem3010005/s1, Chapter S1: Detailed characterization of the catalytic materials; Figure S1: gas-phase rig and analytical systems used to carry out experiments; Figures S2–S10: catalytic material XRD patterns and NH3- and CO2-TPD profiles; Figure S11: DRIFTS spectra of pyridine adsorbed at various temperature over m-ZrO2-HT; Figure S12: Blank run as a function of time on stream; Figure S13: Catalytic activity of m-ZrO2-HT for the ketonization of PA to 3-P as a function of the time on stream. Reaction conditions: temperature = 350 °C, PA = 30 mol % in N2, time factor = W/F = 0.1 s·g/mL; Figure S14: Powder XRD pattern of t-ZrO2-PR before and after the reaction. [66,67,68,69,70,71,72,73,74,75,76,77,78] are cited in the supplementary materials.
Author Contributions
Conceptualization, T.T. and F.C.; methodology, J.D.M.; catalyst synthesis, L.B. and A.B.; catalyst characterization, J.D.M., N.S., and A.F., writing—original draft preparation, J.D.M. and T.T.; writing—review and editing, C.L., A.F., and F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data concerning this study are reported in this article or in the supporting informations.
Acknowledgments
The INSTM (Consorzio Interuniversitario per la Scienza e Tecnologia dei Materiali), Florence, is acknowledged for the post-doc research grant to J.D.M.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, R.E.; Mabee, W.; Saddler, J.N.; Taylor, M. An overview of second generation biofuel technologies. Bioresour. Technol. 2010, 101, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Dale, B.E. Biofuels, Land Use Change, and Greenhouse Gas Emissions: Some Unexplored Variables. Environ. Sci. Technol. 2009, 43, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of Wood/Biomass for Bio-oil: A Critical Review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Czernik, S.; Bridgewater, A. Overview of applications of biomass fast pyrolysis oil. Energy Fuels 2004, 18, 590–598. [Google Scholar] [CrossRef]
- Bridgewater, A. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Xiu, S.; Shahbazi, A. Bio-oil production and upgrading research: A review. Renew. Sustain. Energy Rev. 2012, 16, 4406–4414. [Google Scholar] [CrossRef]
- Elliot, D. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar] [CrossRef]
- Wildschut, J.; Mahfud, F.H.; Venderbosch, R.; Heeres, H.J. Hydrotreatment of Fast Pyrolysis Oil Using Heterogeneous Noble-Metal Catalysts. Ind. Eng. Chem. Res. 2009, 48, 10324–10334. [Google Scholar] [CrossRef]
- Pham, T.N.; Sooknoi, T.; Crossley, S.P.; Resasco, D.E. Ketonization of Carboxylic Acids: Mechanisms, Catalysts, and Implications for Biomass Conversion. ACS Catal. 2013, 3, 2456–2473. [Google Scholar] [CrossRef]
- Tabanelli, T. Unrevealing the hidden link between sustainable alkylation and hydrogen transfer processes with alcohols. Curr. Opin. Green Sustain. Chem. 2021, 29, 100449. [Google Scholar] [CrossRef]
- Bridgewater, A. Production of high grade fuels and chemicals from catalytic pyrolysis of biomass. Catal. Today 1996, 29, 285–295. [Google Scholar] [CrossRef]
- Wan, S.; Pham, T.; Zhang, S.; Lobban, L.; Resasco, D.; Mallinson, R. Direct catalytic upgrading of biomass pyrolysis vapors by a dual function Ru/TiO2 catalyst. AIChE J. 2013, 59, 2275–2285. [Google Scholar] [CrossRef]
- Aguado, R.; Olazar, M.; José, M.J.S.; Aguirre, A.G.; Bilbao, J. Pyrolysis of Sawdust in a Conical Spouted Bed Reactor. Yields and Product Composition. Ind. Eng. Chem. Res. 2000, 39, 1925–1933. [Google Scholar] [CrossRef]
- Zapata, P.A.; Faria, J.; Ruiz, M.P.; Resasco, D.E. Condensation/Hydrogenation of Biomass-Derived Oxygenates in Water/Oil Emulsions Stabilized by Nanohybrid Catalysts. Top. Catal. 2012, 55, 38–52. [Google Scholar] [CrossRef]
- Nie, L.; Resasco, D.E. Improving carbon retention in biomass conversion by alkylation of phenolics with small oxygenates. Appl. Catal. A Gen. 2012, 447–448, 14–21. [Google Scholar] [CrossRef]
- Jia, N.; Zhao, H.; Yang, T.; Ibatullin, T.; Gao, J. Experimental Measurements of Bitumen–Water Aquathermolysis during a Steam-Injection Process. Energy Fuels 2016, 30, 5291–5299. [Google Scholar] [CrossRef]
- Vakhin, A.V.; Aliev, F.A.; Mukhamatdinov, I.I.; Sitnov, S.A.; Kudryashov, S.I.; Afanasiev, I.S.; Petrashov, O.V.; Nurgaliev, D.K. Extra-Heavy Oil Aquathermolysis Using Nickel-Based Catalyst: Some Aspects of In-Situ Transformation of Catalyst Precursor. Catalysts 2021, 11, 189. [Google Scholar] [CrossRef]
- Aliev, F.A.; Mukhamatdinov, I.I.; Sitnov, S.A.; Ziganshina, M.R.; Onishchenko, Y.V.; Sharifullin, A.V.; Vakhin, A.V. In-Situ Heavy Oil Aquathermolysis in the Presence of Nanodispersed Catalysts Based on Transition Metals. Processes 2021, 9, 127. [Google Scholar] [CrossRef]
- De Maron, J.; Eberle, M.; Cavani, F.; Basile, F.; Dimitratos, N.; Maireles-Torres, P.J.; Rodriguez-Castellón, E.; Tabanelli, T. Continuous-Flow Methyl Methacrylate Synthesis over Gallium-Based Bifunctional Catalysts. ACS Sustain. Chem. Eng. 2021, 9, 1790–1803. [Google Scholar] [CrossRef]
- Pestman, R.; Koster, R.; Van Duijne, A.; Pieterse, J.; Ponec, V. Reactions of Carboxylic Acids on Oxides: 2. Bimolecular Reaction of Aliphatic Acids to Ketones. J. Catal. 1997, 168, 265–272. [Google Scholar] [CrossRef]
- Yakerson, V.I. Mechanism of thermal decomposition of salts of carboxylic acids. Bull. Acad. Sci. USSR Div. Chem. Sci. 1963, 12, 914–921. [Google Scholar] [CrossRef]
- Yamada, Y.; Segawa, M.; Sato, F.; Kojima, T.; Sato, S. Catalytic performance of rare earth oxides in ketonization of acetic acid. J. Mol. Catal. A Chem. 2011, 346, 79–86. [Google Scholar] [CrossRef]
- Nagashima, O.; Sato, S.; Takahashi, R.; Sodesawa, T. Ketonization of carboxylic acids over CeO2-based composite oxides. J. Mol. Catal. A Chem. 2005, 227, 231–239. [Google Scholar] [CrossRef]
- Pham, T.; Shi, D.; Sooknoi, T.; Resasco, D. Aqueous-phase ketonization of acetic acid over Ru/TiO2/carbon catalysts. J. Catal. 2012, 295, 169–178. [Google Scholar] [CrossRef]
- Pulido, A.; Oliver-Tomas, B.; Renz, M.; Boronat, M.; Corma, A. Ketonic Decarboxylation Reaction Mechanism: A Combined Experimental and DFT Study. ChemSusChem 2013, 6, 141–151. [Google Scholar] [CrossRef]
- Ignatchenko, A. Density Functional Theory Study of Carboxylic Acids Adsorption and Enolization on Monoclinic Zirconia Surfaces. J. Phys. Chem. C 2011, 115, 16012–16018. [Google Scholar] [CrossRef]
- Renz, M. Ketonization of Carboxylic Acids by Decarboxylation: Mechanism and Scope. Eur. J. Org. Chem. 2005, 2005, 979–988. [Google Scholar] [CrossRef]
- Gonzales, F.; Munuera, G.; Prieto, J. Mechanism of ketonization of acetic acid on anataseTiO2 surfaces. J. Chem. Soc. Faraday Trans. 1978, 74, 1547. [Google Scholar]
- Neunhoeffer, O.; Paschke, P. Über den Mechanismus der Ketonbildung aus Carbonsäuren. Chem. Ber. 1939, 72, 919. [Google Scholar] [CrossRef]
- Bamberger, E. Notiz über das Verhalten von Essigsäureanhydrid bei hoher Temperatur. Chem. Ber. 1910, 43, 3517. [Google Scholar] [CrossRef]
- Boekaerts, B.; Sels, B. Catalytic advancements in carboxylic acid ketonization and its perspectives on biomass valorization. App. Catal. B 2021, 283, 119607. [Google Scholar] [CrossRef]
- Gliński, M.; Kijeński, J.; Jakubowski, A. Ketones from monocarboxylic acids: Catalytic ketonization over oxide systems. Appl. Catal. A Gen. 1995, 128, 209–217. [Google Scholar] [CrossRef]
- Gliński, M.; Zalewski, G.; Burno, E.; Jerzak, A. Catalytic ketonization over metal oxide catalysts. XIII. Comparative measurements of activity of oxides of 32 chemical elements in ketonization of propanoic acid. Appl. Catal. A Gen. 2014, 470, 278–284. [Google Scholar] [CrossRef]
- Schommer, C.; Ebel, K.; Dockner, T.; Irgang, M.; Hoelderich, W.; Rust, H. Preparation of Ketones. U.S. Patent 4,950,763, 21 August 1990. [Google Scholar]
- Beavers, W.; Ignatchenko, A.; Liu, Z.; Ashcroft, C.; White, T. Catalyst and Process for the Preparation of Unsymmetrical Ketones. U.S. Patent 2007/0100166 A1, 3 May 2007. [Google Scholar]
- Hussmann, G. Preparation of Ketones from Aliphatic Carboxylic Acids. U.S. Patent 4,754,074, 28 June 1988. [Google Scholar]
- Jahangiri, H.; Osatiashtiani, A.; Bennett, J.A.; Isaacs, M.A.; Gu, S.; Lee, A.F.; Wilson, K. Zirconia catalysed acetic acid ketonisation for pre-treatment of biomass fast pyrolysis vapours. Catal. Sci. Technol. 2018, 8, 1134–1141. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Iglesia, E. Experimental and Theoretical Evidence for the Reactivity of Bound Intermediates in Ketonization of Carboxylic Acids and Consequences of Acid–Base Properties of Oxide Catalysts. J. Phys. Chem. C 2017, 121, 18030–18046. [Google Scholar] [CrossRef]
- Minachev, K.H.; Atat’yan, O.K.; Markov, M.A. Conversion of butyraldehyde on the oxides of rare-earth elements. Russ. Chem. Bull. 1978, 27, 2437–2442. [Google Scholar] [CrossRef]
- Ding, S.; Wang, H.; Han, J.; Zhu, X.; Ge, Q. Ketonization of Propionic Acid to 3-Pentanone over CexZr1–xO2 Catalysts: The Importance of Acid–Base Balance. Ind. Eng. Chem. Res. 2018, 57, 17086–17096. [Google Scholar] [CrossRef]
- Dou, H.; Roman-Leshkov, Y. Highly Active Oxide Catalyst for the Catalytic Ketonization of Carboxylic Acids. U.S. Patent 8,748,670 B1, 10 June 2014. [Google Scholar]
- Murkute, A.D.; Jackson, J.; Miller, D.J. Supported mesoporous solid base catalysts for condensation of carboxylic acids. J. Catal. 2011, 278, 189–199. [Google Scholar] [CrossRef]
- Lu, F.; Jiang, B.; Wang, J.; Huang, Z.; Liao, Z.; Yang, Y. Insights into the improvement effect of Fe doping into the CeO2 catalyst for vapor phase ketonization of carboxylic acids. Mol. Catal. 2018, 444, 22–33. [Google Scholar] [CrossRef]
- Stonkus, W.; Yuskovets, J.; Leite, L.; Fleisher, M.; Edolfa, K.; Liepina, I.; Mishnev, A.; Shmidlers, A. Vapor-phase ketonization of aliphatic acids on a chromite catalyst. Russ. J. Gen. Chem. 2011, 81, 1523–1528. [Google Scholar] [CrossRef]
- Jiang, B.; Xi, Z.; Lu, F.; Huang, Z.; Yang, Y.; Sun, J.; Liao, Z.; Wang, J.; Yang, Y. Ce/MgAl mixed oxides derived from hydrotalcite LDH precursors as highly efficient catalysts for ketonization of carboxylic acid. Catal. Sci. Technol. 2019, 9, 6335–6344. [Google Scholar] [CrossRef]
- Fufachev, E.V.; Weckhuysen, B.M.; Bruijnincx, P.C. Toward Catalytic Ketonization of Volatile Fatty Acids Extracted from Fermented Wastewater by Adsorption. ACS Sustain. Chem. Eng. 2020, 8, 11292–11298. [Google Scholar] [CrossRef]
- Kuriacose, J.; Swaminathan, R. Studies on the ketonization of acetic acid on chromia: 1. The Adsorbate-catalyst interaction. J. Catal. 1969, 14, 348–354. [Google Scholar] [CrossRef]
- Panchenko, V.; Zaytseva, Y.; Simonov, M.; Simakova, I.; Paukshtis, E. DRIFTS and UV–vis DRS study of valeric acid ketonization mechanismover ZrO2 in hydrogen atmosphere. J. Mol. Catal. A Chem. 2014, 388–389, 133–140. [Google Scholar] [CrossRef]
- Pestman, R.; Koster, R.; Pieterse, J.; Ponec, V. Reactions of Carboxylic Acids on Oxides: 1. Selective Hydrogenation of Acetic Acid to Acetaldehyde. J. Catal. 1997, 168, 255–264. [Google Scholar] [CrossRef]
- Zaytseva, Y.A.; Panchenko, V.N.; Simonov, M.N.; Shutilov, A.A.; Zenkovets, G.A.; Renz, M.; Simakova, I.; Parmon, V.N. Effect of Gas Atmosphere on Catalytic Behaviour of Zirconia, Ceria and Ceria–Zirconia Catalysts in Valeric Acid Ketonization. Top. Catal. 2013, 56, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Sushkevich, V.; Ordomsky, V.; Ivanova, I. Synthesis of isoprene from formaldehyde and isobutene over phosphate catalysts. Appl. Catal. A Gen. 2012, 441–442, 21–29. [Google Scholar] [CrossRef]
- Vásquez, P.B.; Tabanelli, T.; Monti, E.; Albonetti, S.; Bonincontro, D.; Dimitratos, N.; Cavani, F. Gas-Phase Catalytic Transfer Hydrogenation of Methyl Levulinate with Ethanol over ZrO2. ACS Sustain. Chem. Eng. 2019, 7, 8317–8330. [Google Scholar] [CrossRef]
- Tabanelli, T.; Paone, E.; Vásquez, P.B.; Pietropaolo, R.; Cavani, F.; Mauriello, F. Transfer Hydrogenation of Methyl and Ethyl Levulinate Promoted by a ZrO2 Catalyst: Comparison of Batch vs. Continuous Gas-Flow Conditions. ACS Sustain. Chem. Eng. 2019, 7, 9937–9947. [Google Scholar] [CrossRef]
- Li, W.-Z.; Huang, H.; Li, H.; Zhang, W.; Liu, H. Facile Synthesis of Pure Monoclinic and Tetragonal Zirconia Nanoparticles and Their Phase Effects on the Behavior of Supported Molybdena Catalysts for Methanol-Selective Oxidation. Langmuir 2008, 24, 8358–8366. [Google Scholar] [CrossRef] [PubMed]
- Turco, M.; Ciambelli, P.; Bagnasco, G.; La Ginestra, A.; Galli, P.; Ferragina, C. TPD study of NH3 adsorbed by different phases of zirconium phosphate. J. Catal. 1989, 117, 355–361. [Google Scholar] [CrossRef]
- Kumar, V.; Naresh, G.; Sudhakar, M.; Anjaneyulu, C.; Bhargava, S.; Tradio, J.; Reddy, V.; Padmasri, A.; Venugopal, A. An investigation on the influence of support type for Ni catalysed vapour phase hydrogenation of aqueous levulinic acid to g-valerolactone. RSC Adv. 2016, 6, 9872–9879. [Google Scholar] [CrossRef]
- Pokrovski, K.; Jung, K.T.; Bell, A. Investigation of CO and CO2 Adsorption on Tetragonal and Monoclinic Zirconia. Langmuir 2001, 17, 4297–4303. [Google Scholar] [CrossRef]
- Busca, G. The surface acidity of solid oxides and its characterization by IR spectroscopic methods. An attempt at systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Estevez, R.; Lopez-Pedrajas, S.; Blanco-Bonilla, F.; Luna, D.; Bautista, F. Production of acrolein from glycerol in liquid phase on heterogeneous catalysts. Chem. Eng. J. 2015, 282, 179–186. [Google Scholar] [CrossRef]
- Antonetti, C.; Melloni, M.; Licursi, D.; Fulignati, S.; Ribechini, E.; Rivas, S.; Parajo, J.; Cavani, F.; Raspolli, A.G. Microwave-assisted dehydration of fructose and inulin to HMFcatalyzed by niobium and zirconium phosphate catalysts. Appl. Cat. B 2017, 206, 364–377. [Google Scholar] [CrossRef]
- Guo, Z.; Theng, D.S.; Tang, K.Y.; Zhang, L.; Huang, L.; Borgna, A.; Wang, C. Dehydration of lactic acid to acrylic acid over lanthanum phosphate catalysts: The role of Lewis acid sites. Phys. Chem. Chem. Phys. 2016, 18, 23746–23754. [Google Scholar] [CrossRef]
- Foraita, S.; Fulton, J.L.; Chase, Z.A.; Vjunov, A.; Xu, P.; Baráth, E.; Camaioni, D.M.; Zhao, C.; Lercher, J.A. Impact of the Oxygen Defects and the Hydrogen Concentration on the Surface of Tetragonal and Monoclinic ZrO2 on the Reduction Rates of Stearic Acid on Ni/ZrO2. Chem. A Eur. J. 2014, 20, 2423–2434. [Google Scholar] [CrossRef]
- Ignatchenko, A.V. Multiscale approach for the optimization of ketones production from carboxylic acids by the decarboxylative ketonization reaction. Catal. Today 2019, 338, 3–17. [Google Scholar] [CrossRef]
- Parida, K.; Mishra, H.K. Catalytic ketonisation of acetic acid over modified zirconia: 1. Effect of alkali-metal cations as promoter. J. Mol. Catal. A Chem. 1999, 139, 73–80. [Google Scholar] [CrossRef]
- Innocenti, G.; Papadopoulos, E.; Fornasari, G.; Cavani, F.; Mefford, A.; Sievers, C. Continuous liquid-phase upgrading of dihydroxyacetone to lactic acid over metal phosphate catalysts. ACS Catal. 2020, 10, 11936–11950. [Google Scholar] [CrossRef]
- Parshetti, G.; Suryadharma, M.; Pham, T.; Mahmood, R.; Balasubramanian, R. Heterogeneous catalyst-assisted thermochemical conversion of food waste biomass into 5-hydroxymethylfurfural. Bioresour. Technol. 2015, 178, 19–27. [Google Scholar] [CrossRef]
- Ochoa, J.V.; Bandinelli, C.; Vozniuk, O.; Chieregato, A.; Malmusi, A.; Recchi, C.; Cavani, F. An analysis of the chemical, physical and reactivity features of MgO–SiO2 catalysts for butadienesynthesis with the Lebedev process. Green Chem. 2016, 18, 1653–1663. [Google Scholar] [CrossRef]
- Tsyganenko, A.; Storozheva, E.; Manoilova, O.; Lesage, T.; Daturi, M.; Lavalley, J. Brønsted acidity of silica silanol groups induced by adsorption of acids. Catal. Lett. 2020, 70, 159–163. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Yabushita, M.; Kim, M.; Hirajama, J.; Motokura, K.; Fukuoka, A.; Nakajima, K. Catalytic conversion of biomass-derived carbohydrates to methyl lactate by acid-base bifunctional γ-Al2O3. ACS Sustain. Chem. Eng. 2018, 6, 8113–8117. [Google Scholar] [CrossRef]
- di Cosimo, J.; Torres, G.; Apesteguia, C. One-step MIBK synthesis: A new process from 2-propanol. J. Catal. 2002, 208, 114–123. [Google Scholar] [CrossRef]
- Santacroce, V.; Bigi, F.; Casnati, A.; Maggi, R.; Storaro, L.; Moretti, E.; Vaccaro, L.; Maestri, G. Selective monomethyl esterification of linear dicarboxylic acids with bifunctional alumina catalysts. Green Chem. 2016, 18, 5764–5768. [Google Scholar] [CrossRef]
- Mu, Q.; Wang, Y. “Synthesis, characterization, shape-preserved transformation, and optical properties of La(OH)3, La2O2CO3, and La2O3 nanorods. J. Alloys Compd. 2011, 509, 396–401. [Google Scholar] [CrossRef]
- Zhang, J.; He, D. Surface properties of Cu/La2O3 and its catalytic performance in the synthesis of glycerol carbonate and monoacetin from glycerol and carbon dioxide. J. Colloid Interface Sci. 2014, 419, 31–38. [Google Scholar] [CrossRef]
- Costa, C.; Anastasiadou, T.; Efstathiou, A. The selective catalytic reduction of nitric oxide with methane over La2O3–CaO systems: Synergistic effects and surface reactivity Studies of NO, CH4, O2, and CO2 by transient techniques. J. Catal. 2000, 194, 250–265. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, N.; Wei, W.; Sun, Y. Catalytic performance of metal oxides for the synthesis of propylene carbonate from urea and 1,2-propanediol. J. Mol. Catal. A: Chem. 2007, 270, 44–49. [Google Scholar] [CrossRef]
- Bachiller-Baeza, B.; Rodriguez-Ramos, I.; Guerrero-Ruiz, A. interaction of carbon dioxide with the surface of zirco-nia polymorphs. Langmuir 1998, 14, 3556–3564. [Google Scholar] [CrossRef]
- Ding, S.; Zhao, J.; Yu, Q. Effect of the zirconia polymorph on vapor-phase ketonization of propionic acid. Catalysts 2019, 9, 768. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).