Abstract
The direct chlorination, bromination and azidation of beta keto esters, 2-acetyl-1-tetralone and methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate is achieved utilizing anion-coordinated hypervalent iodine benziodazoles derived from hypervalent iodine macrocycles. This reaction, which introduces the halogen, azido or cyano group at the alpha carbon atom of beta keto esters, is accomplished in chloroform at 60 °C and results in the formation of a chiral center. Depending on the structure of the benziodazole reagent, the reaction can have mild enantioselectivity. The reaction between 2-acetyl-1-tetralone and phenylalanine-derived hypervalent iodine benziodazoles results in the chlorinated product with 26% enantiomeric excess.
1. Introduction
Azide, cyano and halogen groups are important functionalities in organic chemistry. For example, the insertion of cyano groups in different organic compounds is pivotal in modern synthetic chemistry as it stands as a significant functional group in several natural products [1], pharmaceuticals [2] and materials [3]. Organoazide compounds have received considerable attention from the scientific community owing to their use in industrial-scale synthesis of heterocycles such as triazole and tetrazole [4]. Additionally, organic azides serve as the energy-rich molecules and flexible intermediates in organic chemistry [5]. Similarly, many pharmaceutical drugs consist of halogenated organic structures [6]. Halogenated organic compounds have emerged as structures of great interest owing to their applications as dyes, flame retardants, imaging agents in medical diagnosis and in materials science [7]. Traditional synthetic methods to prepare cyano, azido and halo compounds often rely on the use of hazardous chemical reagents like sodium cyanide, sodium azide and H-X (X = F, Cl, Br). These reactions not only pose safety risks in the laboratory but also cause harmful effects on the environment upon disposal. Therefore, there is a growing need to find alternative strategies as safer, sustainable and cost-effective approaches for the large-scale production of these functionalized compounds.
In general, nucleophilic substitution of RX (X = leaving group) with inorganic cyanide, azide and halide has been widely used in research laboratories as well as in industry to synthesize organic cyanide, organic azide and organic halide, respectively. In addition, significant achievements have been made in recent decades towards nucleophilic addition of cyanides to electrophiles for the synthesis of cyano compounds; the reaction of the corresponding aliphatic amine with triflyl azide (TfN3) to synthesize organic azides [8]; and reactions involving N-bromosuccinimide (NBS), N-fluorobenzenesulfonimide (NFSI) and diethylaminosulfur trifluoride (DAST) as halogenating agents to synthesize organic halides [7]. As an alternative, cyanation, azidation and halogenation of α-functionalization of carbonyl compounds can also lead to the synthesis of large numbers of interesting synthetic building blocks and molecules [9]. Several groups have reported the direct halogenation, cyanation and azidation of beta keto esters using hypervalent iodine reagents in an enantioselective manner [9,10]. However, very little is known about the benziadazole-based tetracoordinated hypervalent iodine species featuring an I-X bond (X = F, Cl, Br, CN, N3) for the direct cyanation, halogenation and azidation of beta keto esters. Herein, we have demonstrated direct azidation and halogenation with a new chiral hypervalent iodine reagent in 2-acetyl-1-tetralone and methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate systems.
2. Materials and Methods
Unless otherwise noted, all reagents were used as received and all reactions were carried out under an argon atmosphere. Column chromatography was performed on a CombiFlash® Rf system with Redisep normal-phase silica columns (Teledyne ISCO Inc., Lincoln, NE, USA). 1H NMR and 13C NMR were recorded on a Varian 400 MHz or JEOL 500 MHz NMR workstation at room temperature, unless otherwise noted.
2.1. General Procedure for the Chlorination, Azidation and Bromination of 2-Acetyl-1-Tetralone
In a 20 mL round-bottom flask, 2-acetyl-1-tetralone (0.23 mmol, 1 eq.) was dissolved in 5 mL CHCl3. A freshly prepared adduct solution (2a-c, 3a-c) was prepared from mixing precursor 1a or 1b (0.20 mmol–0.6 mmol of monomer, 2.6 eq.) and tetrabutylammonium salt (0.60 mmol, 2.6 eq.) in 5 mL CHCl3. The adduct solutions were added at room temperature to the round-bottom flask. The reaction mixture was sonicated to make the solution homogenous and then stirred at 60 °C for 4-48 h. After completion of the reaction, the solvent was evaporated to give a solid that was purified using column chromatography with 90:10 hexane/ethyl acetate to give 6a-c.
1-(2-chloro-1-hydroxy-1,2,3,4-tetrahydronapthalen-2-yl) ethenone (6a): 2-Acetyl 1-tetralone (43.3 mg, 0.23 mmol), 1a (234 mg, 0.20 mmol) and tetrabutyl ammonium chloride (99 mg, 0.60 mmol) gave 6a (29 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 7.8 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 7.3 Hz, 1H), 3.33–3.20 (m, 1H), 3.09–3.00 (m, 1H), 2.95–2.87 (m, 1H), 2.52 (s, 3H) and 2.44–2.37 (m, 1H). 13C NMR (125 MHz, CDCl3) 201.9, 189.9, 142.9, 134.6, 130.1, 128.9, 128.8, 127.3, 73.9, 33.5, 27.7 and 25.7. HRMS(ES+): m/z for C12H12O2Cl. Calc: 223.0526, found: 223.0527.
1-(2-azido-1-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl) ethenone (6b) 2-Acetyl 1-tetralone (43.3 mg, 0.23 mmol), 1a (234 mg, 0.20 mmol) and tetrabutyl ammonium azide (170 mg, 0.60 mmol) gave 6b (32 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 7.9 Hz, 1H), 7.56 (t, J = 7.5 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.27 (d, J = 8.5 Hz, 1H), 3.22–3.10 (m, 1H), 3.00–2.90 (m, 1H), 2.59–2.51 (m, 1H), 2.30 (s, 3H) and 2.21–2.15 (m, 1H). 13C NMR (125 MHz, CDCl3) 202.4, 191.0, 143.3, 134.8, 130.4, 128.8, 128.4, 127.3, 75.5, 30.5, 26.5 and 24.9. HRMS(ES+): m/z for C12H12O2N [M-N2+H] + Calc: 202.0868, found: 208.0873.
1-(2-Bromo-1-hydroxy-1,2,3,4-tetrahydro-naphthalen-2-yl)-ethanone (6c) 2-Acetyl 1-tetralone (43.3 mg, 0.23 mmol), 1a (234 mg, 0.10 mmol) and tetrabutyl ammonium bromide (194 mg, 0.60 mmol) gave 6c (25 mg, 40%). 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 7.9 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 3.27–3.15 (m, 1H), 3.10–3.02 (m, 1H), 2.89–2.80 (m, 1H), 2.61 (s, 3H) and 2.52–2.44 (m, 1H). 13C NMR (125 MHz, CDCl3) 200.7, 189.7, 142.7, 134.4, 129.9, 128.8, 128.8, 127.2, 68.3, 33.8, 28.3 and 26.8. HRMS(ES+): m/z for C12H12O2Br [M+H] + Calc: 267.0021, found: 267.0021.
2-acetyl-1-oxo-1,2,3,4-tetrahydronaphthalene-2-carbonitrile (6d) 2-Acetyl 1-tetralone (43.3 mg, 0.23 mmol), 1a (234 mg, 0.20 mmol) and tetrabutyl ammonium cyanide (161 mg, 0.60 mmol) gave 6d (15 mg, 30%). 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 7.8 Hz, 1H), 7.59 (td, J = 7.5, 1.4 Hz, 1H), 7.40 (d, J = 7.6 Hz, 1H), 7.38–7.33 (m, 1H), 3.40 (ddd, J = 17.5, 13.3, 4.4 Hz, 1H), 3.22 (ddd, J = 17.2, 4.9, 2.7 Hz, 1H), 2.84 (td, J = 13.5, 4.9 Hz, 1H), 2.36 (ddd, J = 13.9, 4.5, 2.6 Hz, 1H) and 1.65 (s, 4H). 13C NMR (125 MHz, CDCl3) 189.3, 143.0, 135.0, 132.5, 129.1, 127.9, 127.3, 117.0, 67.0, 54.4, 30.3, 29.7 and 28.0. HRMS(EI+): m/z for C13H11O2N [M] + Calc: 213.0789, found: 213.0787.
2.2. General Procedure for the Chlorination, Azidation and Bromination of the Methyl 1-Oxo-2,3-Dihydro-1H-Indene-2-Carboxylate
Methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate was prepared following a previously reported procedure [10]. Methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate (0.16 mmol, 1 eq) was dissolved in 5 mL of chloroform (CHCl3) in a 20 mL round-bottom flask. Separately, freshly prepared adduct solution 3a-c was obtained by mixing precursor 1a or 1b (0.14 mmol, 2.6 eq) with a tetrabutylammonium salt (0.86 mmol) in 5 mL of CHCl3. The resulting adduct solution was then transferred to the round-bottom flask at room temperature. The reaction mixture was sonicated to ensure homogeneity and subsequently stirred at 60 °C for 4-48 h. After completion of the reaction, the solvent was evaporated under reduced pressure to yield a crude solid. Purification by column chromatography used a 90:10 hexane/ethyl acetate.
Methyl 2-chloro-1-oxo-2,3-dihydro-1H-indene-2-carboxylate 7a 1-oxo-2,3-dihydro-1H-indene-2-carboxylate (31.2 mg, 0.16 mmol), 1b (165 mg, 0.14 mmol) and tetrabuyl ammonium chloride (240 mg, 0.86 mmol) gave 7a (28 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 7.3 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 6.7 Hz, 2H), 4.11 (d, J = 17.1 Hz, 1H), 3.82 (s, 3H) and 3.57 (d, J = 17.9 Hz, 1H). 13C NMR (125 MHz, CDCl3) 195.1; matched previous report [10].
Methyl 2-azido-1-oxo-2,3-dihydro-1H-indene-2-carboxylate 7b 1-oxo-2,3-dihydro-1H-indene-2-carboxylate (31.2 mg, 0.16 mmol), 1b (149 mg, 0.14 mmol) and tetrabutyl ammonium azide (246 mg, 0.86 mmol) gave 7b (33 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 7.6 Hz, 1H), 7.69 (t, J = 7.2 Hz, 1H), 7.46 (t, J = 7.7 Hz, 2H), 3.81 (s, 3H), 3.68 (d, J = 17.3 Hz, 1H) and 3.05 (d, J = 17.4 Hz, 1H). Matched previous report [10].
Methyl 2-bromo-1-oxo-2,3-dihydro-1H-indene-2-carboxylate 7c 1-oxo-2,3-dihydro-1H-indene-2-carboxylate (40.6 mg, 0.21 mmol), 1b (165 mg, 0.14 mmol) and tetrabutyl ammonium bromide (278 mg, 0.86 mmol) gave 7c (19 mg, 33%). 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.0 Hz, 1H), 7.70 (t, J = 7.5 Hz, 1H), 7.51–7.45 (m, 2H), 4.23 (d, J = 18.4 Hz, 1H), 3.84 (s, 3H) and 3.69 (d, J = 18.1 Hz, 1H). Matched previous report [10].
2.3. Chiral Column Separation
A Chiralpak IB N-5 (particle size 5 μm, 250 × 4.6 mm ID) column equipped with the IB N-5 guard column was used to perform chiral separations of compounds. HPLC-grade hexane and isopropanol (99:1) were used as the mobile phase. Samples were dissolved in the same solvent mixture and 10 μL was injected from the sample vial into the system. Separation was run at 1.0 mL/min.
3. Results
Our group synthesized amino acid-based hypervalent iodine macrocycles (HIMs) [11] 1a and 1b and confirmed the structure by NMR spectroscopy, mass spectrometry and X-ray crystallography (Figure 1) [12,13,14]. Upon the addition of tetra butyl ammonium (TBA) salts of fluoride, chloride, bromide, azide and cyanide to an HIM, the anion selectively bound to the iodine center, resulting in the disassembly of the HIM trimer into a distinct HIM monomer (Figure 2). This new species was identified as a tetracoordinated HIM monomer, where iodine bound to the anion via an iodine–halogen bond. The formation of the monomer upon the addition of TBA(Cl) was further confirmed by obtaining the single crystal that was solved using X-ray diffraction (Figure 2) [12]. From the X-ray analysis, we observed one adduct per tetrabutylammonium cation.
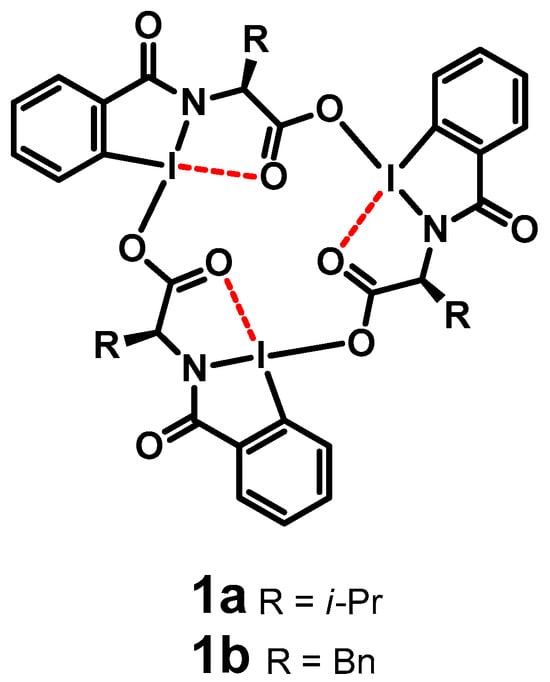
Figure 1.
Hypervalent iodine macrocycles (HIMs) prepared from amino acid-based benziodazoles. Dashed line represents the secondary bonding character. (adapted from Refs. [11,12]).
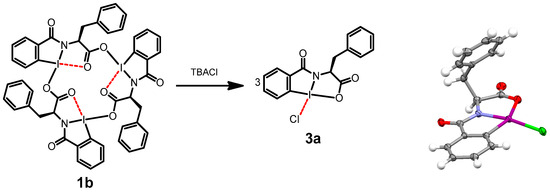
Figure 2.
(Left) Formation of tetracoordinated hypervalent iodine adduct upon the addition of tetrabutylammonium chloride to HIMs in chloroform. (Right) Crystal structure of adduct with tetrabutyl ammonium cation removed for clarity. Dashed line represents the secondary bonding character (adapted from Ref. [12]).
During the preliminary investigations of the anion-coordinated system, it was observed that the macrocycle could be reformed upon the addition of a coordinating metal such as silver nitrate. The chloride anion was removed from the hypervalent iodine system as a precipitate of silver chloride. Building upon this observation of the addition and removal of anions, respectively, we hypothesized that anion-coordinated HIM monomers could be used to directly transfer the anion to the alpha carbon of beta keto esters. In addition, the chiral nature of the reagent suggested asymmetric induction may be possible. Other TBA salts including TBA-Br and TBA-N3 were found to lead to similar adducts including 2a–c and 3a–c (Scheme 1).
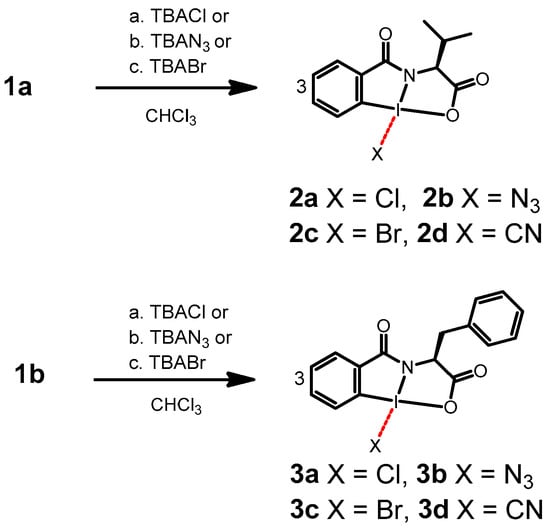
Scheme 1.
(Top) Synthesis of adduct 2a–d from Val HIM 1a. (Bottom) Synthesis of adduct 3a–d from Phe HIM 1b. Dashed line represents the secondary bonding character.
Past reports have shown benziodoxol(on)e-based hypervalent iodine reagents can be used to transfer azide, cyanide and acetylene onto 1,3-dicarbonyl compounds [9,10]. With this precedent and our previous results regarding reversibility in the HIM system (Figure 2), we were motivated to explore our own adduct to investigate its ability to transfer halogens and azides to the carbon center in 1,3-dicarbonyl compounds. We further hypothesized that if the iodine center in adducts remained in a chiral environment, direct chlorination, bromination and azidation could be achieved with some enantioselectivity.
To test our hypothesis, we initially chose 2-acetyl-1-tetralone (4) and methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate (5) as model substrates to establish the direct chlorination reaction with adduct 2a (Scheme 2). Upon reaction of 4 in chloroform at 60 °C with 2a, successful formation of the chlorinated product 6a (56%) was observed within 4 h (Table 1, entry 4). For initial screening of the reaction, chloroform was used as a solvent as macrocycles 1a and 1b were stable in chloroform, a fact supported by the successful isolation of single crystals of the macrocycle from chloroform in previous experiments. The reaction was performed at 60 °C, based on our separate experiments that revealed compounds 1a and 1b decomposed when maintained at temperatures higher than 60 °C for a prolonged duration. Furthermore, reactions at room temperature did not lead to appreciable product (Table 1, entry 3). To ensure that the chloride transfer was enabled by adduct 2a, a control experiment was performed between the 2-acetyl-1-tetralone in the presence of tetrabutylammonium chloride in chloroform at 60 °C for 4 h and no reaction was observed (Table 1, entry 2).
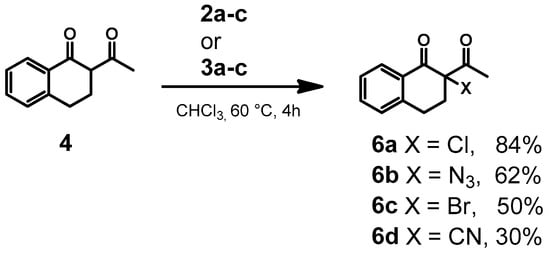
Scheme 2.
Transfer of anions from hypervalent iodine adducts 2a–c or 3a–c to 2-acetyl-1-tetralone.

Table 1.
Reaction optimization (The bold is to describe the compound number).
With this initial success, other anion-based adducts were employed. For example, the reaction of 4 with 2b and 2c in chloroform at 60 °C for 4 h yielded azide and bromide products 6b and 6c with 60% and 40% yield, respectively (Table 1, entries 5 and 6). We investigated other substrates such as methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate 5 for the anion transfer (Scheme 3). Our results showed that the reaction between 5 and 2a–c or 3a–c also resulted in the desired anion-transferred products with a moderate to good yield (Table 1, entries 11–13). To optimize conditions, we screened the reactions in different equivalents of adducts 2a–c or 3a–c as shown in Table 1. When 1.3 equivalents of the adduct (from both 1a and 1b) were used, the reactions with 4 proceeded with moderate yields of 40–60% within 4 h. However, increasing the adduct to 2.6 equivalents and extending the reaction time to 48 h led to significantly improved yields, ranging from 37 to90% for chloride, bromide and azide (Table 1, entries 7–9). The transfer of cyanide to both substrates 4 and 5 was also investigated with the optimal conditions (Table 1, entries 10 and 14). Unfortunately, the reaction was met with limited success with only 30% yield for the six-membered 6d and no detectable product 7d was found. Clearly, there are issues with the transfer of this ion and it is believed to be from the instability of the final product. While these reactions were successful with the TBA salt in chloroform, we were interested to know if cheaper inorganic salts such as NaCl, NaBr and NaN3 could also be used. To utilize these salts, a DMSO and water solvent mixture was employed. While the desired product was found under these reaction conditions, the yields were considerably lower (16–20%, Table 1, entries 15–17). The poor yields observed with polar DMSO may be due to the decomposition of the hypervalent iodine reagent. This is supported by the fact that NMR samples of macrocycle 1a or 1b, prepared in DMSO, showed impurities after 2 days when the same sample was reanalyzed. This matches the fact that DMSO is known to be oxidized by hypervalent iodine reagents.
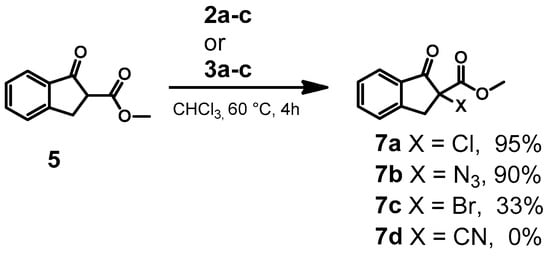
Scheme 3.
Transfer of anions from hypervalent iodine adducts 2a–c or 3a–c to methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate.
The newly synthesized products were characterized by NMR spectroscopy and mass spectrometry. After confirming the purity of the products, we further explored whether the anion transfer from adducts 2a–c or 3a–c to substrates 4 or 5 can occur in an enantioselective manner. A chiral column was used to perform chiral separations of compounds. For a preliminary reaction of 4 with valine-based 2b, two peaks were observed at similar retention times and of the same area and suggested that the reaction gave a 1:1 racemic mixture of compound 6b (Supporting Information). We observed similar racemic mixtures when using other valine-based reagents 2a–c. However, we observed promising results in case of phenylalanine-based reagents. As an example, modification of 4 with 3a led to a non-racemic mixture with an enantiomeric excess (e.e.) of 26% (Figure 3). This experiment confirmed that the transfer of anions from tetracoordinate hypervalent iodine based adducts to beta keto esters can proceed in a mildly enantioselective manner. While the e.e. was not exceedingly high, it suggested larger non-natural amino acids that could provide a larger steric field around the reaction site and could provide even better control of the enantioselectivity.
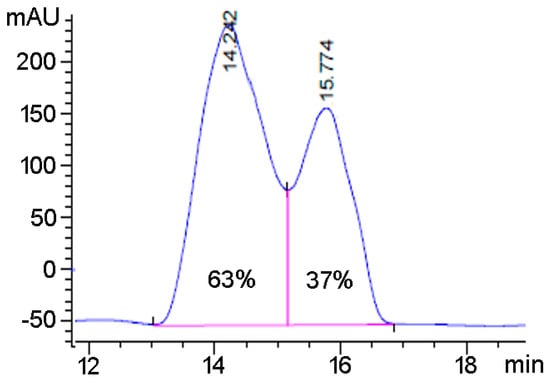
Figure 3.
Chiral chromatogram of enantioenriched 6a upon functionalization with 3a. Percentage listed is the enantiomeric excess based on Area1: 21,039, Area 2: 12,305.
4. Conclusions
In conclusion, we have demonstrated a direct transfer of anions from an anion-associated hypervalent iodine reagent. Specifically, these freshly prepared adducts were employed to facilitate anion transfer within the 2-acetyl-1-tetralone and methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate. The anion transfer from the phenylalanine–HIM adducts was achieved in a mildly enantiomerically selective manner and suggested tuning of the sterics around the reagent could lead to better selectivities.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/org6040052/s1, Chiral HPLC of 6b prepared from 2b, and 6a prepared from 2a, NMR of compounds.
Author Contributions
Conceptualization, K.N.P.; methodology, M.D.P., T.A., K.P., S.A. and E.J., investigation, M.D.P., T.A., K.P., S.A. and E.J., writing—original draft preparation, M.D.P.; writing—review and editing, K.N.P.; project administration, K.N.P.; funding acquisition, K.N.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Foundation (#2003654) and the American Chemical Society Petroleum Research Fund (61467-ND1).
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author(s).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Scotti, C.; Barlow, J.W. Natural Products Containing the Nitrile Functional Group and Their Biological Activities. Nat. Prod. Commun. 2022, 17, 1934578X221099973. [Google Scholar] [CrossRef]
- Bonatto, V.; Lameiro, R.F.; Rocho, F.R.; Lameira, J.; Leitão, A.; Montanari, C.A. Nitriles: An Attractive Approach to the Development of Covalent Inhibitors. RSC Med. Chem. 2023, 14, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, C.; Burešová, Z.; Bureš, F.; Liu, J. Cyano-Capped Molecules: Versatile Organic Materials. J. Mater. Chem. A 2023, 11, 3753–3770. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Thirupathi, N.; Wei, F.; Tung, C.-H.; Xu, Z. Divergent Synthesis of Chiral Cyclic Azides via Asymmetric Cycloaddition Reactions of Vinyl Azides. Nat. Commun. 2019, 10, 3158. [Google Scholar] [CrossRef] [PubMed]
- Hernandes, M.Z.; Cavalcanti, S.M.; Moreira, D.R.; de Azevedo Junior, W.F.; Leite, A.C. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Cantillo, D.; Kappe, C.O. Halogenation of Organic Compounds Using Continuous Flow and Microreactor Technology. React. Chem. Eng. 2017, 2, 7–19. [Google Scholar] [CrossRef]
- Barral, K.; Moorhouse, A.D.; Moses, J.E. Efficient Conversion of Aromatic Amines into Azides: A One-Pot Synthesis of Triazole Linkages. Org. Lett. 2007, 9, 1809–1811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Qiu, J.; Kong, D.; Gao, Y.; Lu, F.; Karmaker, P.G.; Chen, F.-X. The Direct Electrophilic Cyanation of β-Keto Esters and Amides with Cyano Benziodoxole. Org. Biomol. Chem. 2014, 13, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Vita, M.V.; Waser, J. Azidation of β-Keto Esters and Silyl Enol Ethers with a Benziodoxole Reagent. Org. Lett. 2013, 15, 3246–3249. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Koposov, A.E.; Smart, J.T.; Tykwinski, R.R.; McDonald, R.; Morales-Izquierdo, A. Secondary Bonding-Directed Self-Assembly of Amino Acid Derived Benziodazoles: Synthesis and Structure of Novel Hypervalent Iodine Macrocycles. J. Am. Chem. Soc. 2001, 123, 4095–4096. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; Arafin, S.; Jones, E.; Du, Y.; Kulkarni, G.C.; Uddin, A.; Woods, T.J.; Plunkett, K.N. Assembly and Disassembly of Supramolecular Hypervalent Iodine Macrocycles via Anion Coordination. J. Org. Chem. 2024, 89, 7437–7445. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; Arafin, S.; Venus, G.; Jones, E.; Du, Y.; Pandey, M.D.; Awais, T.; Wang, L.; Plunkett, K.N. Pi-Extended Hypervalent Iodine Macrocycles and Their Supramolecular Assembly with Buckminsterfullerene. J. Mater. Chem. C 2025, 13, 842–848. [Google Scholar] [CrossRef]
- Pandey, K.; Orton, L.X.; Venus, G.; Hussain, W.A.; Woods, T.; Wang, L.; Plunkett, K.N. Supramolecular Assembly of Hypervalent Iodine Macrocycles and Alkali Metals. Beilstein J. Org. Chem. 2025, 21, 1095–1103. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).