
Unveiling the Relationship between Structure and Anticancer Properties of Permethylated Anigopreissin A: A Study with Thirteen Analogues

 ,
,  ,
,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
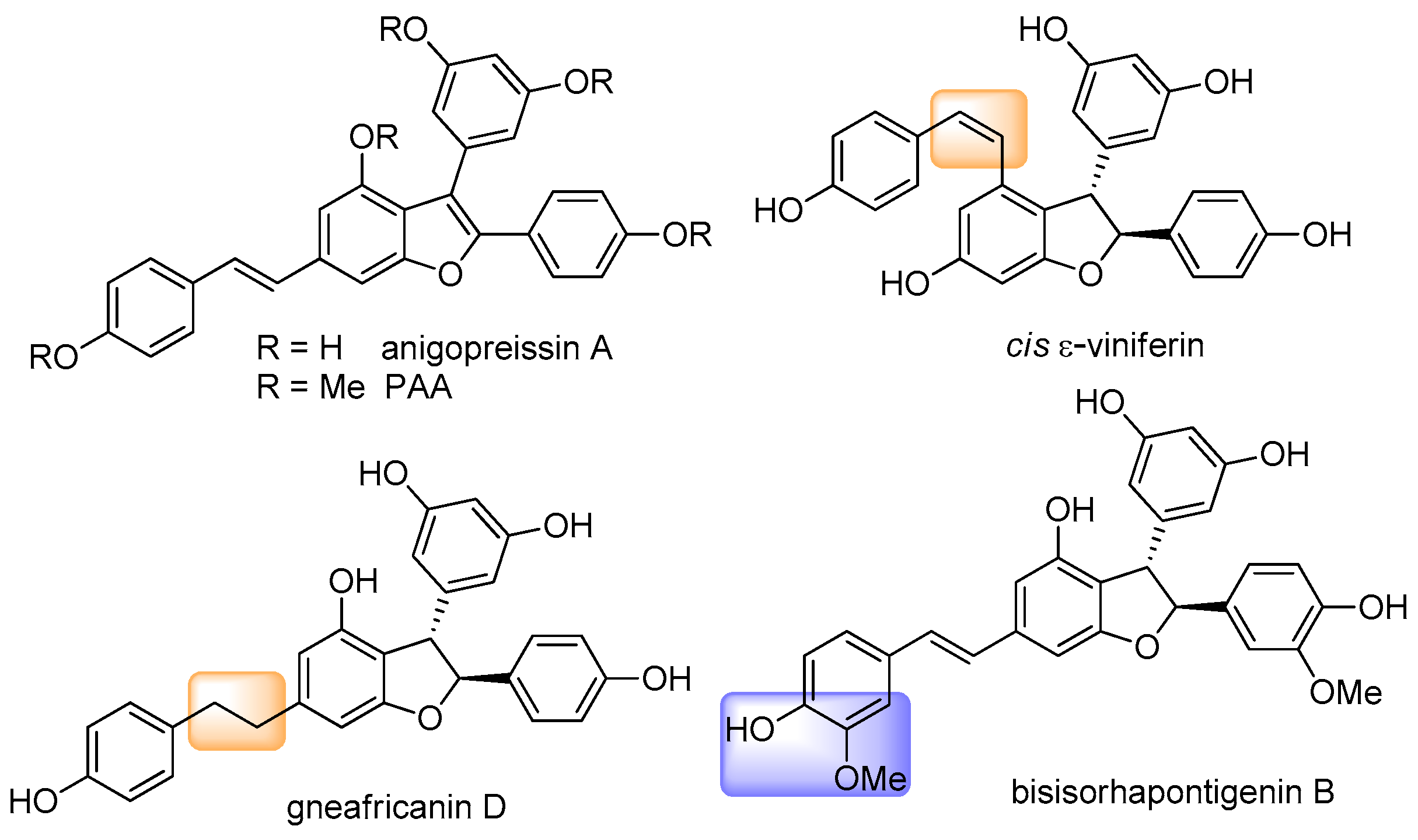
1. Introduction
2. Materials and Methods
2.1. General Procedures
2.1.1. General Procedure of Wittig Olefination for the Preparation of Compound 5a, 6b, 6c, 7, and 8
2.1.2. Reduction of the Styryl Double Bond for Preparation of Compound 5b
2.1.3. Julia–Kocienski Reaction for Preparation of Compound 6a
2.1.4. General Procedure for the Suzuki Reaction for the Preparation of Compounds 33, 9a–9f
2.2. Cell Culture, Treatments, and Viability Assay
2.2.1. Cell Viability Assay
2.2.2. Statistical Analysis
2.3. Computational Details
3. Results and Discussion
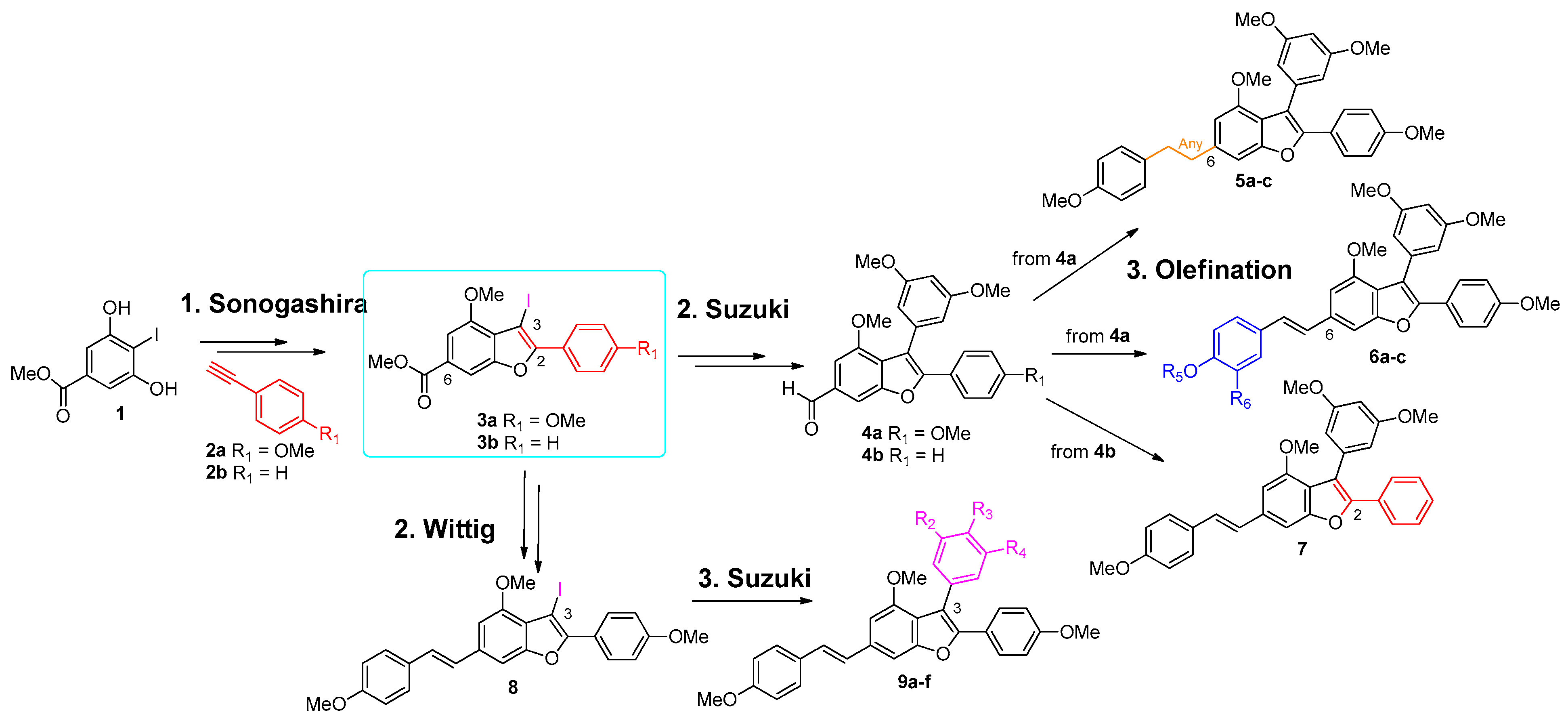
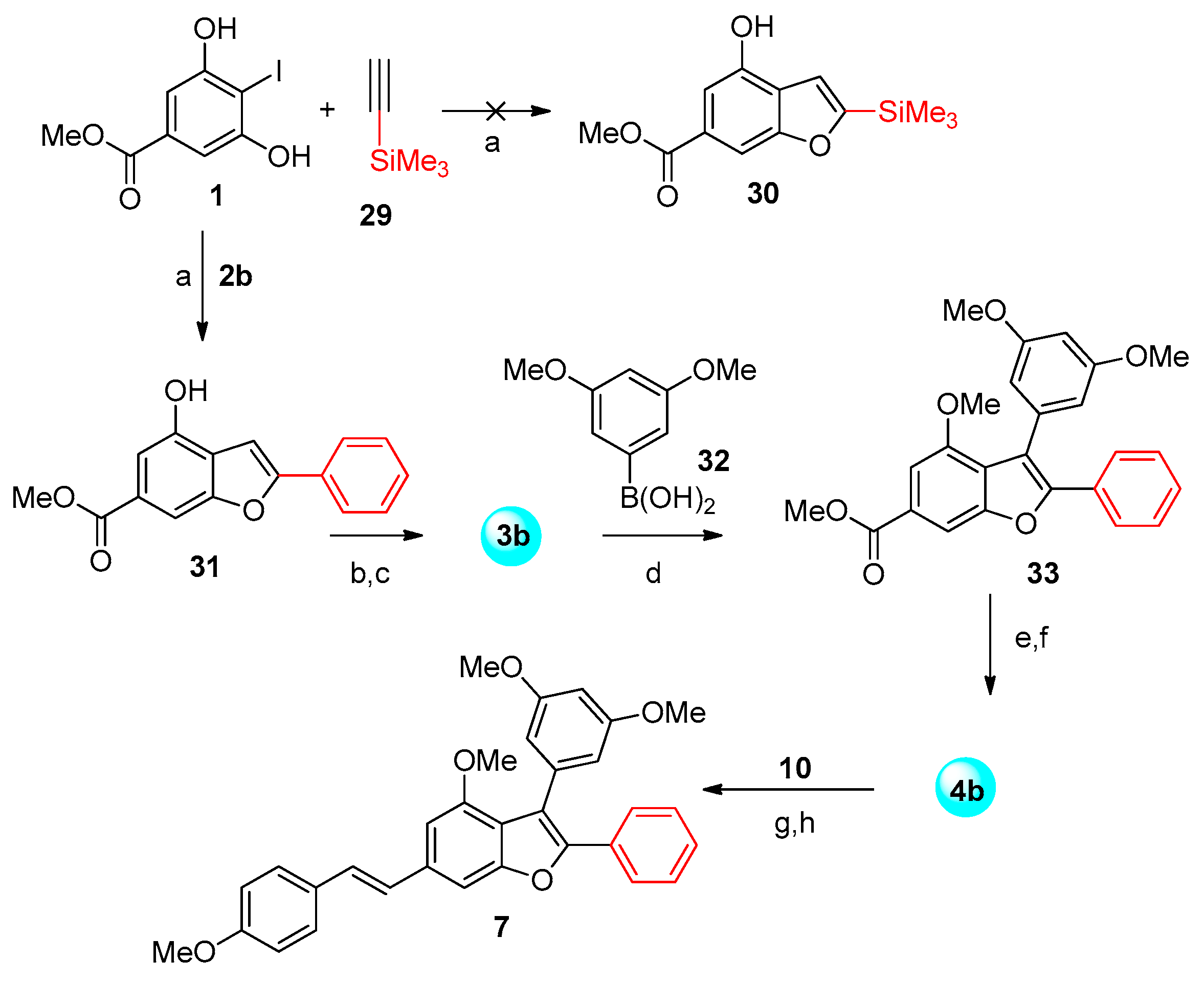
3.1. Synthesis of the Thirteen Analogues
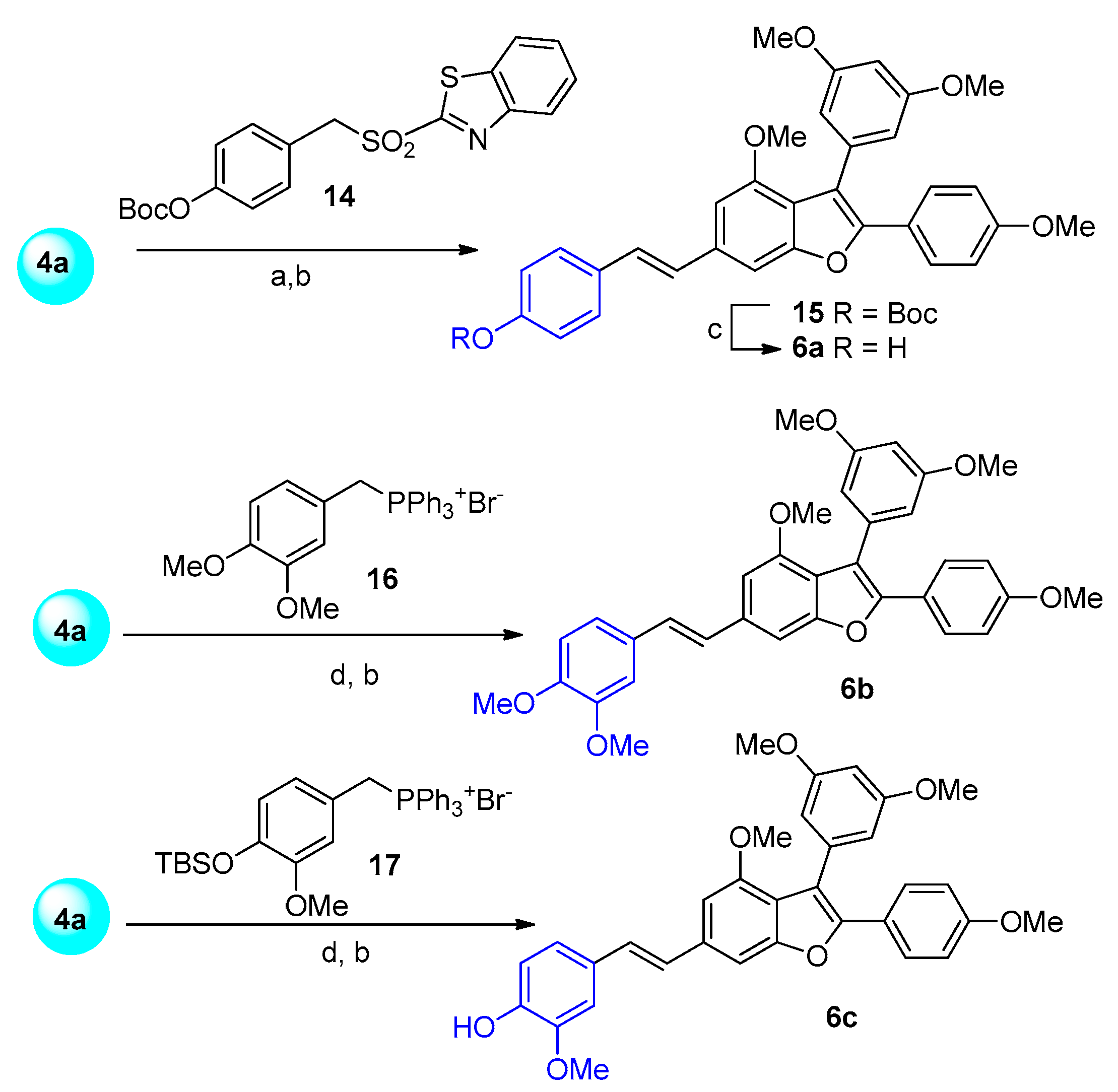
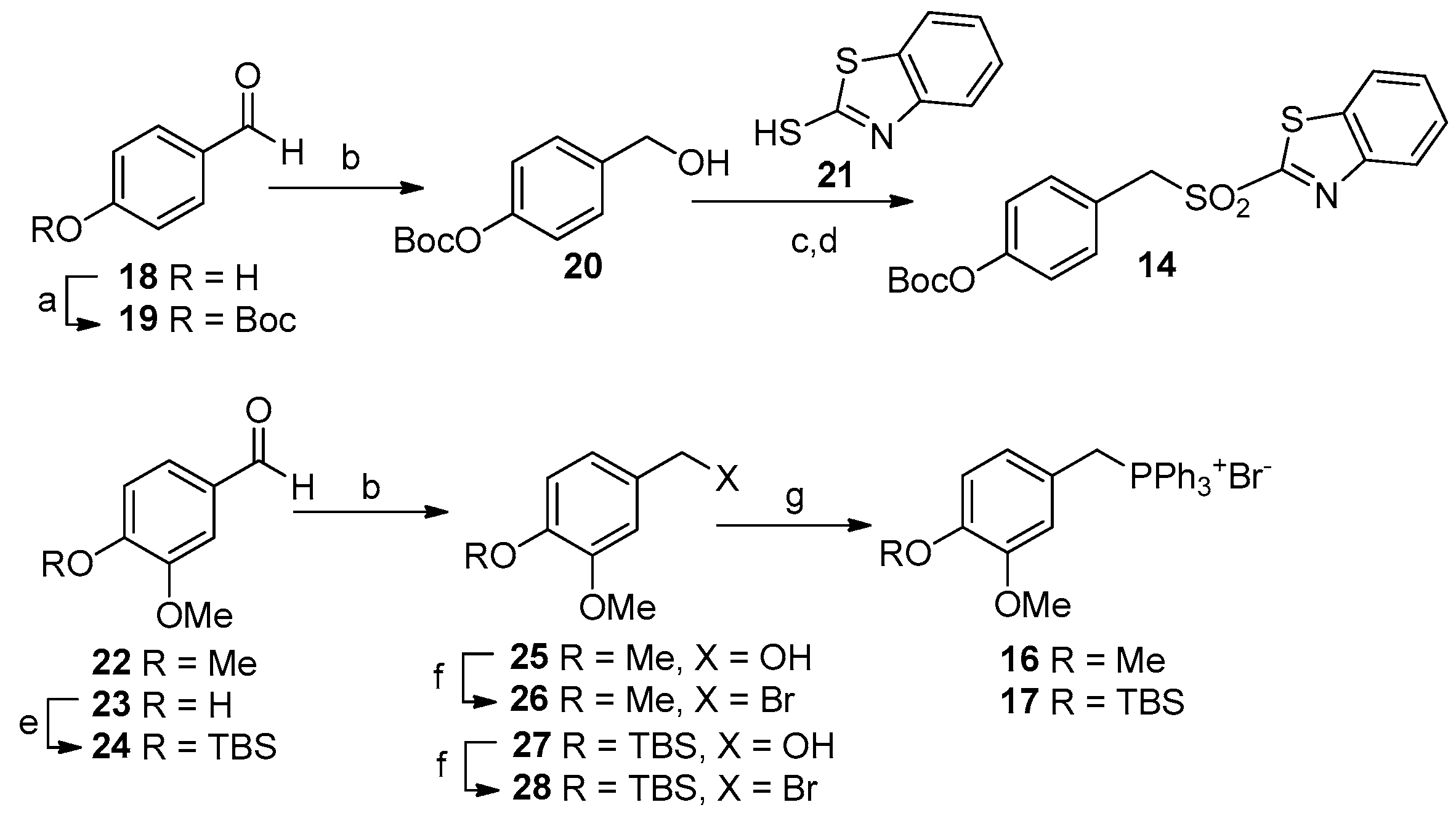
3.1.1. Synthesis of Analogues 5a–c and 6a–c
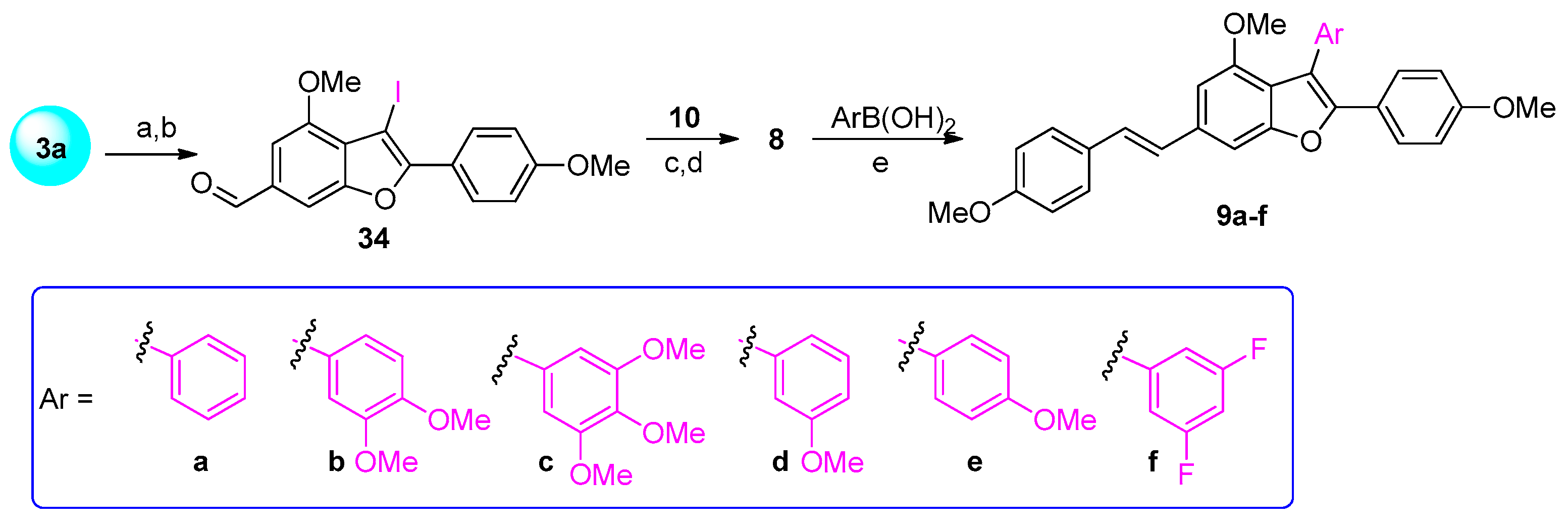
3.1.2. Synthesis of Analogues 7 and 9a–f
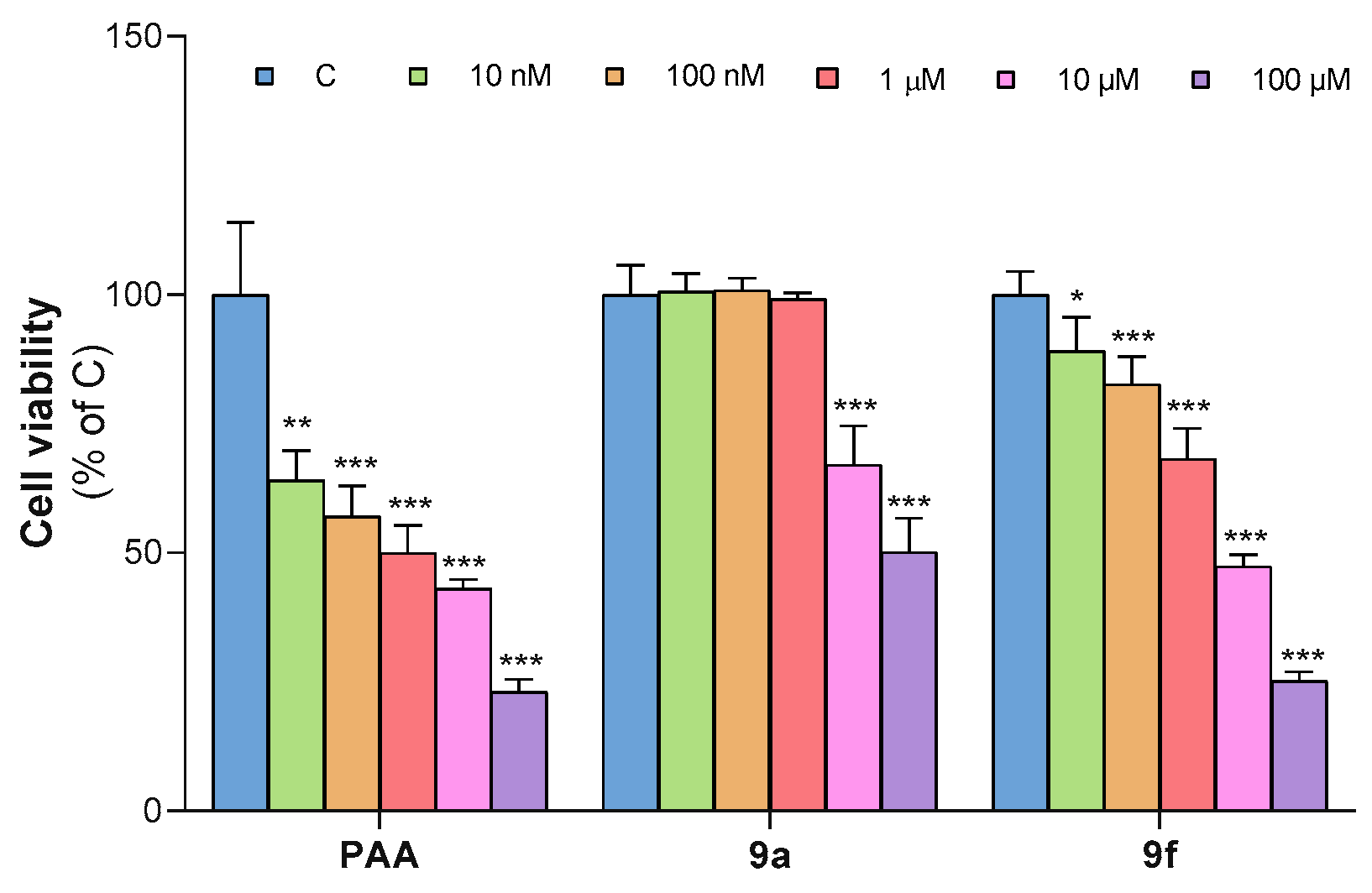
3.2. Antiproliferative Effect of the 13 Analogues Compared to PAA
3.2.1. Antiproliferative Effect on HepG2 Cells
3.2.2. Antiproliferative Effect on Alexander Cells
3.3. Computational Studies
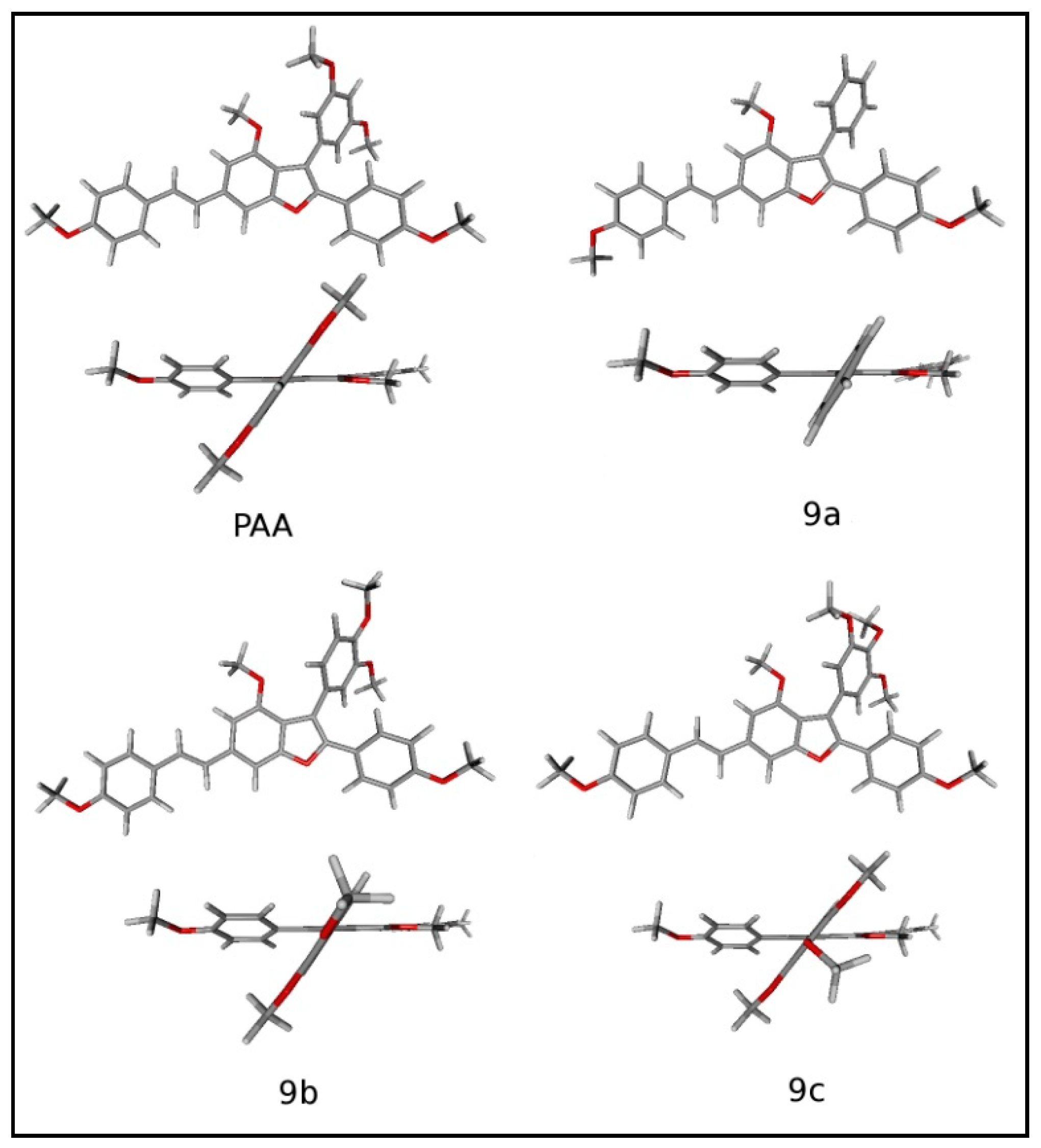
3.3.1. Structure Optimisations
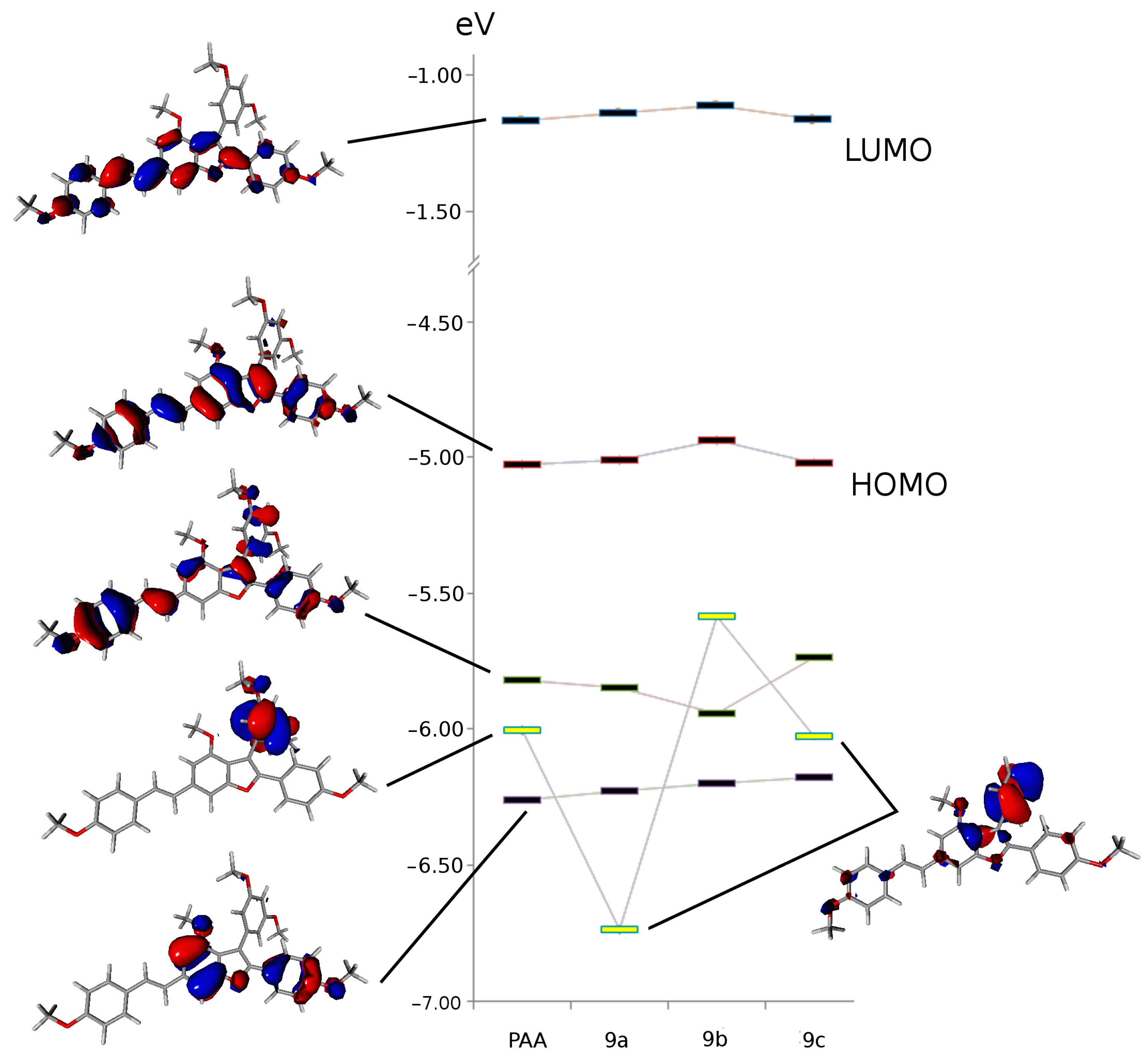
3.3.2. Conformational Study and Electronic Properties
3.4. Structure–Properties Relationship Conclusion
- The trans configuration of the double bond is essential for the cytotoxicity of PAA, as the cis isomer (5a) and the analogues with the ethylene bridge (5b) or a triple bond (5c) were inactive.
- The presence of only one methoxy group in the para position on the styryl ring is important since additional methoxy substituents (as in 6b and 6c) or a hydroxyl group instead of the methoxy group all resulted in inactive compounds.
- The para-methoxy substituent on the C-2 ring is fundamental for the biological activity of PAA since its absence (as in analogue 7) resulted in an inactive species.
- The C-3 ring is the only portion of the molecule that can be modulated to produce other active species, such as compound 9c, with a plain unsubstituted phenyl ring, or analogue 9f, with two fluorine atoms, even if both analogues were less active than PAA.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary Phytochemicals and Cancer Chemoprevention: A Review of the Clinical Evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef] [PubMed]
- Keylor, M.H.; Matsuura, B.S.; Stephenson, C.R.J. Chemistry and Biology of Resveratrol-Derived Natural Products. Chem. Rev. 2015, 115, 8976–9027. [Google Scholar] [CrossRef] [PubMed]
- Vang, O. Resveratrol: Challenges in Analyzing Its Biological Effects. Ann. N. Y. Acad. Sci. 2015, 1348, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Bala, A.E.A.; Kollmann, A.; Ducrot, P.-H.; Majira, A.; Kerhoas, L.; Leroux, P.; Delorme, R.; Einhorn, J. Cis Ε-viniferin: A New Antifungal Resveratrol Dehydrodimer from Cyphostemma crotalarioides Roots. J. Phytopathol. 2000, 148, 29–32. [Google Scholar] [CrossRef]
- Lin, M.; Yao, C.-S. Natural Oligostilbenes. In Studies in Natural Products Chemistry, 1st ed.; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 33, pp. 601–644. [Google Scholar] [CrossRef]
- Kloypan, C.; Jeenapongsa, R.; Sri-in, P.; Chanta, S.; Dokpuang, D.; Tip-pyang, S.; Surapinit, N. Stilbenoids from Gnetum macrostachyum Attenuate Human Platelet Aggregation and Adhesion. Phytother. Res. 2012, 26, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, D.; Schneider, B. A Resveratrol Dimer from Anigozanthos preissii and Musa cavendish. Phytochemistry 1996, 43, 471–473. [Google Scholar] [CrossRef]
- Brkljača, R.; White, J.M.; Urban, S. Phytochemical Investigation of the Constituents Derived from the Australian Plant Macropidia fuliginosa. J. Nat. Prod. 2015, 78, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Chiummiento, L.; Funicello, M.; Lopardo, M.T.; Lupattelli, P.; Choppin, S.; Colobert, F. Concise Total Synthesis of Permethylated Anigopreissin A, a New Benzofuryl Resveratrol Dimer. Eur. J. Org. Chem. 2012, 2012, 188–192. [Google Scholar] [CrossRef]
- Convertini, P.; Tramutola, F.; Iacobazzi, V.; Lupattelli, P.; Chiummiento, L.; Infantino, V. Permethylated Anigopreissin A Inhibits Human Hepatoma Cell Proliferation by Mitochondria-Induced Apoptosis. Chem.-Biol. Interact. 2015, 237, 1–8. [Google Scholar] [CrossRef]
- Marsico, M.; Santarsiero, A.; Pappalardo, I.; Convertini, P.; Chiummiento, L.; Sardone, A.; Di Noia, M.A.; Infantino, V.; Todisco, S. Mitochondria-Mediated Apoptosis of HCC Cells Triggered by Knockdown of Glutamate Dehydrogenase 1: Perspective for Its Inhibition through Quercetin and Permethylated Anigopreissin A. Biomedicines 2021, 9, 1664. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Herraiz-Cobo, J.; Albericio, F.; Álvarez, M. The Larock Reaction in the Synthesis of Heterocyclic Compounds. In Advances in Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; Volume 116, pp. 1–35. [Google Scholar] [CrossRef]
- Chao, J.; Li, H.; Cheng, K.-W.; Yu, M.-S.; Chang, R.C.-C.; Wang, M. Protective Effects of Pinostilbene, a Resveratrol Methylated Derivative, against 6-Hydroxydopamine-Induced Neurotoxicity in SH-SY5Y Cells. J. Nutr. Biochem. 2010, 21, 482–489. [Google Scholar] [CrossRef]
- Chimento, A.; Santarsiero, A.; Iacopetta, D.; Ceramella, J.; De Luca, A.; Infantino, V.; Parisi, O.I.; Avena, P.; Bonomo, M.G.; Saturnino, C.; et al. A Phenylacetamide Resveratrol Derivative Exerts Inhibitory Effects on Breast Cancer Cell Growth. Int. J. Mol. Sci. 2021, 22, 5255. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H.J. Molden: A pre- and post-processing program for molecular and electronic structures. Comput.-Aided Mol. Design 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Antonioletti, R.; Bonadies, F.; Ciammaichella, A.; Viglianti, A. Lithium Hydroxide as Base in the Wittig Reaction. A Simple Method for Olefin Synthesis. Tetrahedron 2008, 64, 4644–4648. [Google Scholar] [CrossRef]
- Marsh, B.J.; Carbery, D.R. One-Pot o-Nitrobenzenesulfonylhydrazide (NBSH) Formation−Diimide Alkene Reduction Protocol. J. Org. Chem. 2009, 74, 3186–3188. [Google Scholar] [CrossRef]
- Fang, Z.; Song, Y.; Sarkar, T.; Hamel, E.; Fogler, W.E.; Agoston, G.E.; Fanwick, P.E.; Cushman, M. Stereoselective Synthesis of 3,3-Diarylacrylonitriles as Tubulin Polymerization Inhibitors. J. Org. Chem. 2008, 73, 4241–4244. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kuang, C.; Yang, Q.; Cheng, X. Cs2CO3-Mediated Synthesis of Terminal Alkynes from 1,1-Dibromo-1-Alkenes. Tetrahedron Lett. 2011, 52, 992–994. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Lister, T.; Denton, R.M.; Gelin, C.F. Total Synthesis of Artochamins F, H, I, and J through Cascade Reactions. Tetrahedron 2008, 64, 4736–4757. [Google Scholar] [CrossRef] [PubMed]
- Schechter, A.; Goldrich, D.; Chapman, J.R.; Uberheide, B.M.; Lim, D. MgBr2·OEt2: A Lewis Acid Catalyst for the O- and N-Boc Protection of Phenols and Amines. Synth. Commun. 2015, 45, 643–650. [Google Scholar] [CrossRef]
- Bonini, C.; Chiummiento, L.; Videtta, V. One-Pot Practical Preparation of Novel Propargylic Aryl and Heteroaryl Sulfides and Sulfones. Synlett 2005, 20, 3067–3070. [Google Scholar] [CrossRef]
- Bonini, C.; Chiummiento, L.; Videtta, V. Direct Preparation of Z-1,3-Enyne Systems with a TMS-Propargylic Sulfone: Application of a One-Pot Julia Olefination. Synlett 2006, 2006, 2079–2082. [Google Scholar] [CrossRef]
- Chankeshwara, S.V.; Chebolu, R.; Chakraborti, A.K. Organocatalytic Methods for Chemoselective O-Tert-Butoxycarbonylation of Phenols and Their Regeneration from the O-t-Boc Derivatives. J. Org. Chem. 2008, 73, 8615–8618. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, V.; Kotapalli, S.S.; Ummanni, R.; Alla, M. Ring Functionalization and Molecular Hybridization of Quinolinyl Pyrazole: Design, Synthesis and Antimycobacterial Activity. ChemistrySelect 2017, 2, 6529–6534. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Ebrahimzadeh, F. Facile Conversion of Alcohols into Their Bromides and Iodides by N-Bromo and N-Iodosaccharins/Triphenylphosphine under Neutral Conditions. Tetrahedron Lett. 2006, 47, 1771–1775. [Google Scholar] [CrossRef]
- Singh, S.B.; Pettit, G.R. Synthesis of (±)-Isocombretastatins A–C. Synth. Commun. 1987, 17, 877–892. [Google Scholar] [CrossRef]
- Chiummiento, L.; D’Orsi, R.; Funicello, M.; Lupattelli, P. Last Decade of Unconventional Methodologies for the Synthesis of Substituted Benzofurans. Molecules 2020, 25, 2327. [Google Scholar] [CrossRef] [PubMed]
- Shabir, G.; Saeed, A.; Zahid, W.; Naseer, F.; Riaz, Z.; Khalil, N.; Muneeba; Albericio, F. Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022). Pharmaceuticals 2023, 16, 1162. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caivano, I.; Santarsiere, A.; Amati, M.; Convertini, P.; Funicello, M.; Lupattelli, P.; Chiummiento, L.; Santarsiero, A. Unveiling the Relationship between Structure and Anticancer Properties of Permethylated Anigopreissin A: A Study with Thirteen Analogues. Organics 2024, 5, 237-251. https://doi.org/10.3390/org5030012
Caivano I, Santarsiere A, Amati M, Convertini P, Funicello M, Lupattelli P, Chiummiento L, Santarsiero A. Unveiling the Relationship between Structure and Anticancer Properties of Permethylated Anigopreissin A: A Study with Thirteen Analogues. Organics. 2024; 5(3):237-251. https://doi.org/10.3390/org5030012
Chicago/Turabian StyleCaivano, Ilaria, Alessandro Santarsiere, Mario Amati, Paolo Convertini, Maria Funicello, Paolo Lupattelli, Lucia Chiummiento, and Anna Santarsiero. 2024. "Unveiling the Relationship between Structure and Anticancer Properties of Permethylated Anigopreissin A: A Study with Thirteen Analogues" Organics 5, no. 3: 237-251. https://doi.org/10.3390/org5030012
APA StyleCaivano, I., Santarsiere, A., Amati, M., Convertini, P., Funicello, M., Lupattelli, P., Chiummiento, L., & Santarsiero, A. (2024). Unveiling the Relationship between Structure and Anticancer Properties of Permethylated Anigopreissin A: A Study with Thirteen Analogues. Organics, 5(3), 237-251. https://doi.org/10.3390/org5030012