Abstract
We investigated the reactivity of different substituted nitrylimine-type three atom components (TACs) in [3+2] cycloaddition (32CAs) reactions with electrophilically activated nitroethenes within molecular electron density theory (MEDT). In parallel research, the molecular mechanism of the considered transformation was examined through analysis of all possible reaction channels and full optimization of all critical structures. In particular, the existence of zwitterionic intermediates on reaction paths was verified. On the basis of the bonding evolution theory (BET), the mechanism of the 32CA reaction between C,N-diphenylnitrylimine and (E)-2-phenyl-1-cyano-1-nitroethene should be treated as a one-step two-stage mechanism.
1. Introduction
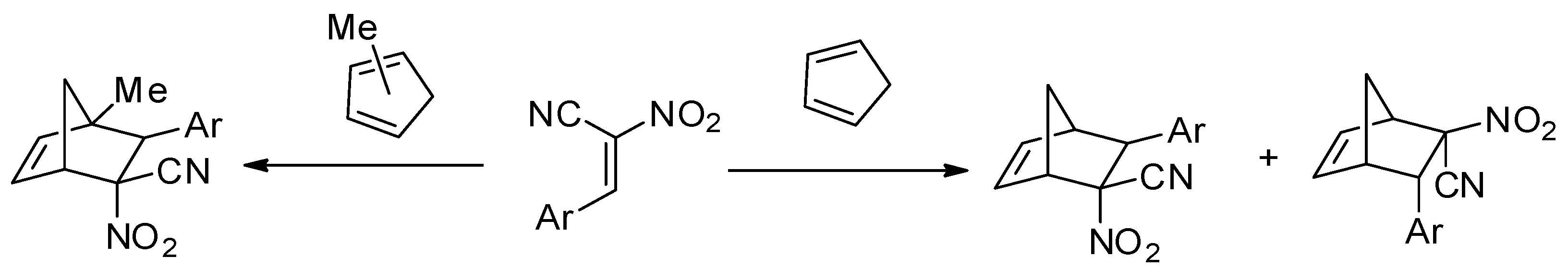
This work is a continuation of our comprehensive, experimental, and theoretical study regarding the synthesis and reactivity of (E)-2-aryl-1-cyano-1-nitroethenes (ACN). Some examples of this group of conjugated nitroalkenes have been known since the second half of the 20th century [1,2]. In recent years, similar-type compounds have been synthetized [3,4]. Unfortunately, knowledge of their chemical properties is still limited. Recently, we detected a series of interesting chemical properties of ACNs regarding their participation in cycloaddition processes. For example, despite the high reactivity of the shielded reaction sites, ACNs react rapidly with cyclopentadiene even at r.t., yielding mixtures of respective endo-nitro and exo-nitro cycloadducts [5] (Scheme 1). In similar reactions involving the mixture of methylcyclopentadienes, of which many are possible, stereoisomeric products are formed [6]. In contrast, less sterically crowded 2-aryl-1-nitroethenes react with cyclopentadiene at temperatures up to 80 °C [7].
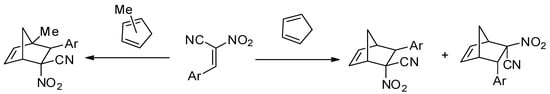
Scheme 1.
Cycloaddition reactions of conjugated dienes with CNAs.
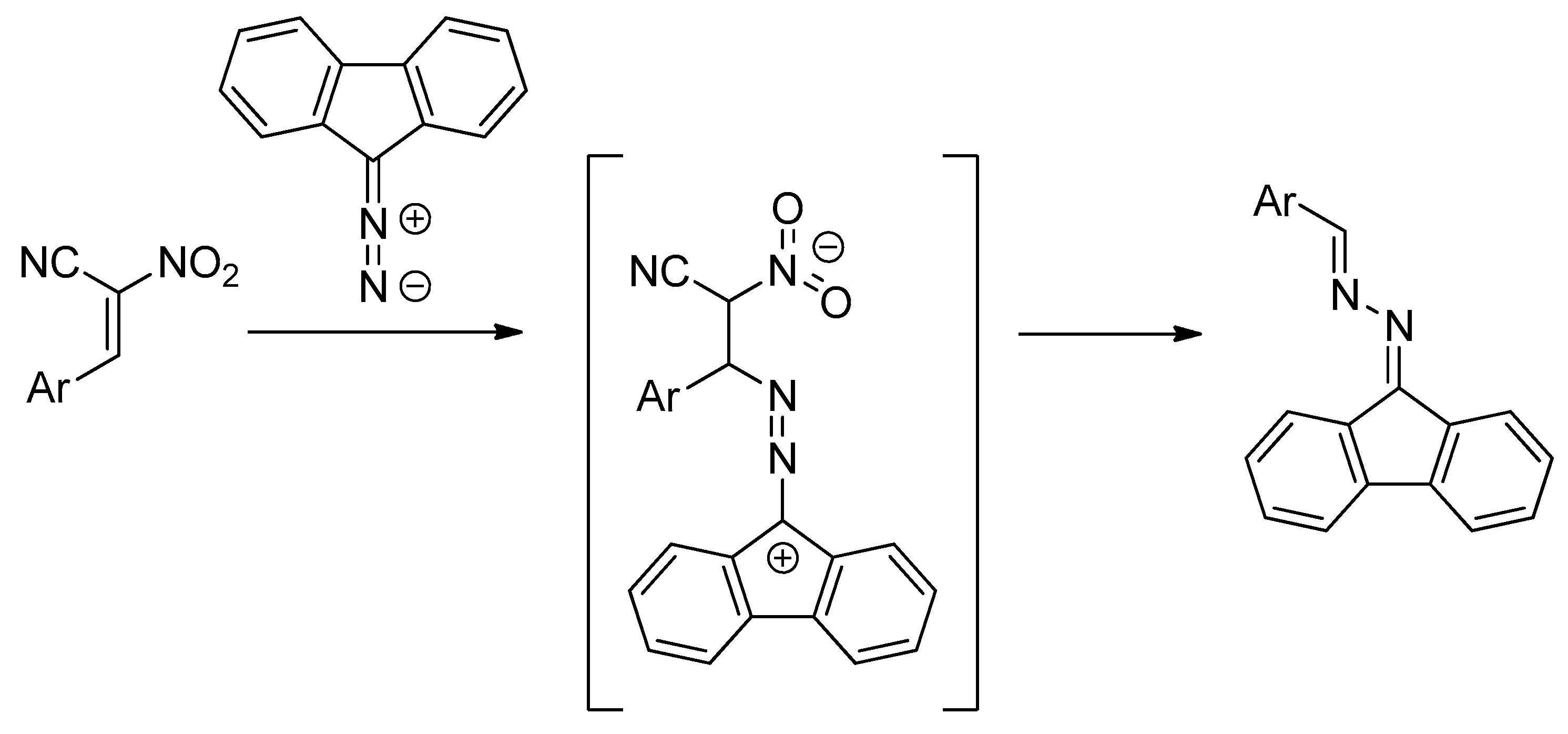
[3+2] cycloaddition (32CA) reactions of ACNs and N-methylazomethine ylide proceed under mild conditions and produce nitropyrrolidines [8]. In contrast, in analogous 32CAs involving diazafluorene, zwitterionic acyclic adducts were formed in the first reaction step. These zwitterions converted spontaneously under the reaction conditions into azine molecular systems [9]. At the same time, other nitroalkenes characterized by similar electrophilicity as ACNs reacted with diazafluorene to produce Δ1-nitropyrazolines (Scheme 2).
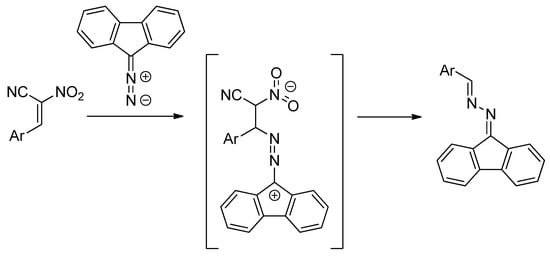
Scheme 2.
The zwitterionic intermediate in reaction of diazafluorene and ACNs.
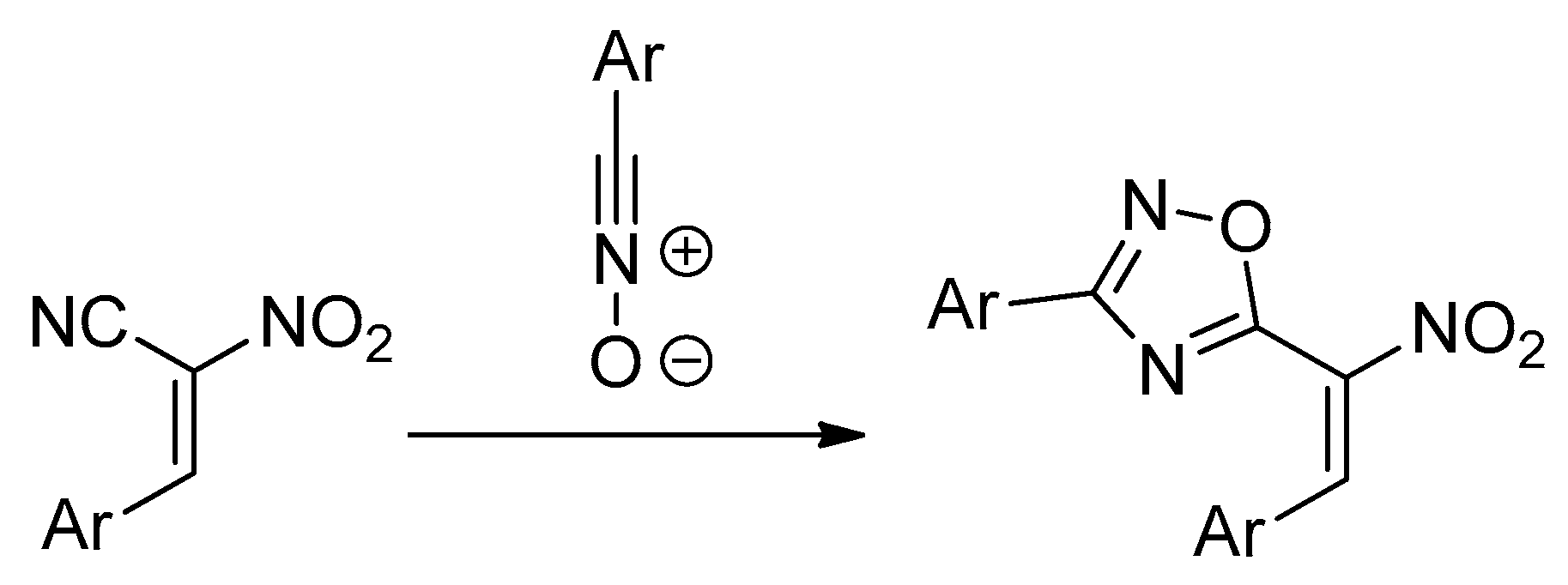
Non-catalyzed 32CA reactions between ACNs and nitrile N-oxides proceed unexpectedly with the participation of a nitrile bond, instead of resulting in the expected >C=C< moiety of nitroalkene [10] (Scheme 3). There are evidently rare cases of 32CAs in the CN bond, which is generally recognized as inactive in the 32CA processes.
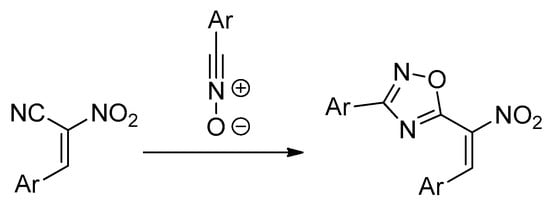
Scheme 3.
32CAs converting to a nitrile CN bond of ACNs.
There is currently no published work regarding the participation of ACNs in 32CA processes involving nitrylimine TAC systems. Our work initiates comprehensive research in this area. We aim to shed light on the selectivity and molecular mechanism of model processes involving different substituted diarylnitrylimines (DNI) and ACNs. This analysis will be help to guide further experimental study in the presented area.
2. Computational Details
The global reactivity descriptors of the addends, namely electronic potential μ, chemical hardness η, global electrophilicity ω and global nucleophilicity N, were approximated in pursuance of the equations defined on the basis of conceptual density functional theory (CDFT) according to the equations recommended by Parr [11] and Domingo [12,13]. In the calculation we used the correlation-exchange functional B3LYP together with the basic level set of 6-31G(d) in the gas phase [12,13,14,15].
The electronic chemical potentials (μ) and chemical hardness (η) were evaluated in terms of one-electron energies of FMO (EHOMO and ELUMO) using the following equations [11,12,13,14,15,16]:
where EHOMO and ELUMO may be approached in terms of the one-electron energies of the frontier MOs respectively HOMO and LUMO. Next, values of μ and η were then used to calculate a global electrophilicity (ω) according to the formula [12,13]:
μ ≈ (EHOMO + ELUMO)/2
η ≈ EHOMO − ELUMO
Ω = μ2/η
The global nucleophilicity (N) can be presented as follow [13]:
where EHOMO (TCE) is the HOMO energy for tetracyanoethylene (TCE); is the reference, because it presents the lowest HOMO (EHOMO (TCE) = −9.368 eV).
N = EHOMO − EHOMO (TCE)
The local electrophilicity (ωk) and the local nucleophilicity (Nk) concentrated on atom k was calculated based on global properties and the Parr function (Pk+ or Pk−), according to the formulas [17]:
ωk = P+k · ω
Nk = P−k · N
For localization of the transition states (TSs) the wb97xd/6-311+g(d) level of theory was applied [18]. All transition states were verified by diagonalization of the Hessian matrix and by analysis of the intrinsic reaction coordinates (IRC). For the simulation of solvents effect, the polarizable continuum model (PCM) [19] was used. Calculations of all critical structures were performed for the temperature T = 298 K and pressure p = 1 atm. Global electron density transfer (GEDT) [20] was calculated according to the formula:
where qA is the net Mulliken charge and the sum takes over all the atoms of nitroalkene.
GEDT = −ΣqA
Indexes of σ-bonds development (I) were calculated according to formula [21]:
where rTSA-B is the distance between the reaction centers A and B at the TS and rPA-B is the same distance at the corresponding product.
IA-B = 1 − [(rTSA-B − rPA-B)/rPA-B)
The electron localization function (ELF) [22,23,24] and BET [25] studies were conducted according to well-known procedure using the TopMod [26] program. This approach to explain the molecular mechanism of various classes of compounds has been the subject of many studies [10,27,28,29,30,31,32,33,34].
3. Results and Discussion
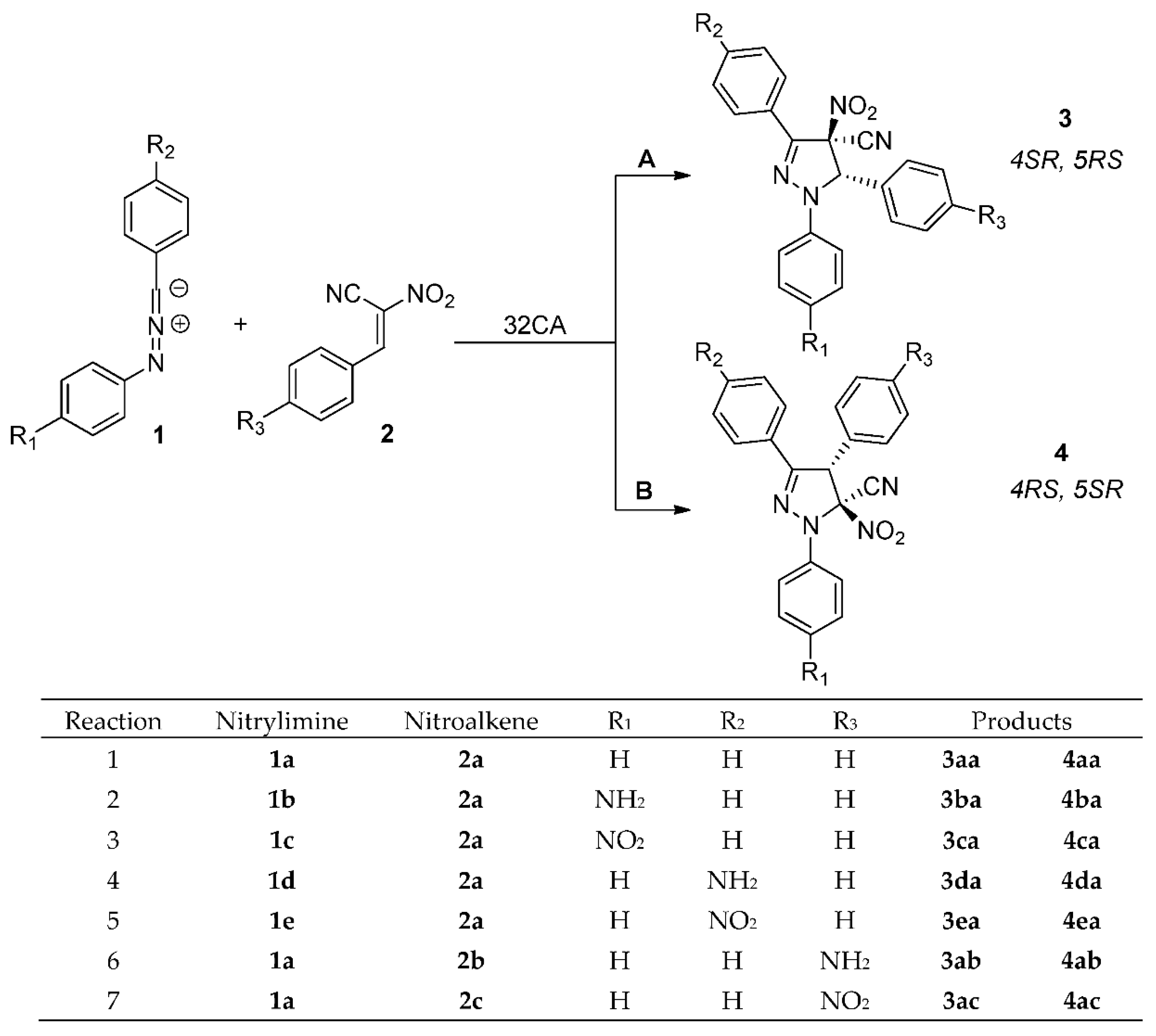
Theoretically possible, regioisomeric channels of the analyzed reactions are presented on Scheme 4. In particular, according to competitive reaction channels, 1,3,5-triaryl-4-cyano-4-nitro-Δ2-pyrazolines 3 and/or 1,3,4-triaryl-5-cyano-5-nitro-Δ2-pyrazolines 4 can be formed in the consequence of interactions between addends. Firstly, we decide to lead an exploration of the nature of these interactions.
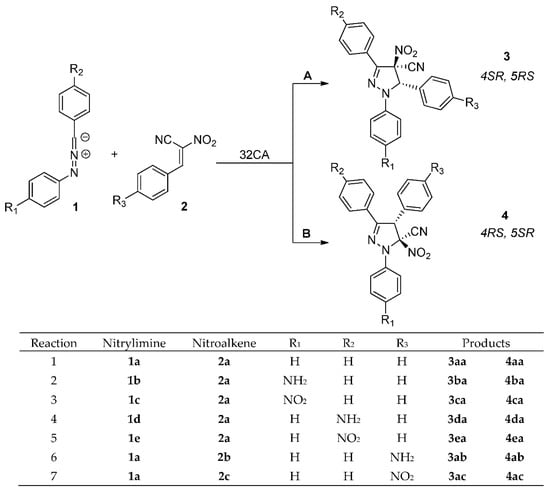
Scheme 4.
Theoretically possible channels of [3+2] cycloadditions between diarylnitrylimines 1a–e and ACNs 2a–c.
3.1. Nature of Intermolecular Interactions of Addents According to the Analysis Based on the CDFT Reactivity Indices of Reagents
3.1.1. Global Reactivity
The Conceptual Density Functional Theory (CDFT) should be considered as a strong implement that helps comprehension the reactivity of components in polar processes such as cycloaddition. All of indices was determined via calculations based on B3LYP/6-31G(d) theory level in the gas phase. It is very useful for estimate electrophilicity and nucleophilicity of the reagents [35,36,37]. Due to this, such indices as global reactivity indices: electronic chemical potential μ, chemical hardness η, global electrophilicity ω, global nucleophilicity N were determined and given in Table 1.

Table 1.
Global electronic properties (electronic chemical potential μ, chemical hardness η, global electrophilicity ω, and nucleophilicity N; all in eV) for the studied addends.
Analysis of the electronic chemical potential μ, can define the direction of the electron density flux between reagents in determined path of reaction. In case of the model reaction between nitrylimine 1a and nitroethene 2a, the electronic chemical potential μ [35,36] of 1a, −3.37 eV, is significantly higher than this of 2a, −5.28 eV, (Table 1). That means, in a course of model reaction 1a with 2a, the electron density flux will take place from nitrylimine 1a to nitroethene 2a. Similar results can be observed for the other reagents, as long as all the nitrylimine components 1b–e can be characterized by substantially higher value of electronic chemical potential than nitroethanes 2b–c (Table 1).
Calculated index of electrophilicity [35,36] ω of C,N-diphenylnitrylimine (1a) is 1.50 eV. In turn, the calculated index of nucleophilicity [13] N for this compound is 3.86 eV (Table 1). These values give the conclusion that model nitrylimine 1a acts similar to moderate electrophile and strong nucleophile in a polar reaction.
Introduction of electron-donating (ED) –NH2 group at para position of the phenyl ring in nitrylimine 1a slightly reduces the electrophilicity ω index of 1b and 1d, 1.15 and 1.29 eV, respectively, slightly increases the nucleophilicity N index to 4.40 eV (1b) and 4.11 eV (1d) (Table 1). In a consequence, both C-phenyl-N-(4-aminophenyl)-nitrylimine (1b) and C-(4-aminophenyl)-N-phenylnitrylimine (1d) can be considered as a moderate electrophiles and also strong nucleophiles.
On the other hand, the presence of electron-withdrawing (EWG) –NO2 group at para position of the phenyl ring in nitrylimine 1a significantly increases the electrophilicity ω index of 1c and 1e, respectively, 2.29 and 2.36 eV, and also slightly reduces the nucleophilicity N index to 3.47 eV (1c) and 3.58 eV (1e) (Table 1). Consequently, C-phenyl-N-(4-nitrophenyl)-nitrylimine (1c) and C-(4-nitrophenyl)-N-phenylnitrylimine (1e) can be classified both as a strong electrophiles and strong nucleophiles. Simultaneously, these nitrylimines will behave as ambiphilic species [38,39].
In turn, calculated phenyl index of nucleophilicity N for this compound is 2.87 eV (Table 1). These values provide the conclusion that model nitroalkene 2a acts similar to strong electrophile and also moderate nucleophile in a polar reaction.
The presence of both ED group such as –NH2 or and EWG group such as –NO2 in at para position of the phenyl ring in nitroalkene 2a significantly changes global electronic properties. Especially, it increases the electrophilicity ω index to 3.42 (2b) and 4.47 eV (2c) and slightly reduces nucleophilicity N index to 1.81 (2b) and 1.14 eV (2c) (Table 1). Therefore, both of (E)-2-(4-aminophenyl)-1-cyano-1-nitroethenes (2b) and (E)-2-(4-nitrophenyl)-1-cyano-1-nitroethenes (2c) can be classified as a marginal nucleophile but remains as a strong electrophile.
Polar cycloaddition reactions require the participation of good electrophiles and good nucleophiles. CDFT reactivity indices show that all of analyzed (E)-2-aryl-1-cyano-1-nitroethenes 2a–c can be classified as evidently strong electrophiles. In turn, the simplest C,N-diphenylnitrylimine (1a) and para amino analogues of diarylnitrylimines 1b and 1d can be classified as moderate electrophiles. It follows that, the difference in electrophilicity Δω index between components NIs and CNAs is significant (Δω > 1 eV) (Table 1). Therefore, it is expected, that the 32CA reaction of nitrylimines 1a, 1b and 1d with nitroethenes 2a–c will have a polar character. In turn, nitro substituted diarylnitrylimines 1c and 1e can be classified as strong electrophiles. These components have an evidently higher electrophilicity ω index than nitrylimines 1a, 1b, 1d (Table 1). It causes, that the difference in electrophilicity Δω between components para nitro NIs (1c, 1e) and CNAs (2a–c) has been significantly lower compared to the previous 32CAs (1a, 1b, 1d + 2a–c) (Δω < 1 eV) (Table 1). It follows that, the 32CA reaction of nitrylimines 1c and 1e with nitroethenes 2a–c will not have a polar character [13,16].
Very recently, organic reactions have been classified as forward electron density flux (FEDF) and reverse electron density flux (REDF) reactions, depending on the direction of the flux of the electron density [38,40]. Non-polar reactions are classified as null electron density flux (NEDF) reactions [38,41]. Thus, the reactions involving nucleophilic nitrylimines 1a, 1b and 1d and electrophilic nitroalkenes 2a–c are classified as FEDF in agreement with the CDFT analysis. On the other hand, the non-polar 32CA reactions of 1c or 1e with all of the nitrylimine components 1a–e should be classified as NEDF.
3.1.2. Local Reactivity
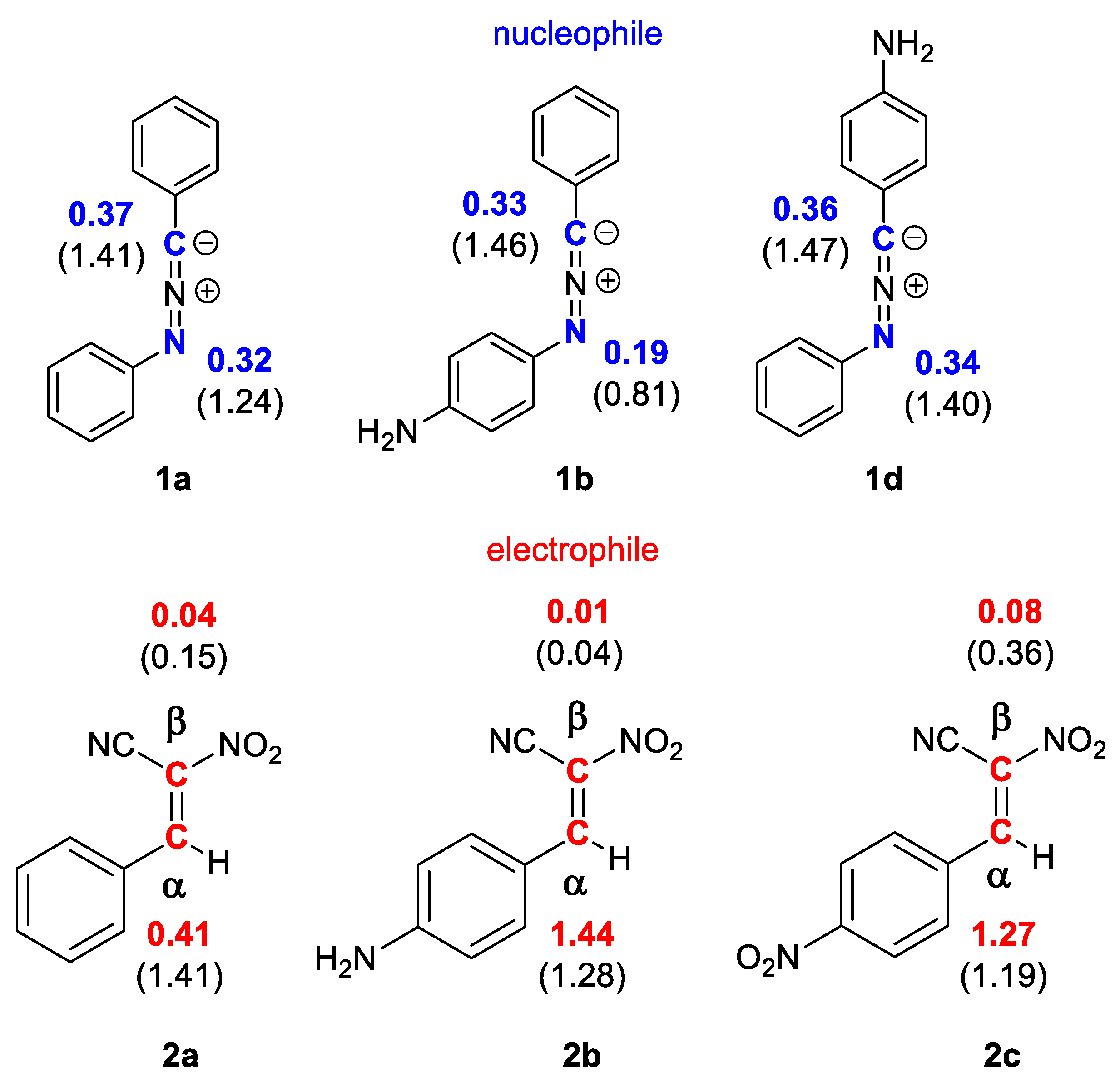
The regioselectivity of polar processes of non-symmetric reagents can be specified through interaction between the most electrophilic center of the electrophile and the most nucleophilic ones of the nucleophile. For this purpose, electrophilic Pk+ and nucleophilic Pk− Parr functions, derived from the changes of spin electron density reached via the GEDT process from the nucleophile to the electrophile, can be used as a powerful tool in the study of the local reactivity [42,43,44]. According to the nucleophilic Pk− Parr functions of nitrylimine components 1a,c,e and the electrophilic Pk+ Parr functions of nitroethanes 2a–c in order to characterize the most nucleophilic and electrophilic centers of the species involved in this polar 32CA reaction were analyzed (Figure 1).
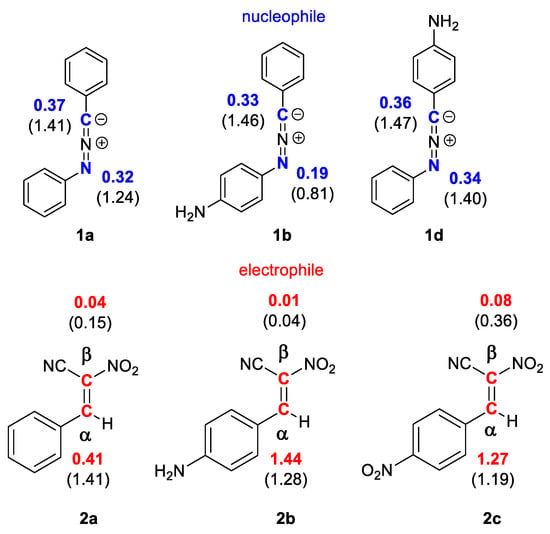
Figure 1.
The local electronic properties of nitrylimines 1a,c,e and nitroethanes 2a–c. The nucleophilic Pk− given in blue and the electrophilic Pk+ given in red; the indexes of local nucleophilicity Nk and local electrophilicity ωk given in brackets.
Analysis of the electrophilic Pk+ Parr functions of (E)-2-phenyl-1-cyano-1-nitroethenes (2a) indicates that the most electrophilic center is situated on carbon atom α, Pk+ = 1.41 eV (Figure 1). The presence of amino or nitro group at para position of the phenyl ring of nitroethenes 2b and 2c does not cause significant changes in local reactivity. It means that for nitroethenes 2b and 2c the most electrophilic center is also located in atom α of carbon, Pk+ = 1.44 eV (2b) and 1.27 eV (2c), respectively (Figure 1). It implies that center will react with the most nucleophilic α center of nitrylimines 1a,c,e.
On the other hand, the analysis of the nucleophilic Pk− Parr functions of the C,N-diphenylnitrylimine (1a) indicates that the carbon atom of –N=N=C– fragment constitutes the most nucleophilic center at molecule, presenting the maximum values at Pk− = 0.32 eV (Figure 1). Same as in the previous example the presence of amino group at para position of the benzene ring for nitroethenes 2c and 2e does not significantly change the values of local reactivities. Therefore, nitrylimines 2c and 2e show a similar reactivity when compared with C,N-diphenylnitrylimine (1a) and presenting the maximum values at Pk− = 0.33 (2c) and 0.36 (2e) eV, respectively (Figure 1).
Summarizing, according to CDFT theory [12,14] the polar reactions of nitrylimines 1a,c,e with nitroalkenes 2a–c will be realized through the interaction of α atom of carbon for 1a,c,e with the carbon atom of –N=N=C– fragment for 2a–c. Therefore, the more preferred reaction channel for analyzed [3+2] cycloaddition reactions are forming 5-nitro-substituted Δ2-isoxazolines 4a,c,e according to path B. The regioselectivity of 32CA between nitrylimines 1a,c,e and nitroalkenes 2a–c cannot be determined using similar approach due to the non-polar nature of these processes [45].
3.2. Reaction Profiles
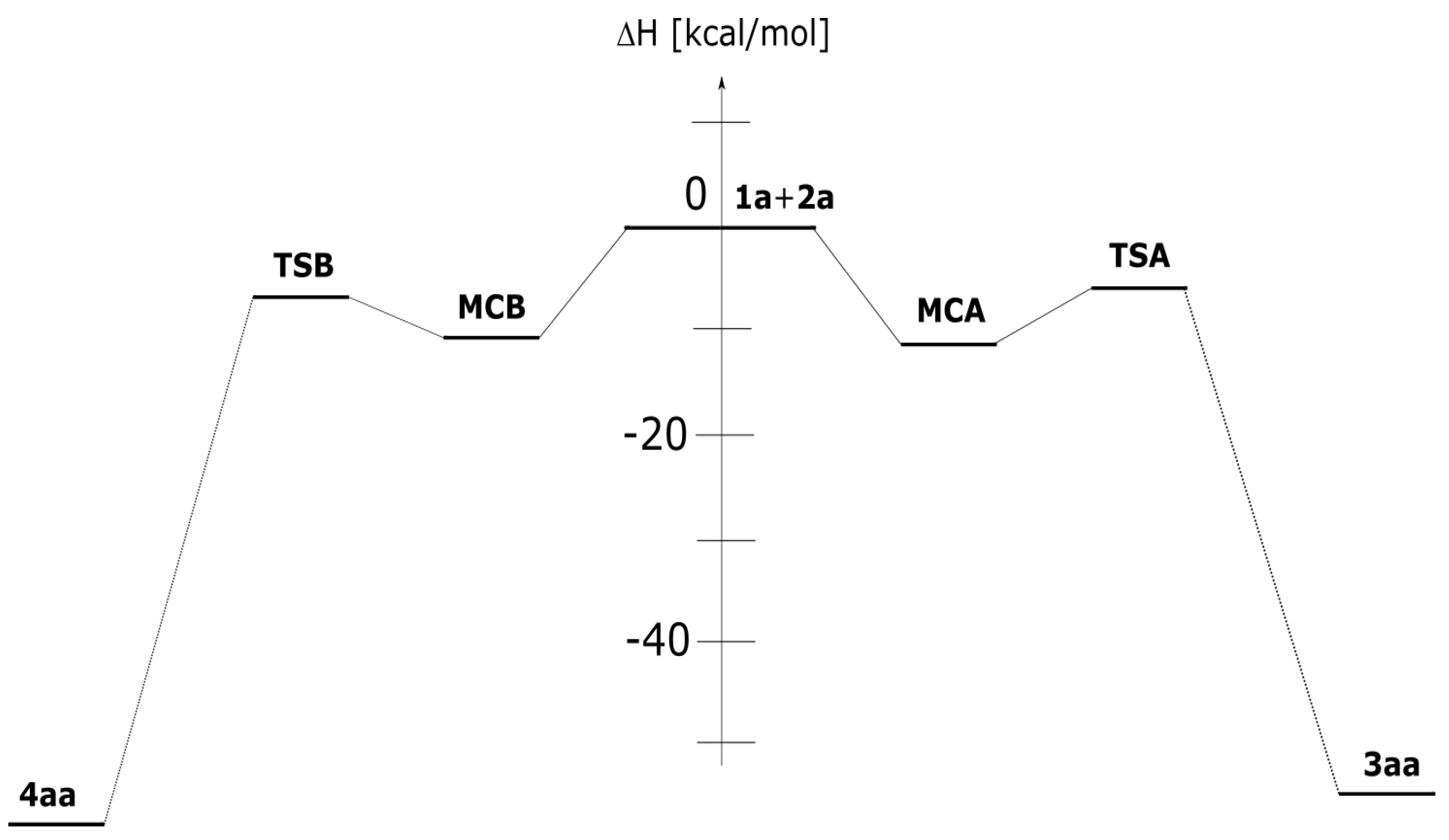
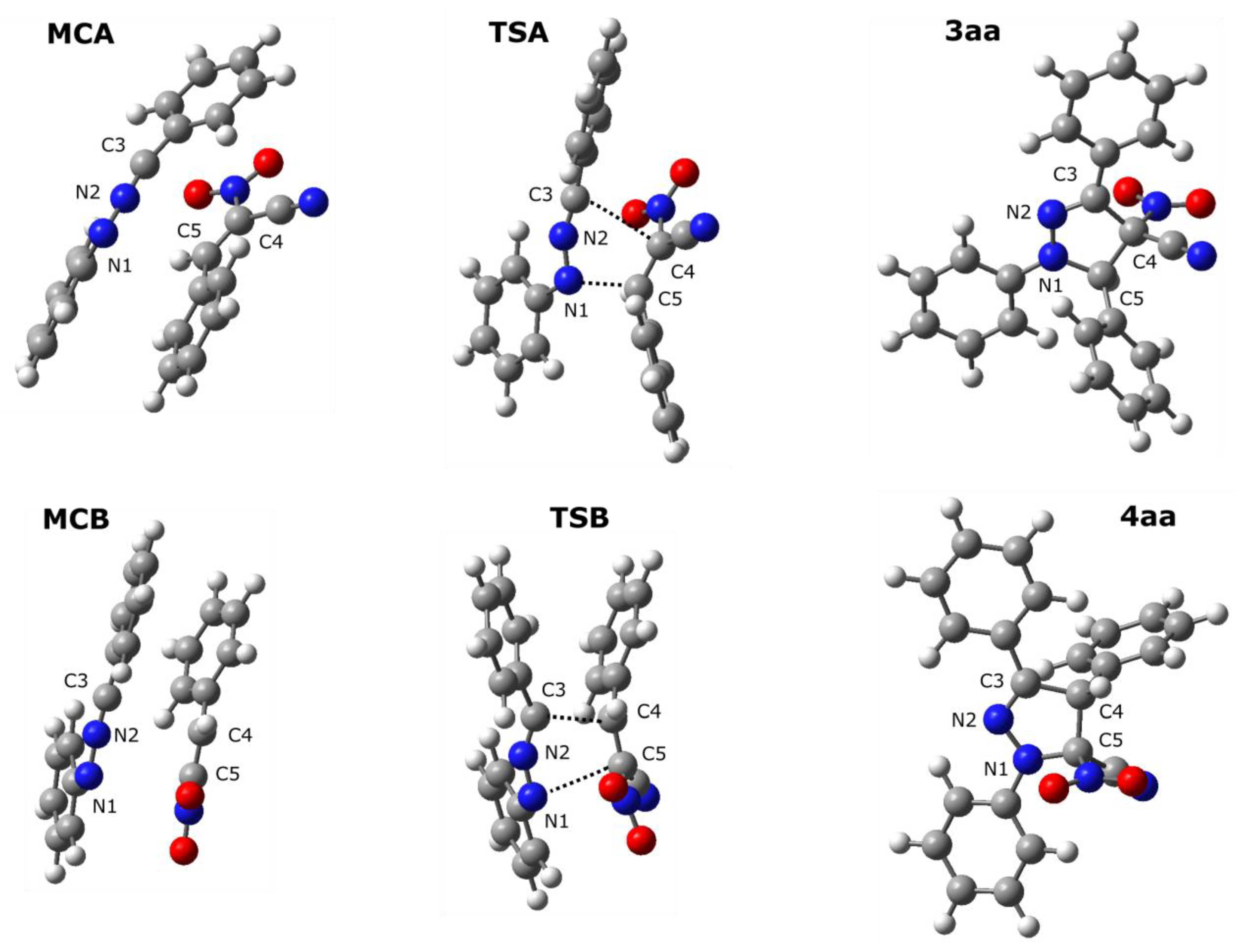
The mechanistic study regarding the title reactions we initiated from the exploration of the model process involving parent nitrylimine 1a and parent CNA 2a. It was found, that in toluene solution, the nature of energy profiles of both considered reaction channels are qualitatively similar (Figure 2). In particular, between valleys of individual reagents and products, two critical points were localized. These are connected with the existence of pre-reaction molecular complexes (MC) as well as transition states (TS). The first transformation of the reaction system is a formation of MC. This stage is characterized by reduction of the enthalpy of the reaction system about 11–12 kcal/mol (Table 2). However, the entropy factors do not determine the possibility of the existence of MCs as stable intermediates (ΔG > 0 kcal/mol). Within MCs, addends are oriented relatively, for the achieving maximally good coulombic interactions between substructures (Table 3, Figure 3). These are not a charge-transfer complexes, which was confirmed by GEDT analysis [20]. Within MCs, reaction centers are oriented for the fashion, which determine further, positive interactions between substructures. Key interatomic distances (Table 4 and Table 5) are characterized by values which are beyond of the typical range for formed sigma-bonds in transition states. The further conversion of MCs, lead on both considered paths to area of the existence of TSs. This requires an overcome of the energetical barrier about 8kcal/mol. It should be mentioned, that both considered paths should be permitted, as allowed from the kinetic point of view. Next, small difference between energies of the activation suggest rather low reaction regioselectivity. Within TSs, great amount of the charge transfer between substructures are observed (0.67e and 0.39e for TSA and TSB, respectively). This confirms expected previously, polar nature of considered cycloadditions. Next, in the framework of TS, the key interatomic distances are reduced substantially, in the comparison for analogous distances within MCs. The formation of new sigma-bonds proceeds evidently according to the asynchronous manner (Table 4 and Table 5). This is typical for polar 32Cas processes involving unsymmetrical substituted components. The asynchronicity of TSs are however not sufficient for the extort the stepwise mechanism of the cycloaddition. The IRC analysis connects without any doubts both TSs, with respective MCs and respective products.
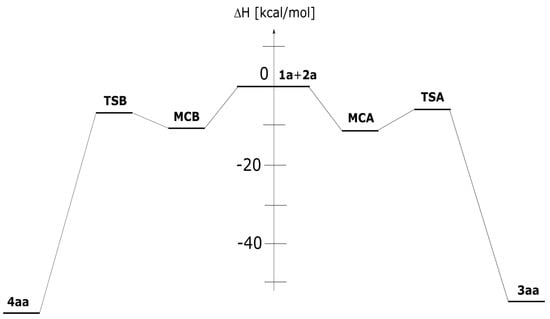
Figure 2.
Enthalpy profile for the 32CA between diarylnitrylimine 1a and can 2a in toluene solution according to the DFT computational study.
Table 2.
DFT kinetic and thermodynamic parameters for the 32Cas of diarylnitrylimines 1a–e and ACNs 2a–c in toluene (ΔH, ΔG are in kcal/mol; ΔS are in cal/mol·K).
Table 3.
DFT kinetic and thermodynamic parameters for the 32Cas of diphenylnitrylimine 1a and (E)-2-phenyl-1-cyano-1-nitroethenes 2a in nitromethane (ΔH, ΔG are in kcal/mol; ΔS are in cal/mol·K).
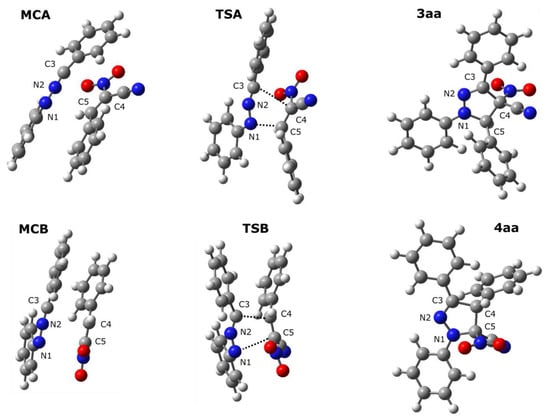
Figure 3.
Views of key structures for the 32CA between diarylnitrylimine 1a acanACN 2a in toluene solution according to the DFT computational study.
Table 4.
Selected parameters for the key structures of the 32CAs of diarylnitrylimines 1a–e and ACNs 2a–c, in toluene, obtained from DFT calculations.
Table 5.
Selected parameters for the key structures of the 32CAs of diarylnitrylimines 1a–e and ACNs 2a–c, in nitromethane, obtained from DFT calculations.
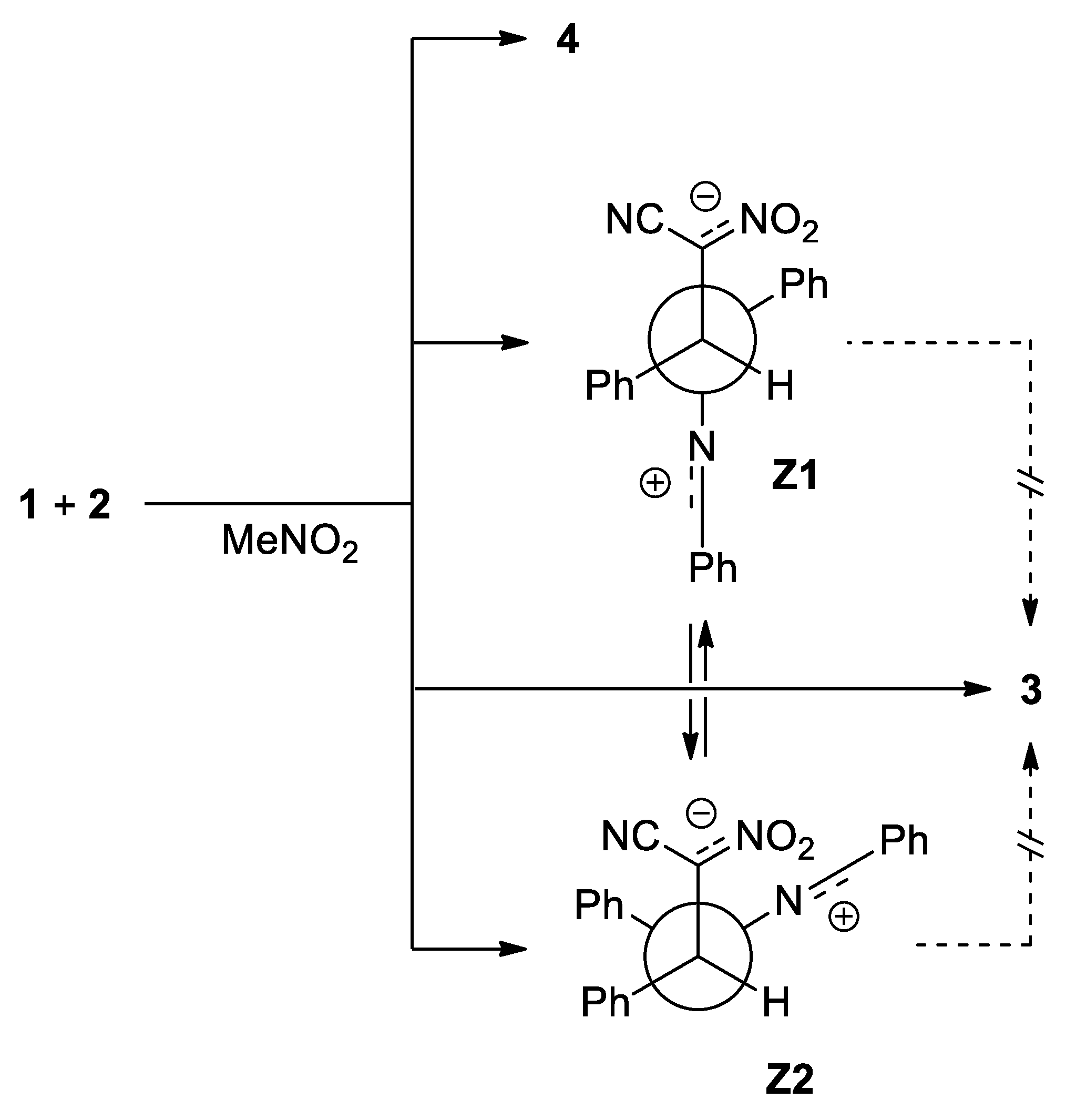
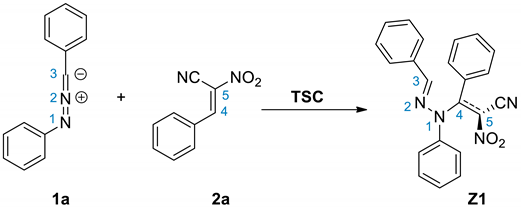
In the second part of our research, we decided to perform a similar study regarding to the 1a+2a process for the simulated presence of polar solvent—nitromethane. It was found, that both, the nature of energy profiles, as well as the quantitative description of all critical points, are close to results obtained for the simulated presence of toluene. Parallel, we detected two alternative channels of the addition reactions between considered reagents, which are not available in the toluene solution (paths C and D) (Scheme 5). In both cases, the first stage of the reaction is—similarly as in the case of 32CAs process—the formation of pre-reaction MC complexes. Their nature is close, to observed for cycloaddition reactions, but their further transformations proceed via different mechanistic scheme. In particular, conversion of mentioned MCs, leads directly to transition states TSC and TCD, respectively. Within these TSs, the distance between reaction centers C5-N1 are reduced to values, which can be considered as the stage of the formation of new sigma-bond. At the same time, the distances C3-C4 are characterized by values which are beyond of the typical range for formed sigma-bonds in transition states. Both localized TSs exhibit evidently polar nature, which is confirmed by analysis of GEDT values (Table 4 and Table 5). The further conversion of TSC and TSD leads directly for acyclic adducts Z1 and Z2, respectively. This was confirmed by IRC analysis. Optimized adducts exhibit zwitterionic nature, which is evidently right due to GEDT values (GEDT > 1e). The direct transformation of these zwitterions into cycloadduct is however impossible, as the consequence of their conformation. The theoretically possible transformations from Z1 or Z2 to pyrazoline systems must proceed via dissociation in to individual reagents, and, in next step, via 32CA processes described above. Only, transformation between Z1 and Z2 zwitterions are possible, according to the simple rotation around C5-N1 bond.
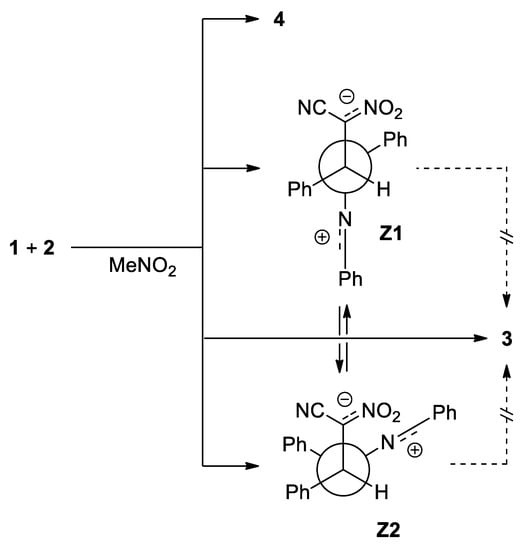
Scheme 5.
The formation and transformations of zwitterionic intermediates derived via reactions between diarylnitrylimine 1a and ACN 2a.
Lastly, we examined the influence of the substituents’ nature into reaction course. It was found that, the molecular reaction mechanism in all cases is close to observed for 32CAs involving diarylnitrylimine 1a and ACN 2a. In particular, in any case the stepwise zwitterionic mechanism was not detected. Only, the qualitative description of energy profile as well as key structures were changing in some range.
3.3. BET Analysis of the 32CA between C,N-diphenylnitrylimine 1a and (E)-2-phenyl-1-cyano-1-nitroethene 2a
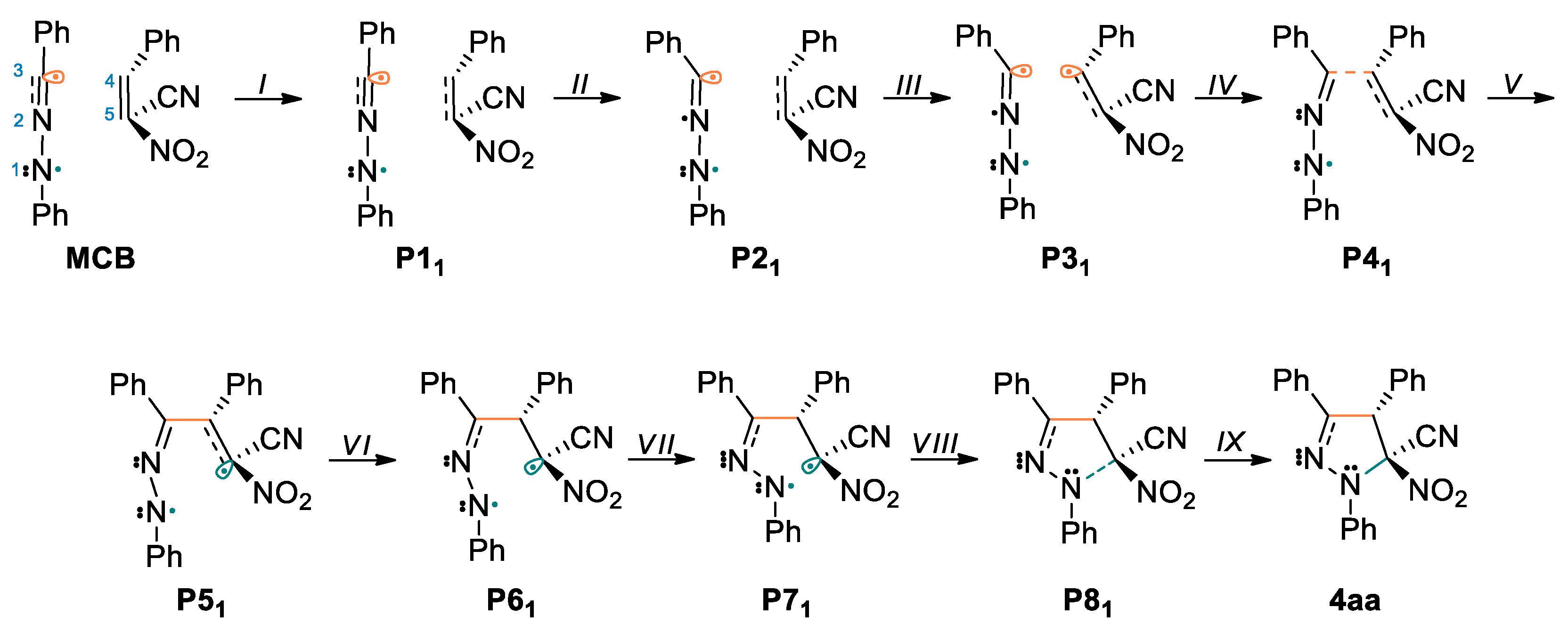
In order to carefully investigate the bonding changes occurring in the 32CA of the 1a and 2a, we decided to conduct a BET analysis. Scheme 6 represents the molecular mechanism by Lewis-like structures resulted from the ELF topological analysis. The most significant ELF basin populations together with attractor positions for a reaction leading to 4aa are gathered in Table 6 and Figure 4.
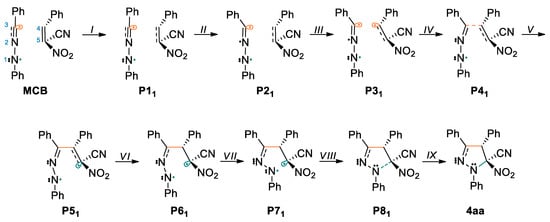
Scheme 6.
The proposed molecular mechanism of the 32CA reaction between 1a and 2a.
Table 6.
BET analysis results for the 32CA reaction of the 1a with 2a. The table also lists the structures 1a, 2a, MCB, TSB and 4aa.
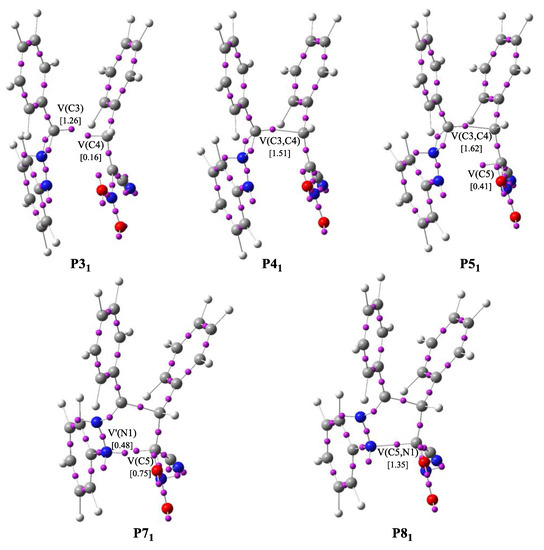
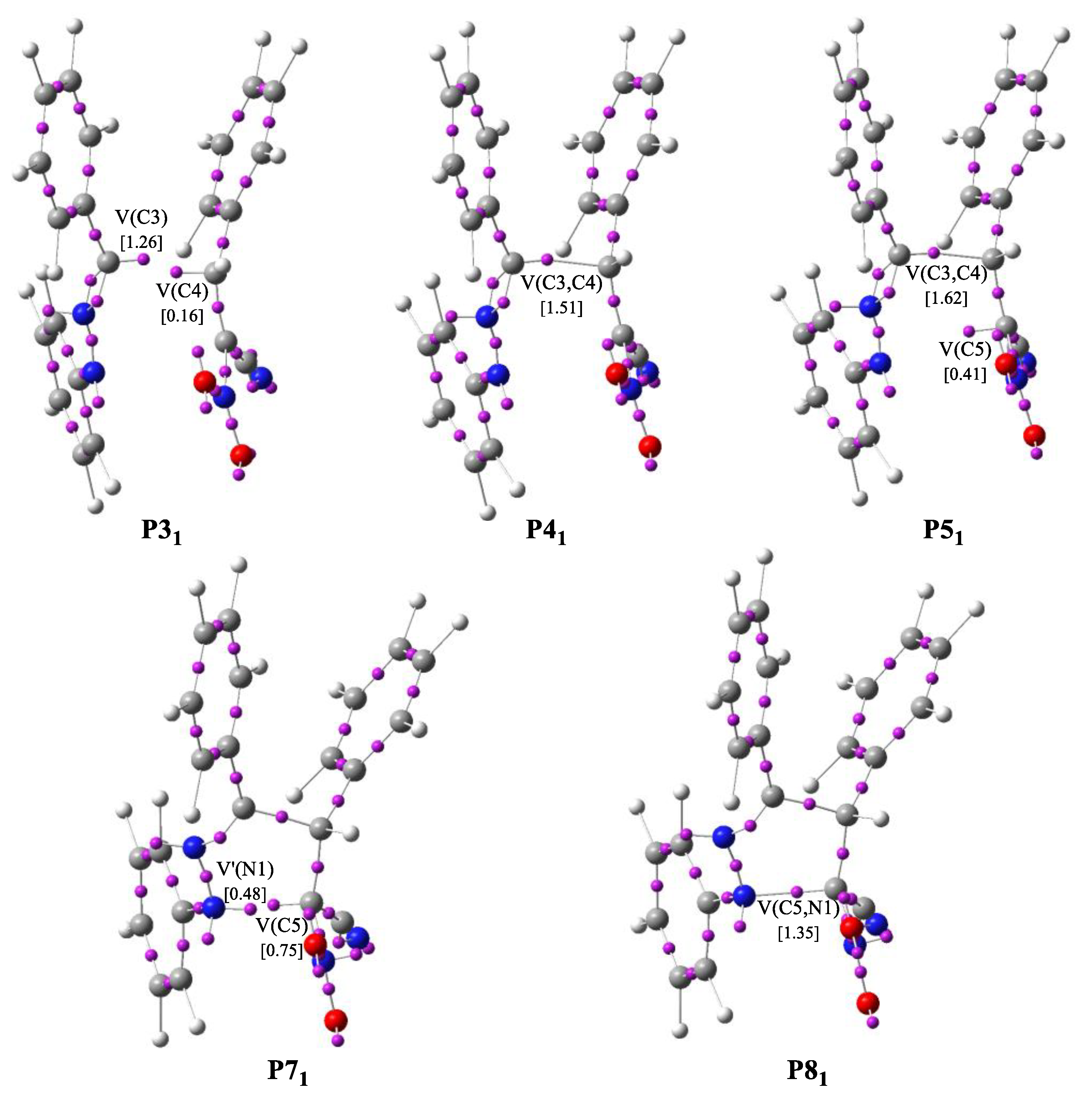
Figure 4.
The main ELF valence basin populations for the points P31–P51, P71 and P81 participating in the 32CA reaction of the 1a and 2a.
The bonding changes along this 32CA between C,N- diphenylnitrylimine 1a and 2a are characterized by nine different topological phases (Table 4). The ELF picture of the first structure MCB is very similar to those of the divided reagents. Phase II begins at P11, where it can be observed that the V(C4,C5) and V’(C4,C5) disynaptic basins current in previous MCB point, have combined into one new V(C4,C5) disynaptic basin with initial population 3.45 e. Along Phase III, which begins at P21, the new V(N2) monosynaptic basin is created with population of 0.94 e. Creation of this monosynaptic basin is related to depopulation of V(N1,N2), V(N2,C3) and V’(N2,C3) disynaptic basins and the growth of the population of V(C3) monosynaptic basin. At this point, we found the TS structure of the 32CA between 1a and 2a (TSB, d(C3-C4) = 2.176 Å and d(C5-N1) = 2.646 Å). Phase IV initiates at the structure P31, where can be noticed the creation of a V(C4) monosynaptic basin with 0.16 e, which is connected with depopulation of V(C4,C5) disynaptic basin. Creation of this monosynaptic basin is associated with formation a pseudoradical [46] center at C4 carbon atom (Figure 4). Along Phase V, which begins at P41, we noticed that pseudoradical centers located on C3 and C4 atoms combined into C3-C4 bonding area with an starting value of 1.41 e. The electron density changes mean that creation of the C3-C4 bond starts with a distance d(C3,C4) = 2.014 Å, by divvying the non-bonding electron densities of the two C3 and C4 centers. Phase VI, starts at P51, and is related to formation a new pseudoradical center at C5 carbon atom with value of 0.41 e (Figure 4 and Table 6). Formation of a new V(C5) monosynaptic basin causes depopulation of V(C4,C5) disynaptic basin. In Phase VII, the V(N2,C3) and V’(N2,C3) disynaptic basins, have merged and a new V(N2,C3) disynaptic basin was created with 3.17 e. This shift is related to disruption of the N2-C3 bond and formation a partial double bond (Scheme 6). At P71 begins Phase VIII, which is associated with creation a V’(N2) monosynaptic basin with starting value of 0.48 e. This shift is caused by division of V(N1) monosynaptic basin, present in P61, for two new V(N1) and V’(N1) monosynaptic basins and depopulation of V(N1,N2) disynaptic basin. Subsequently we observed in the last Phase IX, which is located between P81 and 4aa (Table 6). In this phase, the creation of the second C5-N1 bond follows through the connection of the non-bonding electron densities of the two V(C5) and V(N1) monosynaptic basins. The C5-N1 bond was formed with starting distance of d(C5,N1) = 1.750 Å.
On the grounds of the BET study, we gather that: (i) the 32CA reaction between C,N- diphenylnitrylimine 1a and (E)-2-phenyl-1-cyano-1-nitroethene 2a can be depicted by nine topologically various phases; (ii) the activation energy of this reaction, 4.0 kcal·mol−1, is related to the formation of C4 pseudoradical center and lone pair at N2 nitrogen atom; (iii) creation of first C3-C4 bond follows in Phase V by way of merging two C3 and C pseudoradical centers; (iv) creation of the C5-N1 bond occurs in the last Phase IX by merging the C5 pseudoradical center and N1 non-bonding lone pair; (v) it is worth nothing that when the C5-N1 bond begins to form, the first bond is fully formed. According to that, we can conclude that the 32CA of the 1a and 2a proceeds according to one-step two-stage mechanism.
3.4. BET Study of the Creation of Acyclic Adduct Z1
We also decided to analyze the bonding changes along the formation acyclic adduct Z1 by doing the BET analysis. The most significant ELF basin populations and attractor positions for structures participating in the studied reaction are collected in Table 7 and Figure 5.
Table 7.
BET analysis results for the formation of acyclic adduct Z1. The table also lists the structures 1a, 2a, MCC, TSC and Z1.
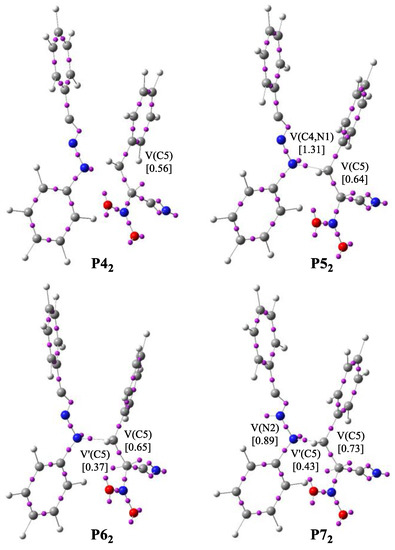
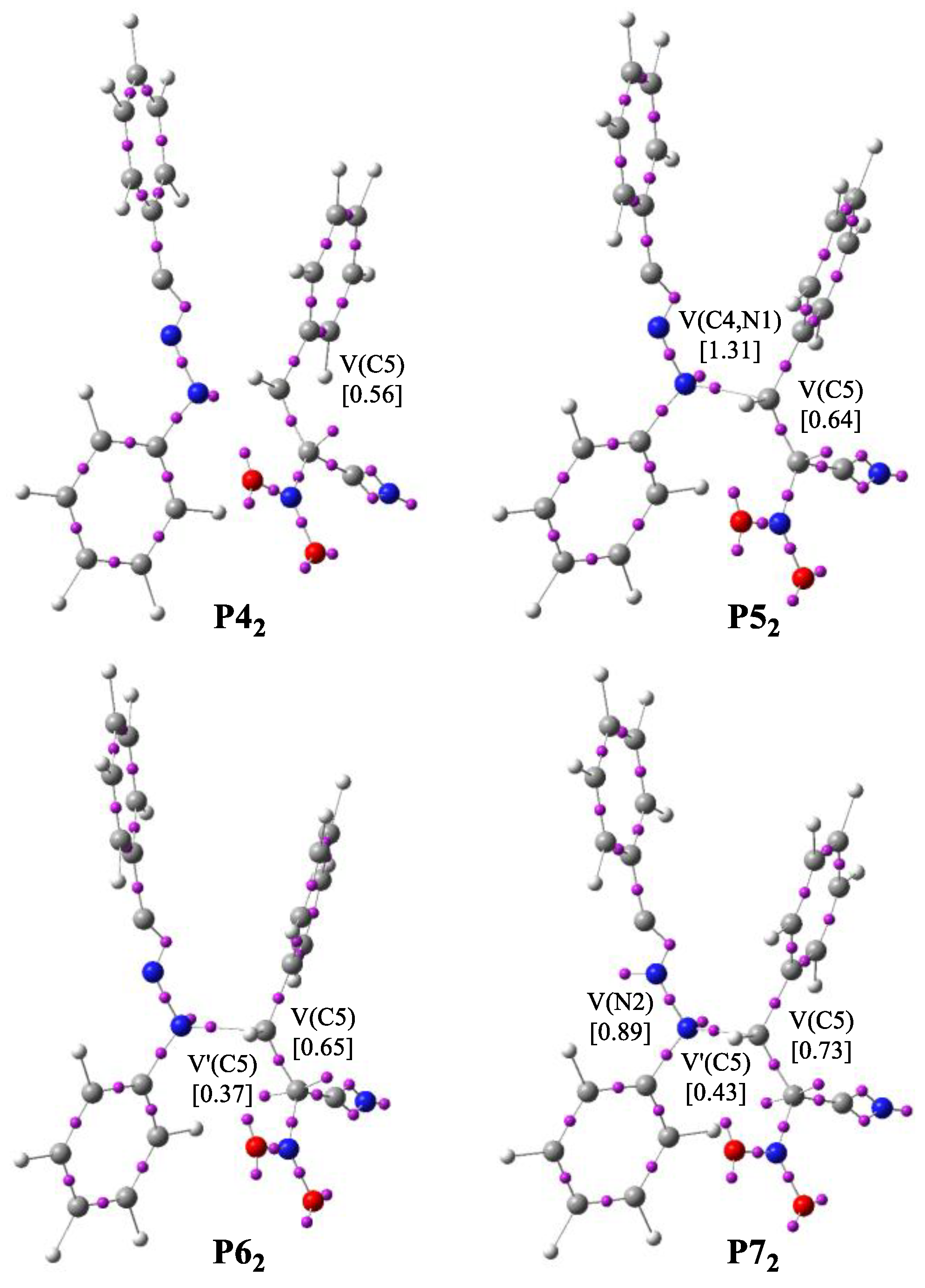
Figure 5.
The main ELF valence basin populations for the points P42–P72 participating in the formation of Z1.
The bonding changes along the formation of acyclic adduct Z1 are described by eight various phases (Table 7). The first point of the IRC is the molecular complex MCC, which presents ELF picture of separated reagents. We observed that the V(N1) monosynaptic and V(N2,C3), V’(N2,C3) and V(C4,C5) disynaptic basins show a slight larger ELF valence basin populations than those shown in the substrates. In turn, in the event of V(N1,N2) and V’(C4,C5) disynaptic and V(C3) monosynaptic basins, we observed the lower values of the ELF basin populations. Phase II begins at structure P12. In this area, the two V(C4,C5) and V’(C4,C5) disynaptic basins merged into one new V(C4,C5) disynaptic basin with starting population of 3.49 e. This topological shift is connected with break of C4-C5 bond in (E)-2-phenyl-1-cyano-1-nitroethene 2a. Along Phase III, the C3 monosynaptic basin disappear and we observed the increase in the population of V(N2,C3) disynaptic basin to about 3.81 e. Along Phase IV, the V(N2,C3) and the V’(N2,C3) disynaptic basins merged into one original V(N2,C3) disynaptic basin integrating 5.87 e. At P32, we notice the TS of the formation of adduct Z1 (TSC, d(C4-N1) = 2.062 Å). Phase V begins at P42 and is related to create a V(C5) monosynaptic basin integrating 0.56 e. This is connected with a creation of pseudoradical center at C5 carbon atom and depopulation of V(C4,C5) disynaptic basin. In Phase VI, we observed the formation a new V(C4,N1) disynaptic basin with population of 1.31 e, created by depopulation of V(N1) monosynaptic and V(C4,C5) disynaptic basins. This topological change is associated with formation of the C4-N1 single bond (Figure 5). The next Phase VII begins at P62 and is associated with creation of a second V’(C5) monosynaptic basin with population of 0.37 e. Forming of pseudoradical center at C5 carbon atom is associated with decrease the population of V(C4,C5) disynaptic basin. The final Phase VIII, placed between P72 and Z1 is related to formation a V(N2) monosynaptic basin through the depopulation of V(N2,C3) disynaptic basin.
On the basis of BET study, we may say that: (i) the molecular mechanism of the formation of acyclic adduct Z1 can be featured by eight various phases; (ii) the activation energy of this reaction, 8.1 kcal·mol−1, is mostly related to breaking of the N2-C3 double bond and formation of C5 pseudoradical center; (iii) formation of C4-N1 single bond takes place in Phase VI by the depopulation of V(N1) monosynaptic and V(C4,C5) disynaptic basins.
4. Conclusions
The DFT computational study shows, that despite of high electrophilic nature of nitroalkenes, the [3+2] cycloaddition reactions between diarylnitrylimines and 2-aryl-1-cyano-1-nitroethenes proceed via single-step mechanism. In a polar nitromethane solution, the cycloaddition process can however compete, with the formation of zwitterionic structures characterized by “extended” conformations. These intermediates cannot be cyclized in to heterocyclic systems via simple one-step reactions, because the key reaction sites are localized on too long relative distance. Their conversion into pyrazoline systems is possible only via the stage of dissociation into individual reagents, and subsequently, one-step cycloaddition.
The topological analysis of the bonding changes associated with 32CA reaction between C,N-diphenylnitrylimine 1a and (E)-2-phenyl-1-cyano-1-nitroethene 2a, can be described by nine topologically different phases. Formation of the first C-C single bond follows by merging of two pseudoradical centers. In turn, the second C-N bond is formed in the last phase of the reaction when the first bond is fully formed. According to that, we can conclude that the 32CA of the 1a and 2a progress according to one-step two-stage mechanism.
Author Contributions
Conceptualization, R.J.; methodology, K.Z., P.W., K.K., A.K.-Z. and A.Ł.-K.; software, R.J., K.Z., P.W., K.K., A.K.-Z. and A.Ł.-K.; formal analysis, R.J., K.Z., K.K. and A.K.-Z.; investigation, R.J., K.Z., P.W., K.K., A.K.-Z. and A.Ł.-K.; writing—original draft preparation, R.J., K.Z., K.K. and A.K.-Z.; writing—review and editing, A.F. and A.O.; visualization, R.J., A.F., A.O., K.K. and A.K.-Z.; supervision, R.J., K.Z., K.K. and A.K.-Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ried, W.; Köhler, E. Reaktionen mit Nitro-acetonitril. Lieb. Ann. Chem. 1956, 598, 145–158. [Google Scholar] [CrossRef]
- Kanishchev, M.I.; Korneeva, N.; Shevelev, S.A. Synthesis of 3-amino-5-benzylamino-4-nitropyrazole. Bull. Acad. Sci. USSR 1986, 35, 2145–2147. [Google Scholar] [CrossRef]
- Boguszewska-Czubara, A.; Kula, K.; Wnorowski, A.; Biernasiuk, A.; Popiołek, Ł.; Miodowski, D.; Demchuk, O.M.; Jasiński, R. Novel functionalized β-nitrostyrenes: Promising candidates for new antibacterial drugs. Saudi Pharm. J. 2019, 27, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Jasiński, R.; Mirosław, B.; Demchuk, O.M.; Babyuk, D.; Łapczuk-Krygier, A. In the search for experimental and quantumchemical evidence for zwitterionic nature of (2E)-3-[4-(dimethylamino)phenyl]-2-nitroprop-2-enenitrile—An extreme example of donor–π–acceptor push–pull molecule. J. Mol. Struct. 2016, 1108, 689–697. [Google Scholar] [CrossRef]
- Łapczuk-Krygier, A.; Ponikiewski, Ł.; Jasiński, R. The crystal structure of (1RS,4RS,5RS,6SR)-5-cyano-5-nitro-6-phenyl-bicyclo[2.2.1]hept-2-ene. Crystallogr. Rep. 2014, 59, 961–963. [Google Scholar] [CrossRef]
- Łapczuk-Krygier, A.; Jasiński, R. New Endo-Nitro-Substituted Norbornene Derivatives and Stereoselective Method for Producing New Endo-Nitro-Substituted Norbornene Derivatives. Patent No. PL 231749, 13 March 2017. [Google Scholar]
- Poos, G.I.; Kleis, J.; Wittekind, R.R.; Rosenau, J.D. Bicyclic Bases. III. Isomeric 2-Amino-3-phenylnorbornanes. J. Org. Chem. 1961, 26, 4898–4904. [Google Scholar] [CrossRef]
- Żmigrodzka, M.; Dresler, E.; Hordyjewicz-Baran, Z.; Kulesza, R.; Jasiński, R. A unique example of noncatalyzed 32CAinvolving (2E)-3-aryl-2-nitroprop-2-enenitriles. Chem. Heterocycl. Compd. 2017, 53, 1161–1162. [Google Scholar] [CrossRef]
- Jasiński, R.; Kula, K.; Kącka, A.; Mirosław, B. Unexpected course of reaction between (E)-2-aryl-1-cyano-1-nitroethenes and diazafluorene: Why is there no 1,3-dipolar cycloaddition? Mon. Chem. 2017, 148, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Woliński, P.; Kącka-Zych, A.; Demchuk, O.M.; Łapczuk-Krygier, A.; Mirosław, B.; Jasiński, R. Clean and molecularly programmable protocol for preparation of bis-heterobiarylic systems via a domino pseudocyclic reaction as a valuable alternative for TM-catalyzed cross-couplings. J. Clean. Prod. 2020, 275, 122086. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, P.; Domingo, L.R.; Duque-Noreña, M.; Chamorro, E. A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J. Mol. Struct. 2009, 895, 86–91. [Google Scholar] [CrossRef]
- Greelings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Zawadzińska, K.; Kula, K. Application of ß-phosphorylated nitroethenes in [3+2] cycloaddition reactions involving benzonitrile N-oxide in the light of DFT computational study. Organics 2021, 2, 26–37. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1.3-dipolar cycloaddition reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Saez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Fox, D.J.; et al. (Eds.) Gaussian 16; Revision, A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry Optimization of Molecular Structures in Solution by the Polarizable Continuum Model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Jasiński, R. A stepwise, zwitterionic mechanism for the 1,3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalysed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett. 2015, 56, 532–535. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Silvi, B.; Colonna, F. Topological Analysis of the Electron Localization Function Applied to Delocalized Bonds. Can. J. Chem. 1996, 74, 1088–1096. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Krokidis, K.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7281. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, K.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Mirosław, B.; Babyuk, D.; Łapczuk-Krygier, A.; Kącka-Zych, A.; Demchuk, O.M.; Jasiński, R. Regiospecific formation of the nitromethyl-substituted 3-phenyl-4,5-dihydroisoxazole via [3+2] cycloaddition. Chem. Mon. 2018, 149, 1877–1884. [Google Scholar] [CrossRef]
- Kula, K.; Dobosz, J.; Jasiński, R.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Mirosław, B.; Demchuk, O.M. [3+2] Cycloaddition of diaryldiazomethanes with (E)-3,3,3-trichloro-1-nitroprop-1-ene: An experimental, theoretical and structural study. J. Mol. Struct. 2020, 1203, 127473. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Domingo, L.R.; Ríos-Gutiérrez, M.; Jasiński, R. Understanding the mechanism of the decomposition reaction of nitroethyl benzoate through the Molecular Electron Density Theory. Theor. Chem. Acc. 2017, 136, 129. [Google Scholar] [CrossRef] [Green Version]
- Kącka-Zych, A.; Jasiński, R. Unexpected molecular mechanism of trimethylsilyl bromide elimination from 2-(trimethylsilyloxy)-3-bromo-3-methyl-isoxazolidines. Theor. Chem. Acc. 2019, 138, 81. [Google Scholar] [CrossRef] [Green Version]
- Woliński, P.; Kącka-Zych, A.; Dziuk, B.; Ejsmont, K.; Łapczuk-Krygier, A.; Dresler, E. The structural aspects of the transformation of 3-nitroisoxazoline-2-oxide to 1-aza-2,8-dioxabicyclo[3.3.0]octane derivatives: Experimental and MEDT theoretical study. J. Mol. Struct. 2019, 1192, 27–34. [Google Scholar] [CrossRef]
- Kącka, A.; Jasiński, R. A dramatic change of kinetic conditions and molecular mechanism of decomposition processes of nitroalkyl carboxylates catalyzed by ethylammonium cations. Comput. Theor. Chem. 2017, 1104, 37–42. [Google Scholar] [CrossRef]
- Kącka-Zych, A. Participation of Phosphorylated Analogues of Nitroethene in Diels–Alder Reactions with Anthracene: A Molecular Electron Density Theory Study and Mechanistic Aspect. Organics 2020, 1, 36–48. [Google Scholar] [CrossRef]
- Kącka-Zych, A. The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3+2) Cycloaddition: A DFT Mechanistic Study. Molecules 2021, 26, 4786. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. Molecular Electron Density Theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, e1319. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Kula, K.; Zawadzinska, K. Local nucleophile-electrophile interactions in [3+2] cycloaddition reactions between benzonitrile N-oxide and selected conjugated nitroalkenes in the light of MEDT computational study. Curr. Chem. Lett. 2021, 10, 9–16. [Google Scholar] [CrossRef]
- Domingo, L.R.; Kula, K.; Ríos-Gutiérrez, M. Unveiling the reactivity of cyclic azomethine ylides in [3+2] cycloaddition reactions within the Molecular Electron Density Theory. Eur. J. Org. Chem. 2020, 5938–5948. [Google Scholar] [CrossRef]
- Mlostoń, G.; Kula, K.; Jasiński, R. A DFT Study on the Molecular Mechanism of Additions of Electrophilic and Nucleophilic Carbenes to Non-Enolizable Cycloaliphatic Thioketones. Molecules 2021, 26, 5562. [Google Scholar] [CrossRef]
- Domingo, L.R.; Kula, K.; Ríos-Gutiérrez, M.; Jasiński, R. Understanding the Participation of Fluorinated Azomethine Ylides in Carbenoid-Type [3+2] Cycloaddition Reactions with Ynal Systems: A Molecular Electron Density Theory Study. J. Org. Chem. 2021, 86, 12644–12653. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A molecular electron density theory study of the participation of tetrazines in aza-Diels–Alder reactions. RSC Adv. 2020, 10, 15394–15405. [Google Scholar] [CrossRef] [Green Version]
- Zawadzińska, K.; Ríos-Gutiérrez, M.; Kula, K.; Woliński, P.; Mirosław, B.; Krawczyk, T.; Jasiński, R. The Participation of 3,3,3-Trichloro-1-nitroprop-1-ene in the [3+2] Cycloaddition Reaction with Selected Nitrile N-Oxides in the Light of the Experimental and MEDT Quantum Chemical Study. Molecules 2021, 26, 6774. [Google Scholar] [CrossRef]
- Mlostoń, G.; Jasiński, R.; Kula, K.; Heimgartner, H. A DFT Study on the Barton-Kellogg Reaction—The molecular mechanism of the formation of thiiranes in the reaction between diphenyldiazomethane and diaryl thioketones. Eur. J. Org. Chem 2020, 176–182. [Google Scholar] [CrossRef]
- Kula, K.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Jasiński, R. Analysis of the possibility and molecular mechanism of carbon dioxide consumption in the Diels-Alder processes. Pure Appl. Chem. 2021, 93, 427–446. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos Gutiérrez, M.; Castellanos Soriano, J. Understanding the Origin of the Regioselectivity in Non-Polar [3+2] Cycloaddition Reactions through the Molecular Electron Density Theory. Organics 2020, 1, 19–35. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the High Reactivity of the Azomethine Ylides in [3+2] Cycloaddition Reactions. Lett. Org. Chem. 2010, 7, 432–439. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).