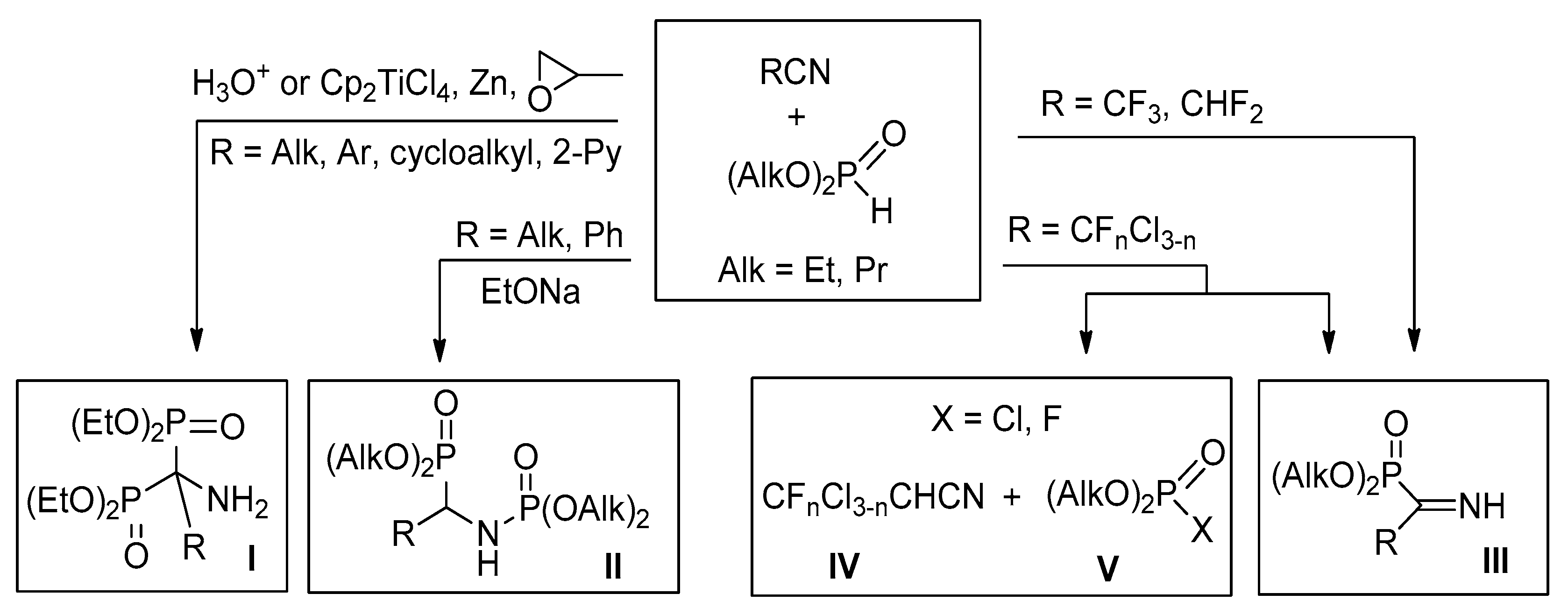
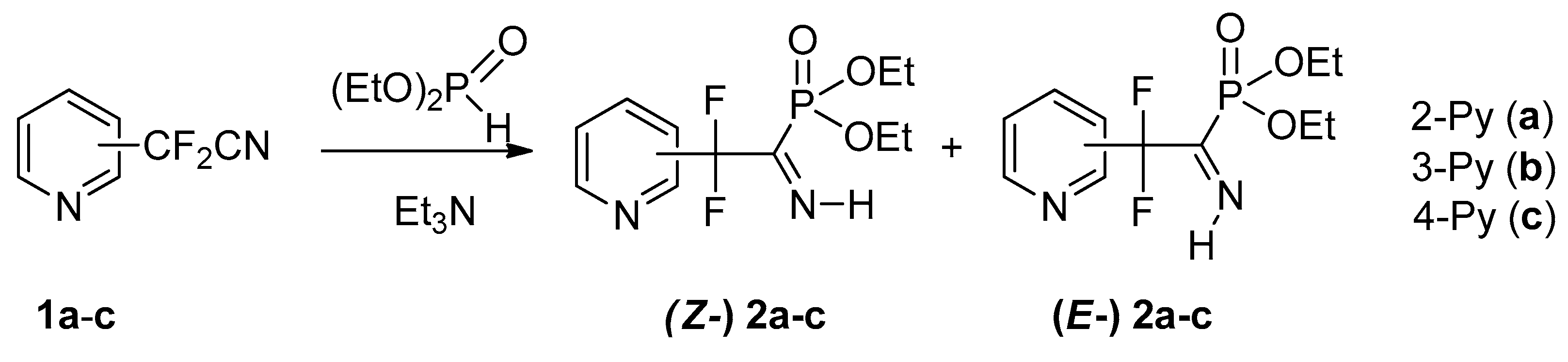
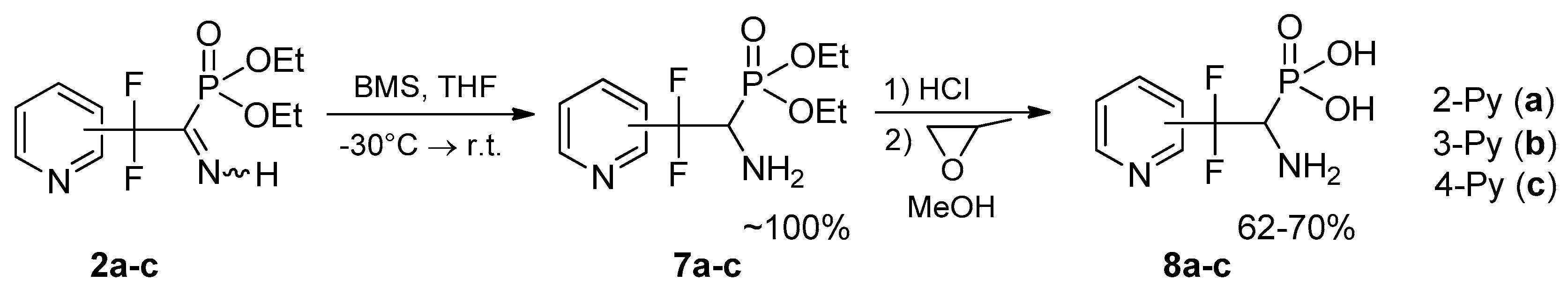
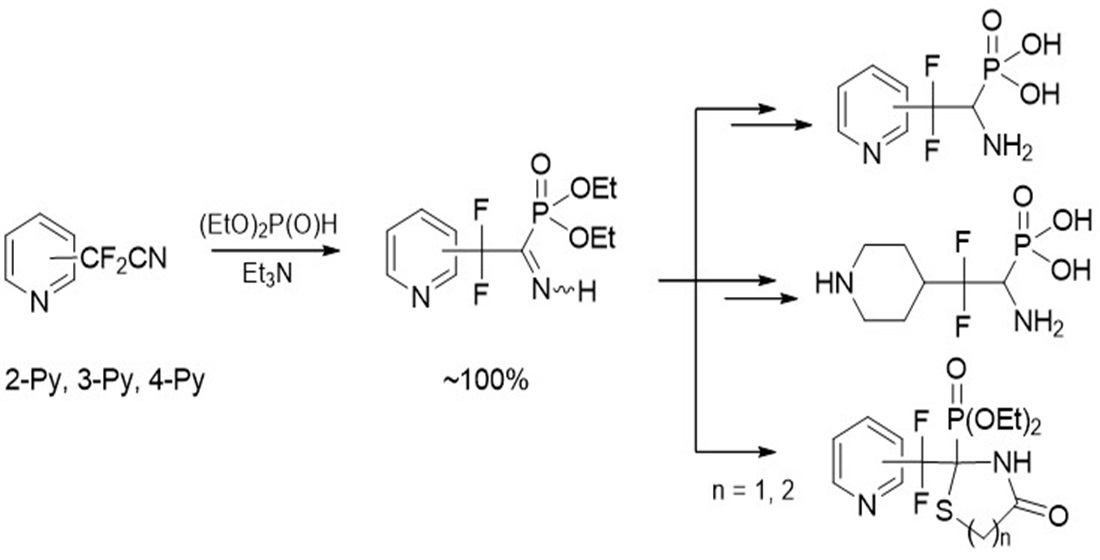
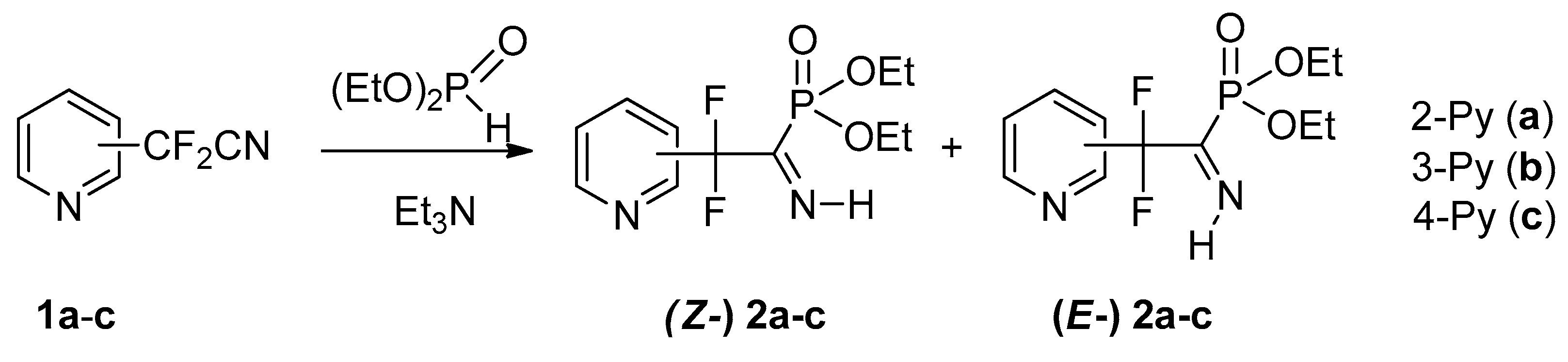
4.1. Synthesis of Diethyl (2,2-Difluoro-1-imino-2-(pyridinyl)ethyl)phosphonates 2a–c: General Procedure
To equimolar mixture of pyridinylacetonitrile 1 (0.45 g, 2.9 mmol) and diethyl phosphite (0.4 g, 2.9 mmol) was added triethylamine (0.58 mmol, 0.059 g). After 12 h for 2b,c and 120 h for 2a triethylamine was removed under vacuum.
4.1.1. Diethyl (2,2-Difluoro-1-imino-2-(pyridin-2-yl)ethyl)phosphonate 2a
Yield 0.85 g (100%); light brown oil. Z/E = 2:1. Z-isomer: 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.33 (t, 3JHH = 7.2 Hz, 6H, CH3), 4.16–4.22 (m, 4H, CH2), 7.40 (t, 3JHH = 7.9 Hz, 1H, Py), 7.74 (d, 3JHH = 7.9 Hz, 1H, Py), 7.84–7.88 (m, 1H, Py), 8.66 (d, 3JHH = 4.9 Hz, 1H, Py), 12.27 (d, 3JHP = 39.9 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.0 (d, 3JCP = 6 Hz, CH3), 63.8 (d, 2JCP = 6 Hz, CH2), 116.1 (td, 1JCF = 246 Hz, 2JCP = 33 Hz, CF2), 120.9 (t, 3JCF = 4 Hz, CPy), 125.1 (s, CPy), 136.9 (s, CPy), 149.3 (s, CPy), 152.8 (t, 2JCF = 28.5 Hz, CCF2), 173.2 (dt, 1JCP = 153 Hz, 2JCF = 33 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.2.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 5.9 (m, 3JPH = 39.9 Hz).
E-isomer: 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.26 (t, 3JHH = 7.1 Hz, 6H, CH3), 4.10–4.15 (m, 4H, CH2), 7.40 (t, 3JHH = 7.7 Hz, 1H, Py), 7.80–7.84 (m, 2H, Py), 8.6 (d, 3JHH = 4.9 Hz, 1H, Py), 11.97 (dt, 3JHP = 61.6 Hz, 4JHF = 4.0 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.2 (d, 3JCP = 6 Hz, CH3), 63.7 (d, 2JCP = 6 Hz, CH2), 113.9 (td, 1JCF = 249 Hz, 2JCP = 42 Hz, CF2), 120.8 (t, 3JCF = 4 Hz, CPy), 125.4 (s, CPy), 137.3 (s, CPy), 149.1 (s, CPy), 151.5 (t, 2JCF = 29 Hz, CCF2), 170.8 (dt, 1JCP = 210 Hz, 2JCF = 30 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −103.5.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 6.6 (m, 3JPH = 61.6 Hz).
Anal. Calc. for C11H15F2N2O3P: C, 45.21; H, 5.17; N, 9.59. Found: C, 45.38; H, 5.29; N, 9.67.
4.1.2. Diethyl (2,2-Difluoro-1-imino-2-(pyridin-3-yl)ethyl)phosphonate 2b
Yield 0.85 g (100%); yellow oil. Z/E = 6:1. Z-isomer: 1H NMR (302 MHz, CDCl3) δ (ppm): 1.27 (t, 3JHH = 7.1 Hz, 6H, CH3), 3.93–4.20 (m, 4H, CH2), 7.29 (dd, 3JHH = 8.1 Hz, 3JHH = 5.1 Hz, Py), 7.81 (d, 3JHH = 8.1 Hz, 1H, Py), 8.63 (d, 3JHH = 5.1 Hz, 1H, Py), 8.74 (s, 1H, Py), 12.17 (d, 3JHP = 39.6 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.1 (d, 3JCP = 6 Hz, CH3), 63.9 (d, 2JCP = 6 Hz, CH2), 117.3 (td, 1JCF = 246 Hz, 2JCP = 35 Hz, CF2), 122.9 (s, CPy), 129.9 (t, 2JCF = 27 Hz, CCF2), 133.8 (t, 3JCF = 6 Hz, CPy), 147.3 (t, 3JCF = 6.5 Hz, CPy), 151.6 (s, CPy), 173.0 (dt, 1JCP = 154 Hz, 2JCF = 35 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −97.5.
31P NMR (81 MHz, CDCl3) δ (ppm): 1.2 (m, 3JPH = 39.6 Hz).
E-isomer: 1H NMR (302 MHz, CDCl3) δ (ppm): 1.15 (t, 3JHH = 6.9 Hz, 6H, CH3), 3.93–4.20 (m, 4H, CH2), 7.30–7.35 (m, 1H, Py), 7.85 (d, 3JHH = 6.7 Hz, 1H, Py), 8.66 (d, 3JHH = 7.2 Hz, 1H, Py), 8.78 (s, 1H, Py), 11.85 (d, 3JHP = 61.2 Hz, 1H, NH).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.5.
31P NMR (81 MHz, CDCl3) δ (ppm): 2.9 (m, 3JPH = 61.2 Hz).
Anal. Calc. for C11H15F2N2O3P: C, 45.21; H, 5.17; N, 9.59. Found: C, 45.46; H, 5.04; N, 9.43.
4.1.3. Diethyl (2,2-Difluoro-1-imino-2-(pyridin-4-yl)ethyl)phosphonate 2c
Yield 0.85 g (100%); light orange oil. Z/E = 7:1. Z-isomer: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.34 (t, 3JHH = 7.1 Hz, 6H, CH3), 4.14–4.27 (m, 4H, CH2), 7.50 (d, 3JHH = 6 Hz, 2H, Py), 8.74 (d, 3JHH = 6 Hz, 2H, Py), 12.21 (d, 3JHP = 39.5 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 6 Hz, CH3), 64.1 (d, 2JCP = 6 Hz, CH2), 116.9 (td, 1JCF = 246 Hz, 2JCP = 34 Hz, CF2), 120.4 (t, 3JCF = 6 Hz, 2CPy), 142.2 (td, 2JCF = 27 Hz, 3JCP = 6 Hz, CCF2), 150.2 (s, 2CPy), 172.9 (dt, 1JCP = 154 Hz, 2JCF = 35 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −99.4.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 0.1 (m, 3JPH = 39.5 Hz).
E-isomer: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.26 (t, 3JHH = 7.1 Hz, 6H, CH3), 4.05–4.13 (m, 4H, CH2), 7.45 (d, 3JHH = 5 Hz, 2H, Py), 8.76 (d, 3JHH = 5 Hz, 2H, Py), 11.81 (d, 3JHP = 60.9 Hz, 1H, NH).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −102.4.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 1.8 (m, 3JPH = 60.9 Hz).
Anal. Calc. for C11H15F2N2O3P: C, 45.21; H, 5.17; N, 9.59. Found: C, 45.03; H, 5.23; N, 9.41.
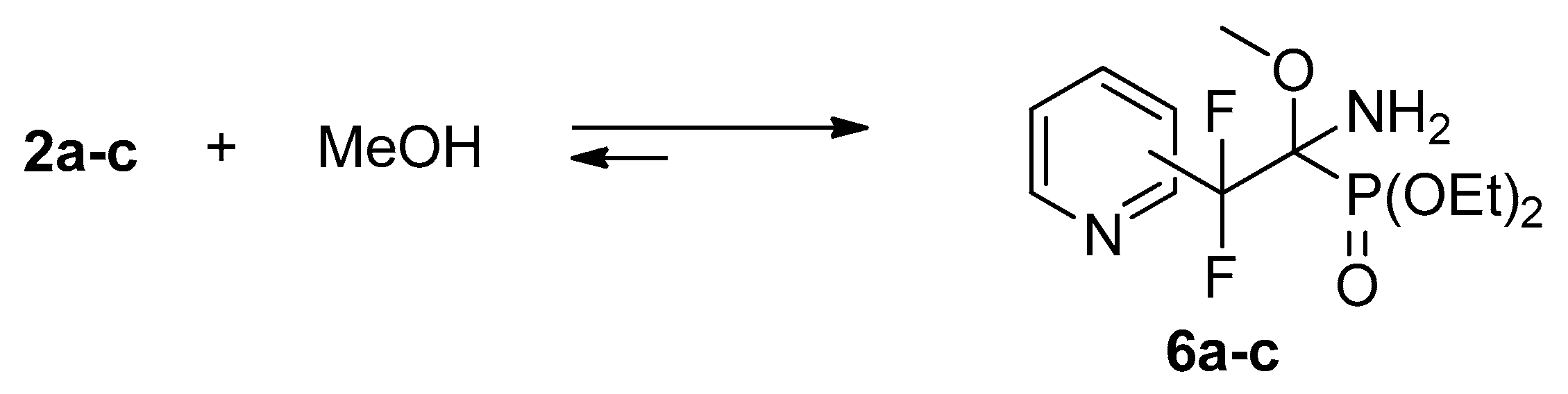
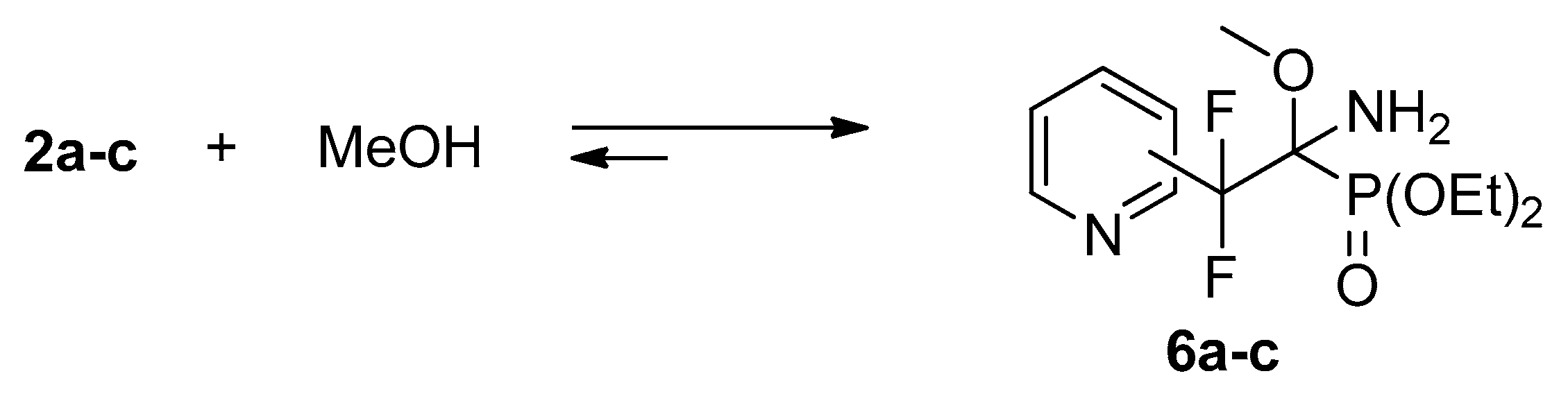
4.2. Reactions of Iminophosphonates 2a–c with Methanol
Iminophosphonates
2a–
c (0.02 g, 0.07 mmol) were dissolved in methanol (0.6 mL) at room temperature. According to
31P,
19F NMR the reaction was completed after 8 h (
2a) or 2 days (
2b,
c). The spectral data are similar to those of methanol adduct with trifluoromethyl analog [
17].
Spectral data for adduct 6a: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.30–1.34 (m, 6H, CH3), 3.21 (s, 3H, OCH3), 4.20–4.30 (m, 4H, CH2), 7.36 (dd, 3JHH = 8.1 Hz, 3JHH = 3.9 Hz, 1H, Py), 7.68 (d, 3JHH = 8.1 Hz, 1H, Py), 7.78 (t, 3JHH = 8.1 Hz, 1H, Py), 8.60 (d, 3JHH = 3.9 Hz, 1H, Py). 19F NMR (470.3 MHz, MeOH) δ (ppm): −103.0 (d, 2JFAFB = 255 Hz), −110.5 (d, 2JFBFA= 255 Hz). 31PNMR (202.3 MHz, MeOH) δ (ppm): 15.1.
Spectral data for adduct 6b: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.09 (t, 3JHH = 7.5 Hz, 3H, CH3), 1.27 (t, 3JHH = 7.6 Hz, 3H, CH3), 2.40 (d, 3JHP = 20 Hz, 2H, NH2), 3.37 (s, 3H, OCH3), 3.94–4.08 (m, 2H, OCH2), 4.13–4.18 (m, 2H, OCH2), 7.30 (dd, 3JHH = 7.3 Hz, 3JHH = 4.2 Hz, 1H, Py), 7.87 (d, 3JHH = 7.3 Hz, 1H, Py), 8.63 (d, 3JHH = 4.2 Hz, 1H, Py), 8.77 (s, 1H, Py). 19F NMR (470.3 MHz, MeOH) δ (ppm): −103.9 (d, 2JFAFB = 257 Hz), −106.6 (dd, 2JFBFA = 257 Hz, 3JFP = 7 Hz). 31PNMR (202.3 MHz, MeOH) δ (ppm): 14.8 (m, 3JPF = 7 Hz).
Spectral data for adduct 6c: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.13 (t, 3JHH = 6.9 Hz, 3H, CH3), 1.30 (t, 3JHH = 6.6 Hz, 3H, CH3), 2.40 (d, 3JHP = 22.5 Hz, 2H, NH2), 3.38 (s, 3H, OCH3), 3.97–4.12 (m, 2H, OCH2), 4.13–4.21 (m, 2H, OCH2), 7.49 (d, 3JHH = 5.3 Hz, 2H, Py), 8.66 (d, 3JHH = 5.3 Hz, 2H, Py). 19F NMR (470.3 MHz, MeOH) δ (ppm): −106.4 (d, 2JFAFB = 254 Hz), −108.4 (dd, 2JFBFA= 254 Hz, 3JFP = 7 Hz). 31PNMR (202.3 MHz, MeOH) δ (ppm): 13.1 (m, 3JPF = 7 Hz).
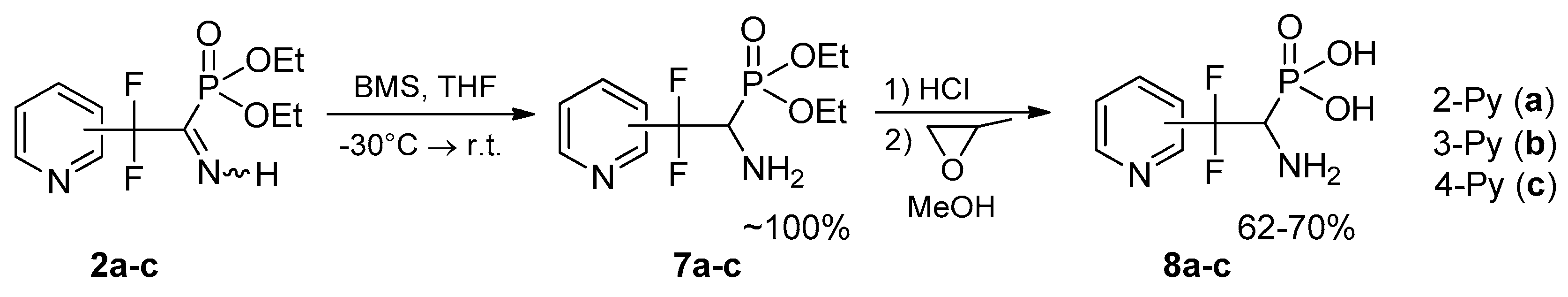
4.3. Synthesis of Diethyl (1-Amino-2,2-difluoro-2-(pyridinyl)ethyl)phosphonates 7a–c: General Procedure
To a solution of respective iminophosphonate 2 (0.65 g, 2.22 mmol) in THF (8 mL) borane-dimethyl sulfide complex (0.25 g, 3.34 mmol) was added dropwise at −30 °C in argon atmosphere. The reaction mixture was kept at this temperature for 0.5 h and allowed to warm to r.t. After that the reaction mixture was quenched with methanol (2 mL) and stirred for 15 min. Then solvents were evaporated under reduced pressure and residue was dried in vacuo.
4.3.1. Diethyl (1-Amino-2,2-difluoro-2-(pyridin-2-yl)ethyl)phosphonate 7a
Yield 0.65 g (100%); yellow crystals; mp 52–53 °C. 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.22 (t, 3JHH = 7.1 Hz, 3H, CH3), 1.25 (t, 3JHH = 7.1 Hz, 3H, CH3), 1.85 (br s, 2H, NH2), 4.18–4.00 (m, 4H, CH2), 4.23 (m, 1H, CH), 7.37 (dd, 3JHH = 7.8, 3JHH = 4.8 Hz, 1H, Py), 7.7 (d, 3JHH = 7.8 Hz, 1H, Py), 7.81 (t, 3JHH = 7.8 Hz, 1H, Py), 8.64 (d, 3JHH = 4.8 Hz, 1H, Py). 13C NMR (125.7 MHz, CDCl3) δ (ppm):16.3 (d, 3JCP = 5.9 Hz, CH3), 16.4 (d, 3JCP = 6.1 Hz, CH3), 53.7 (dt, 1JCP = 154.9 Hz, 2JCF = 28 Hz, CH), 62.7 (d, 2JCP = 6.9Hz, CH2), 63.0 (d, 2JCP = 6.7 Hz, CH2), 120.2 (td, 1JCF = 248 Hz, 2JCP = 5 Hz, CF2), 121.0 (t, 3JCF = 4.8 Hz, CPy), 124.9 (s, CPy), 137.0 (s, CPy), 149.2 (s, CPy), 153.6 (t, 2JCF = 29 Hz, CCF2). 19F NMR (470.3 MHz, CDCl3) δ (ppm): -103.8 (ddd, 2JFAFB = 256.5 Hz, 3JFH = 15.4 Hz, 3JFP = 15.4 Hz), −106.3 (dd, 2JFBFA = 256.5 Hz, 3J = 15.1 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 19.8. Anal. Calc. for C11H17F2N2O3P: C, 44.90; H, 5.82; N, 9.52. Found: C, 44.67; H, 5.73; N, 9.41.
4.3.2. Diethyl (1-Amino-2,2-difluoro-2-(pyridin-3-yl)ethyl)phosphonate 7b
Yield 0.65 g (100%); white-yellow oil. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.13–1.43 (m, 6H, CH3), 1.85 (br s, 2H, NH2), 3.62 (m, 1H, CH), 4.00–4.25 (m, 4H, CH2), 7.54–7.64 (m, 1H, Py), 8.15 (d, 3JHH = 8.1 Hz, 1H, Py), 8.68 (d, 3JHH = 5.8 Hz, 1H, Py), 8.82 (s, 1H, Py). 13C NMR (125.7 MHz, DMSO-d6) δ (ppm): 16.0 (d, 3JCP = 5 Hz, CH3), 16.2 (d, 3JCP = 5 Hz, CH3), 53.8 (dt, 1JCP = 150 Hz, 2JCF = 29 Hz, CH), 62.0 (d, 2JCP = 6 Hz, CH2), 62.5 (d, 2JCP = 6 Hz, CH2), 119.8 (td, 1JCF = 248 Hz, 2JCP = 6 Hz, CF2), 125.8 (s, CPy), 133.6 (td, 2JCF = 28 Hz, 3JCP = 4 Hz, CCF2), 138.0 (t, 3JCF = 6 Hz, CPy), 144.8 (t, 3JCF = 6 Hz, CPy), 148.4 (s, CPy). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.0 (ddd, 2JFAFB = 259.1 Hz, 3JFH = 11.8 Hz, 3JFP = 11.8 Hz), −100.8 (ddd, 2JFBFA = 259.1 Hz, 3JFH = 12.4Hz, 3JFP = 12.4Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 18.8. Anal. Calc. for C11H17F2N2O3P: C, 44.90; H, 5.82; N, 9.52. Found: C, 44.71; H, 5.74; N, 9.39.
4.3.3. Diethyl (1-Amino-2,2-difluoro-2-(pyridin-4-yl)ethyl)phosphonate 7c
Yield 0.65 g (100%); white-yellow powder; mp 105–107 °C. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.12–1.48 (m, 6H, CH3), 1.81 (br s, 2H, NH2), 3.60 (m, 1H, CH), 4.00–4.23 (m, 4H, CH2), 7.70 (d, 3JHH = 6 Hz, 2H, Py), 8.70 (d, 3JHH = 6 Hz, 2H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 5.9 Hz, CH3), 16.4 (d, 3JCP = 5.9 Hz, CH3), 53.4 (dt, 1JCP = 151.3 Hz, 2JCF = 29.2 Hz, CH), 63.2 (d, 2JCP = 6.8 Hz, CH2), 63.5 (d, 2JCP = 6.8 Hz, CH2), 119.0 (td, 1JCF = 248 Hz, 2JCP = 5.7 Hz, CF2), 123.0 (t, 3JCF = 5.9 Hz, CPy), 146.5 (t, 2JCF = 28.2 Hz, CCF2), 147.5 (s, CPy). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −102.4 (ddd, 2JFAFB = 249.4 Hz, 3JFH = 11.4 Hz, 3JFP = 11.4 Hz), −102.7 (ddd, 2JFBFA = 249.4 Hz, 3JFH = 11.8 Hz, 3JFP = 11.8 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 17.95 (m, 2JPH = 18.2, 3JPF = 8.6 Hz). Anal. Calc. for C11H17F2N2O3P: C, 44.90; H, 5.82; N, 9.52. Found: C, 44.78; H, 5.68; N, 9.43.
4.4. Synthesis of (1-Amino-2,2-difluoro-2-(pyridinyl)ethyl)phosphonic Acids 8a–c: General Procedure
To respective aminophosphonate 7 (0.3 g, 1.02 mmol) was added concentrated HCl (2 mL), the reaction mixture was heated at 110 °C for 2.5 h. After that HCl/H2O was evaporated, the residue was dissolved in MeOH (2 mL), and treated with propylene oxide (0.35 g, 6.12 mmol). After 24 h the precipitate formed was separated by filtration, washed with MeOH (2 mL) and dried in vacuo.
4.4.1. (1-Amino-2,2-difluoro-2-(pyridin-2-yl)ethyl)phosphonic Acid 8a
Yield 0.17 g (70%); white powder; mp 235–237 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 4.13–4.30 (m, 1H, CH), 7.58–7.64 (m, 1H, Py), 7.86 (d, 3JHH = 8.2 Hz, 1H, Py), 8.04 (t, 3JHH = 8.2 Hz, 1H, Py), 8.64 (d, 3JHH = 4.9 Hz, 1H, Py). 13C NMR (75.8 MHz, D2O) δ (ppm): 53.8 (dt, 1JCP = 126.5 Hz, 2JCF = 27.8 Hz, CH), 119.1 (td, 1JCF = 247 Hz, 2JCP = 6.0 Hz, CF2), 121.4 (s, CPy), 126.3 (s, CPy), 138.7 (s, CPy), 149.3 (s, CPy), 151.3 (t, 2JCF = 27.5 Hz, CCF2). 19F NMR (470.3 MHz, D2O) δ (ppm): −93.6 (d, 2JFAFB = 257 Hz), −110.0 (dd, 2JFBFA = 257 Hz, 3J = 21.5 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 3.9. Anal. Calc. for C7H9F2N2O3P: C, 35.31; H, 3.81; N, 11.76. Found: C, 35.13; H, 3.66; N, 11.55.
4.4.2. (1-Amino-2,2-difluoro-2-(pyridin-3-yl)ethyl)phosphonic Acid 8b
Yield 0.15 g (62%); white powder; mp 210–125 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 3.62 (m, 1H, CH), 7.66 (dd, 3JHH = 8.2 Hz, 3JHH = 5.1 Hz, 1H, Py), 8.18 (d, 3JHH = 8.2 Hz, 1H, Py), 8.72 (d, 3JHH = 5.1 Hz, 1H, Py), 8.83 (s, 1H, Py). 13C NMR (75.8 MHz, D2O) δ (ppm): 54.8 (dt, 1JCP = 132.7 Hz, 2JCF = 24.1 Hz, CH), 119.3 (t, 1JCF = 248 Hz, CF2), 124.5 (s, CPy), 130.2 (td, 2JCF = 26 Hz, 3JCP = 4.6 Hz, CCF2), 136.3 (t, 3JCF = 5.7 Hz, CPy), 145.6 (t, 3JCF = 7.9 Hz, CPy), 150.4 (s, CPy). 19F NMR (470.3 MHz, D2O) δ (ppm): −86.1 (dd, 2JFAFB = 253 Hz, 3J = 24.7 Hz), −110.1 (dd, 2JFBFA = 253 Hz, 3J = 24.5 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 2.8 (d, 2JPH = 24.3 Hz). Anal. Calc. for C7H9F2N2O3P: C, 35.31; H, 3.81; N, 11.76. Found: C, 35.04; H, 3.72; N, 11.59.
4.4.3. (1-Amino-2,2-difluoro-2-(pyridin-4-yl)ethyl)phosphonic Acid 8c
Yield 0.16 g (67%); white powder; mp 218–220 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 4.08 (m, 1H, CH), 7.73 (d, 3JHH = 5.3 Hz, 2H, Py), 8.72 (d, 3JHH = 5.3 Hz, 2H, Py). 13C NMR (75.8 MHz, D2O) δ (ppm): 54.3 (ddd, 1JCP = 129.6 Hz, 2JCFA = 27.6 Hz, 2JCFB = 20.1 Hz, CH), 119.0 (td, 1JCF = 247.3 Hz, 2JCP = 2.8 Hz, CF2), 121.4 (dd, 3JCFA = 7.3 Hz, 3JCFB = 4.8 Hz, CPy), 143.3 (t, 2JCF = 26.2 Hz, CCF2), 148.8 (s, CPy). 19F NMR (470.3 MHz, D2O) δ (ppm): −89.8 (d, 2JFAFB = 250 Hz), −110.2 (dd, 2JFBFA = 250 Hz, 3J = 23.3 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 3.3. Anal. Calc. for C7H9F2N2O3P: C, 35.31; H, 3.81; N, 11.76. Found: C, 35.12; H, 3.77; N, 11.63.
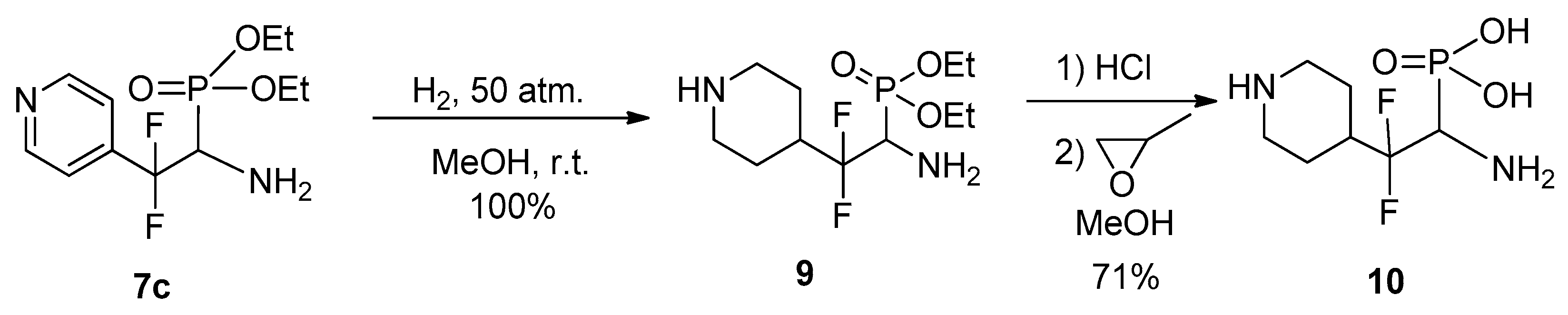
4.5. Diethyl (1-Amino-2,2-difluoro-2-(piperidin-4-yl)ethyl)phosphonate 9
To a solution of aminophosphonate 7c (0.3 g, 1.02 mmol) in MeOH (10 mL) was added Rh/Al2O3 (0.1 g, 10%), the mixture was hydrogenated under 50 atm. of H2 for 10 h. After completion of the reaction, the catalyst was filtered off, the solvent was evaporated to give 0.31 g of 9 (yield 100%) as colorless oil. 1H NMR (302 MHz, DMSO-d6) δ (ppm): 1.09–1.29 (m, 6H, CH3), 1.30–1.93 (m, 4H, CH2), 2.69–2.85 (m, 2H, CH2), 2.87–3.07 (m, 1H), 3.15–3.30 (m, 2H, CH2), 3.47 (m, 1H, CHP), 3.93–4.17 (m, 4H, OCH2), 8.39 (s, 1H, NH). 13C NMR (75.8 MHz, DMSO-d6) δ (ppm): 16.1 (d, 3JCP = 5.2 Hz, CH3), 16.2 (d, 3JCP = 5.2 Hz, CH3), 21.4 (t, 3JCF = 4.1 Hz, CF2CHCH2), 22.0 (t, 3JCF = 4.7 Hz, CF2CHCH2), 37.3 (td, 2JCF = 23.7 Hz, 3JCP = 4.2 Hz, CHCF2), 42.8 (s, 2CH2), 50.7 (dt, 1JCP = 147.3 Hz, 2JCF = 28.4 Hz, CHP), 61.8 (d, 2JCP = 6.8 Hz, OCH2), 62.1 (d, 2JCP = 6.7 Hz, OCH2), 124.0 (td, 1JCF = 248 Hz, 2JCP = 30 Hz, CF2).19F NMR (470.3 MHz, DMSO-d6) δ (ppm): −111.2 (m), −111.5 (m). 31P NMR (202.3 MHz, DMSO-d6) δ (ppm): 21.3. Anal. Calc. for C11H23F2N2O3P: C, 44.00; H, 7.72; N, 9.33. Found: C, 44.21; H, 7.65; N, 9.28.
4.6. (1-Amino-2,2-difluoro-2-(piperidin-4-yl)ethyl)phosphonic Acid 10
To aminophosphonate 9 (0.29 g, 0.97 mmol) was added concentrated HCl (1 mL), the reaction mixture was heated at 110 °C for 2.5 h. After completion of reaction HCl/H2O was evaporated, the residue was dissolved in MeOH (1 mL), and treated with propylene oxide (0.23 g, 4 mmol). After 24 h the precipitate formed was separated by filtration, washed with MeOH (2 mL) and dried to give 0.17 g of 10 (yield 71%) as white powder; mp 250 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 1.65–1.91 (m, 4H), 2.19 (t, 2JHH = 14.9 Hz, 2H), 3.07 (t, 2JHH = 14.1 Hz, 2H), 3.48–3.58 (m, 2H), 3.70–3.78 (m, 1H, CH). 13C NMR (150.8 MHz, D2O) δ (ppm): 27.4 (s, CH2), 28.3 (s, CH2), 37.4 (t, 2JCF = 22.4 Hz, CHCF2), 42.8 (s, CH2), 43.1 (s, CH2), 52.0 (d, 1JCP = 125.6 Hz, CH), 122.5 (t, 1JCF = 248 Hz, CF2).19F NMR (470.3 MHz, D2O) δ (ppm): −107.3 (ddd, 2JFAFB = 246 Hz, 3JFH = 26.5 Hz, 3JFP = 26.5 Hz), −111.6 (dd, 2JFBFA = 246 Hz, 3J = 26.5 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 3.5. Anal. Calc. for C7H15F2N2O3P: C, 34.43; H, 6.19; N, 11.47. Found: C, 34.17; H, 6.07; N, 11.63.
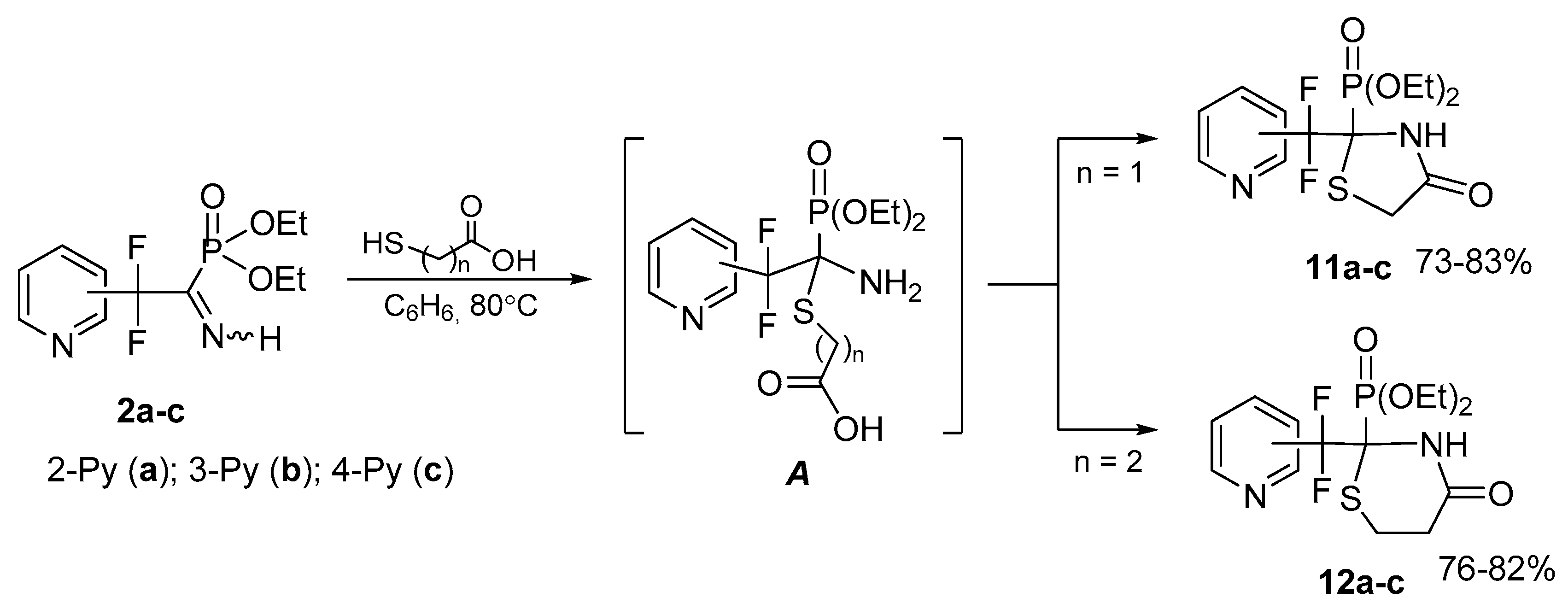
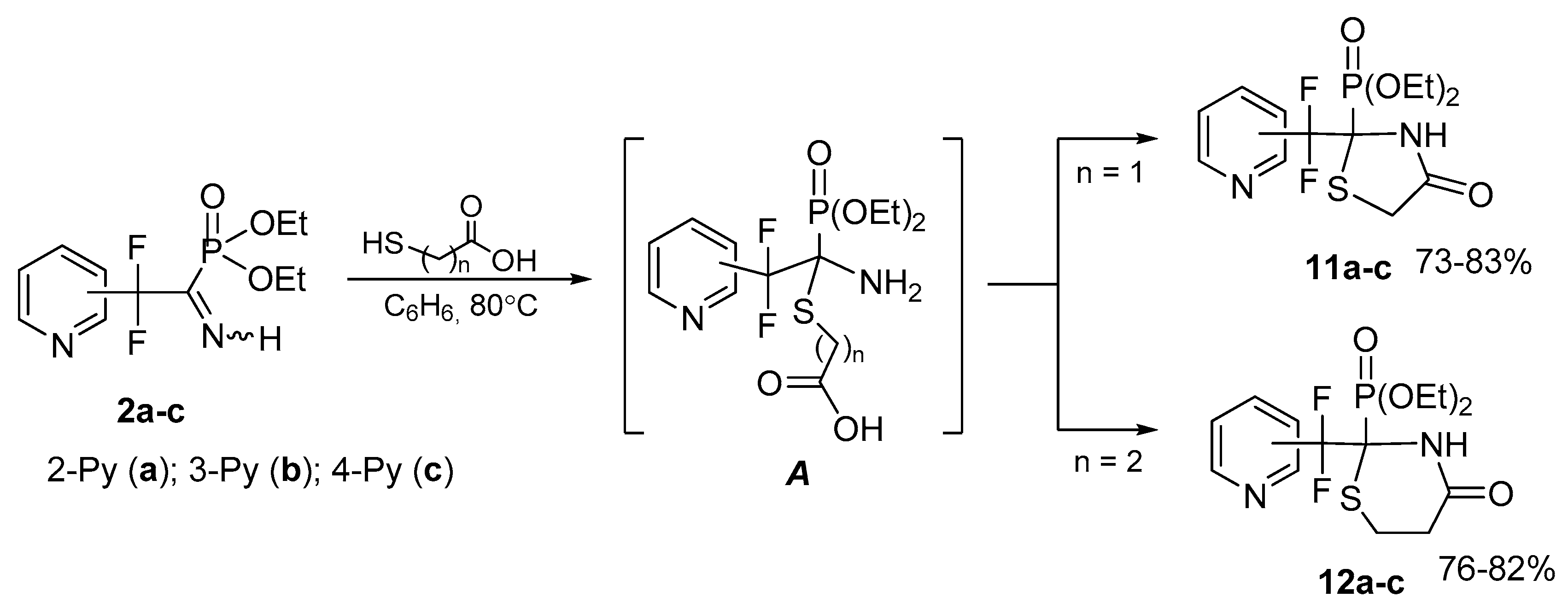
4.7. Synthesis of Diethyl (2-(Difluoro(pyridinyl)methyl)-4-oxothiazolidin-2-yl)phosphonates 11a–c: General Procedure
To solution of respective iminophosphonate 2 (0.12 g, 0.41 mmol) in benzene (1 mL) was added mercaptoacetic acid (0.04 g, 0.41 mmol). The reaction mixture was refluxed for 2 h, the solvent was evaporated in vacuum and the residue was triturated with hexane.
4.7.1. Diethyl (2-(Difluoro(pyridin-2-yl)methyl)-4-oxothiazolidin-2-yl)phosphonate 11a
Yield 0.11 g (73%); yellow oil. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.11–1.30 (m, 3JHH = 7.1 Hz, 6H, CH3), 3.44 (dd, 2JHAHB = 15.1 Hz, 4JHAP = 5.8 Hz, 1H, SCHAHB), 3.6 (d, 2JHBHA = 15.1 Hz, 1H, SCHAHB), 3.93–4.22 (m, 4H, OCH2), 7.45 (dd, 3JHH = 7.9 Hz, 3JHH = 4.9 Hz, 1H, Py), 7.74 (d, 3JHH = 7.9 Hz, 1H, Py), 7.86 (t, 3JHH = 7.9 Hz, 1H, Py), 8.20 (s, 1H, NH), 8.65 (d, 3JHH = 4.9 Hz, 1H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 5.9 Hz, CH3), 16.4 (d, 3JCP = 5.6 Hz, CH3), 32.1 (s, SCH2), 64.8 (d, 2JCP = 7.7 Hz, OCH2), 65.2 (d, 2JCP = 7.6 Hz, OCH2), 68.5 (dt, 1JCP = 170.7 Hz, 2JCF = 32.6 Hz, CP), 116.0 (td, 1JCF = 254 Hz, 2JCP = 21 Hz, CF2), 121.7 (t, 3JCF = 4.5Hz, CPy),125.6 (s, CPy), 137.5 (s, CPy), 149.0 (s, CPy), 152.2 (t, 2JCF = 30Hz, CCF2), 174.1 (d, 3JCP = 5.2 Hz, C(O)). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −98.5 (dd, 2JFAFB = 260 Hz, 3JFP = 12 Hz), −108.4 (d,2JFBFA = 260 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm):13.5. Anal. Calc. for C13H17F2N2O4PS: C, 42.62; H, 4.68; N, 7.65; S, 8.75. Found: C, 42.44; H, 4.53; N, 7.51; S, 8.62.
4.7.2. Diethyl (2-(Difluoro(pyridin-3-yl)methyl)-4-oxothiazolidin-2-yl)phosphonate 11b
Yield 0.125 g (83%); white powder; mp 162–164 °C. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.30 (t, 3JHH = 6.8 Hz, 6H, CH3), 3.05 (dd, 2JHAHB = 15.3 Hz, 4JHAP = 5.9 Hz, 1H, SCHAHB), 3.39 (d, 2JHBHA = 15.3 Hz, 1H, SCHAHB), 4.09–4.45 (m, 4H, OCH2), 7.38 (dd, 3JHH = 8.0 Hz, 3JHH = 4.9 Hz, 1H, Py), 7.99 (d, 3JHH = 8.0 Hz, 1H, Py), 8.73 (d, 3JHH = 4.9 Hz, 1H, Py), 8.73 (s, 1H, NH), 8.91 (s, 1H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 6 Hz, CH3), 16.4 (d, 3JCP = 6 Hz, CH3), 32.1 (s, SCH2), 65.0 (d, 2JCP = 8 Hz, OCH2), 65.1 (d, 2JCP = 8 Hz, OCH2), 68.0 (dt, 1JCP = 167 Hz, 2JCF = 32 Hz, CP), 120.7 (td, 1JCF = 255 Hz, 2JCP = 12 Hz, CF2), 122.7 (s, CPy), 128.2 (td, 2JCF = 27 Hz, 3JCP = 4 Hz, CCF2), 136.0 (t, 3JCF = 7 Hz, CPy), 148.6 (t, 3JCF = 5 Hz, CPy), 151.4 (s, CPy), 174.8 (d, 3JCP = 7 Hz, C(O)). 19F NMR (188 MHz, CDCl3) δ (ppm): −96.7 (d, 2JFAFB = 255.5 Hz), −101.9 (d, 2JFBFA = 255.5 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm):13.4. Anal. Calc. for C13H17F2N2O4PS: C, 42.62; H, 4.68; N, 7.65; S, 8.75. Found: C, 42.47; H, 4.73; N, 7.41; S, 8.58.
4.7.3. Diethyl (2-(Difluoro(pyridin-4-yl)methyl)-4-oxothiazolidin-2-yl)phosphonate 11c
Yield 0.116 g (77%); white powder; mp 80–82 °C. 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.31 (t, 3JHH = 7 Hz, 6H, CH3), 3.1 (dd, 2JHAHB = 15.2 Hz, 4JHAP = 5.6 Hz, 1H, SCHAHB), 3.4 (d, 2JHBHA = 15.2 Hz, 1H, SCHAHB), 4.12–4.33 (m, 4H, OCH2), 7.58 (d, 3JHH = 5.3 Hz, 2H, Py), 8.26 (s, 1H, NH), 8.73 (d, 3JHH = 5.3 Hz, 2H, Py).13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 5.7 Hz, CH3), 16.4 (d, 3JCP = 5.7 Hz, CH3), 32.1 (s, SCH2), 65.0 (d, 2JCP = 7.3 Hz, OCH2), 65.2 (d, 2JCP = 7.3 Hz, OCH2), 67.6 (dt, 1JCP = 167.1 Hz, 2JCF = 34.2 Hz, CP), 120.2 (td, 1JCF = 253 Hz, 2JCP = 11.6 Hz, CF2), 122.4 (t, 3JCF = 5.6 Hz, 2CPy), 140.7 (td, 2JCF = 27.6 Hz, 3JCP = 2.8 Hz, CCF2), 149.3 (s, 2CPy), 174.8 (d, 3JCP = 6.5 Hz, CO). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −98.9 (d, 2JFAFB = 252 Hz), −104.0 (d,2JFBFA = 252 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 13.2.Anal. Calc. for C13H17F2N2O4PS: C, 42.62; H, 4.68; N, 7.65; S, 8.75. Found: C, 42.39; H, 4.56; N, 7.48; S, 8.64.
4.8. Synthesis of Diethyl (2-(Difluoro(pyridinyl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonates 12a–c: General Procedure
To a solution of respective iminophosphonate 2 (0.13 g, 0.44 mmol) in benzene (1 mL) was added 3-mercaptopropionic acid (0.05 g, 0.44 mmol). The reaction mixture was refluxed for 8 h, the solvent was evaporated in vacuum and the residue was triturated with Et2O.
4.8.1. Diethyl (2-(Difluoro(pyridine-2-yl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonate 12a
Yield 0.13 g (76%); yellow oil. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.13 (t, 3JHH = 7.1 Hz, 3H, CH3), 1.25 (t, 3JHH = 7.1 Hz, 3H, CH3), 2.73–2.84 (m, 2H, CH2), 2.91–3.01 (m, 1H, CH), 3.22–3.35 (m, 1H, CH), 3.96–4.28 (m, 4H, 2OCH2), 7.46 (dd, 3JHH = 7.9 Hz, 3JHH = 5.1 Hz, 1H), 7.70 (d, 3JHH = 7.9 Hz, 1H), 7.86 (t, 3JHH = 7.9 Hz, 1H), 8.09 (s, 1H, NH), 8.70 (d, 3JHH = 5.1 Hz, 1H). 13C NMR (125.7 MHz, CDCl3) δ (ppm):16.2 (d, 3JCP = 6 Hz, CH3), 16.3 (d, 3JCP = 5 Hz, CH3), 23.5 (s, SCH2), 33.3 (s, CH2CO), 64.7 (d, 2JCP = 7.7 Hz, OCH2), 65.3 (d, 2JCP = 7.6 Hz, OCH2), 66.6 (dm, 1JCP = 168.7 Hz, CP), 116.0 (td, 1JCF = 256.2 Hz, 2JCP = 15 Hz, CF2), 122.1 (s, CPy),125.5 (s, CPy), 137.2 (s, CPy), 148.8 (s, CPy), 152.2 (td, 2JCF = 30 Hz, 3JCP = 2.5 Hz, CCF2), 170.9 (d, 3JCP = 3.1 Hz, CO). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −99 (dd, 2JFAFB = 255 Hz, 3JFP = 9 Hz), −106.4 (d,2JFBFA = 255 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 13.2 (m, 3J = 9 Hz). Anal. Calc. for C14H19F2N2O4PS: C, 44.21; H, 5.04; N, 7.37; S, 8.42. Found: C, 44.39; H, 4.91; N, 7.43; S, 8.34.
4.8.2. Diethyl (2-(Difluoro(pyridine-3-yl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonate 12b
Yield 0.14 g (82%); white powder; mp 158–160 °C.1H NMR (400 MHz, CDCl3) δ (ppm): 1.31 (t, 3JHH = 7.2 Hz, 6H, CH3), 2.32–2.44 (m, 2H, CH2), 2.53–2.68 (m, 1H, CH2), 2.80–2.89 (m, 1H, CH2), 4.10–4.35 (m, 4H, OCH2), 6.72 (s, 1H, NH), 7.37 (dd, 3JHH = 8.1 Hz, 3JHH = 4.9 Hz, 1H, Py), 7.92 (d, 3JHH = 8.1Hz, 1H, Py), 8.72 (d, 3JHH = 4.9 Hz, 1H, Py), 8.87 (s, 1H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.40 (d, 3JCP = 5.7 Hz, CH3), 16.43 (d, 3JCP = 5.6 Hz, CH3), 23.4 (s, SCH2), 33.4 (s, CH2CO), 65.0 (d, 2JCP = 7.5 Hz, OCH2), 65.2 (d, 2JCP = 7.5 Hz, OCH2), 65.5 (dt, 1JCP = 161.2 Hz, 2JCF = 29.5 Hz, CP), 120.5 (td, 1JCF = 255.7 Hz, 2JCP = 7.4 Hz, CF2), 122.5 (s, CPy), 128.3 (td, 2JCF = 26.7 Hz, 3JCP = 3.6 Hz, CCF2), 135.9 (t, 3JCF = 5.9 Hz, CPy), 148.9 (t, 3JCF = 6.4 Hz, CPy), 151.5 (s, CPy), 170.4 (d, 3JCP = 3.1Hz, C(O)). 19F NMR (470.3 MHz, CDCl3) δ (ppm): -98.8 (d, 2JFAFB = 253.6 Hz), −102.2 (d,2JFBFA = 253.6 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 13.5. Anal. Calc. for C14H19F2N2O4PS: C, 44.21; H, 5.04; N, 7.37; S, 8.42. Found: C, 44.08; H, 4.97; N, 7.48; S, 8.31.
4.8.3. Diethyl (2-(Difluoro(pyridine-4-yl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonate 12c
Yield 0.13 g (76%); white powder; mp 110 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.27–1.38 (m, 3JHH = 7.4 Hz, 6H, CH3), 2.41–2.53 (m, 2H, CH2), 2.56–2.67 (m, 1H, CH), 2.84–2.92 (m, 1H, CH), 4.19–4.31 (m, 4H, OCH2), 6.39 (s, 1H, NH), 7.65 (d, 3JHH = 5.1 Hz, 2H, Py), 8.76 (d, 3JHH = 5.1Hz, 2H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.4 (br d, 3JCP = 5.7 Hz, CH3), 23.4 (s, SCH2), 33.4 (s, CH2CO), 65.2 (d, 2JCP = 7.5 Hz, OCH2), 65.2 (dt, 1JCP = 161.4 Hz, 2JCF = 30.8 Hz, CP), 65.3 (d, 2JCP = 7.5 Hz, OCH2), 119.9 (td, 1JCF = 257 Hz, 2JCP = 8 Hz, CF2), 122.4 (t, 3JCF = 5.7 Hz, 2CPy), 140.8 (td, 2JCF = 28.1 Hz, 3JCP = 4.8 Hz, CCF2), 149.3 (s, 2CPy), 170.6 (d, 3JCP = 2.7 Hz, CO). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.4 (d, 2JFAFB = 251 Hz), −103.5 (d,2JFBFA = 251 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 12.9. Anal. Calc. for C14H19F2N2O4PS: C, 44.21; H, 5.04; N, 7.37; S, 8.42. Found: C, 44.48; H, 4.88; N, 7.28; S, 8.28.


 and
and 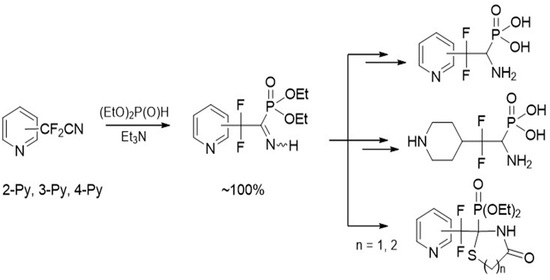


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}