
Application of β-Phosphorylated Nitroethenes in [3+2] Cycloaddition Reactions Involving Benzonitrile N-Oxide in the Light of a DFT Computational Study



Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
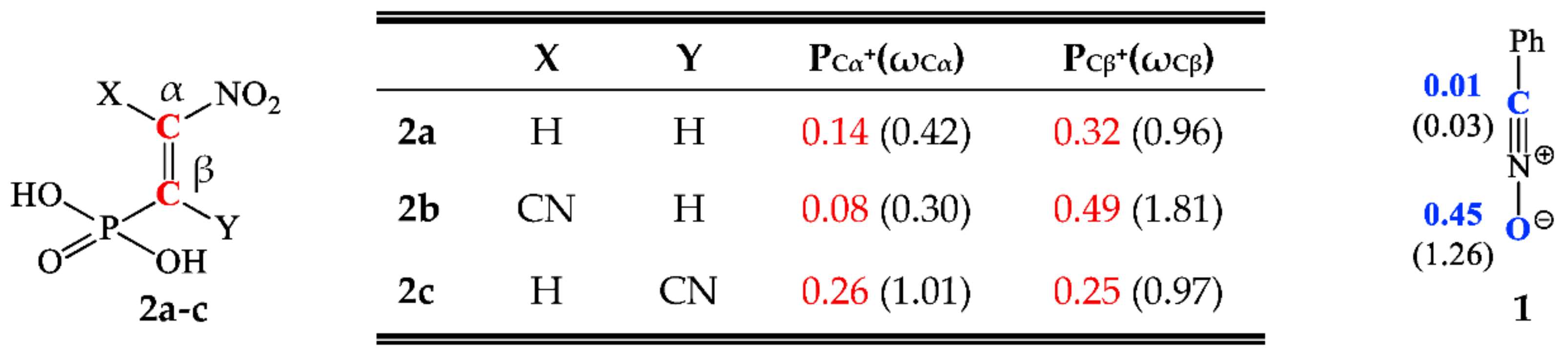
3.1. Analysis of the Electronic Properties of Addents and Their Intermolecular Interactions according to CDFT
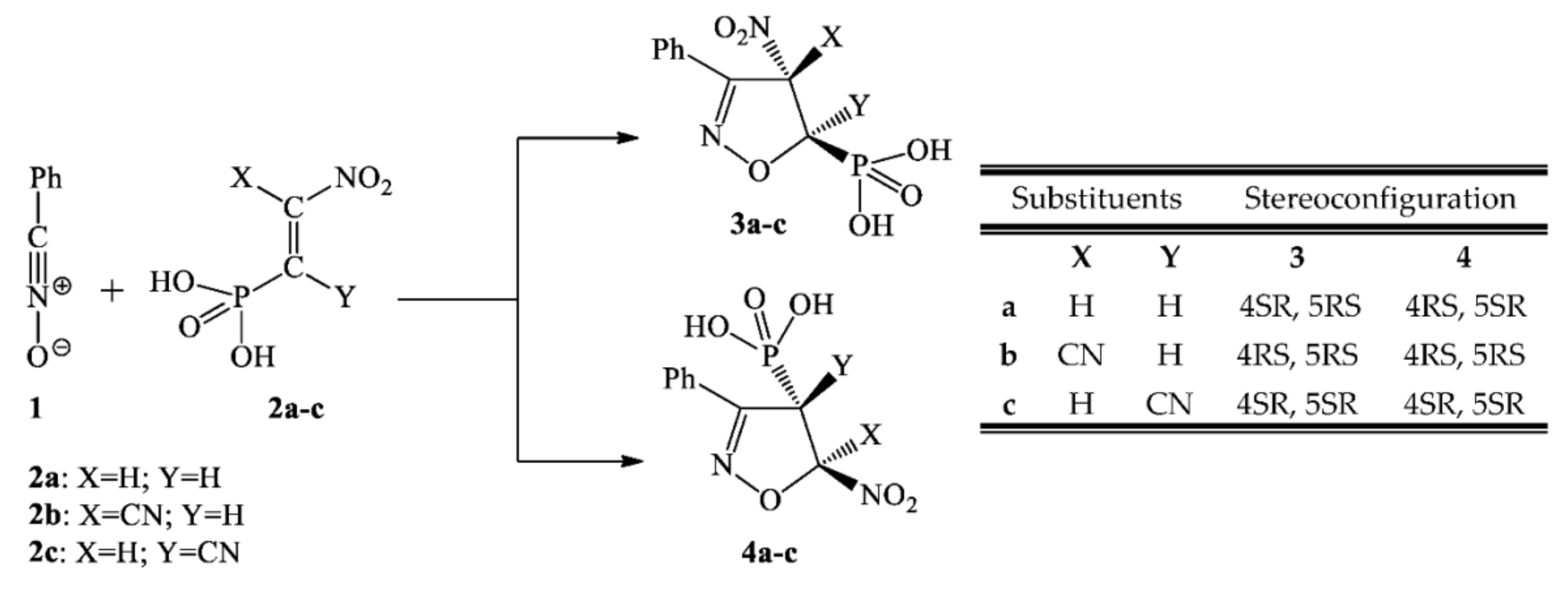
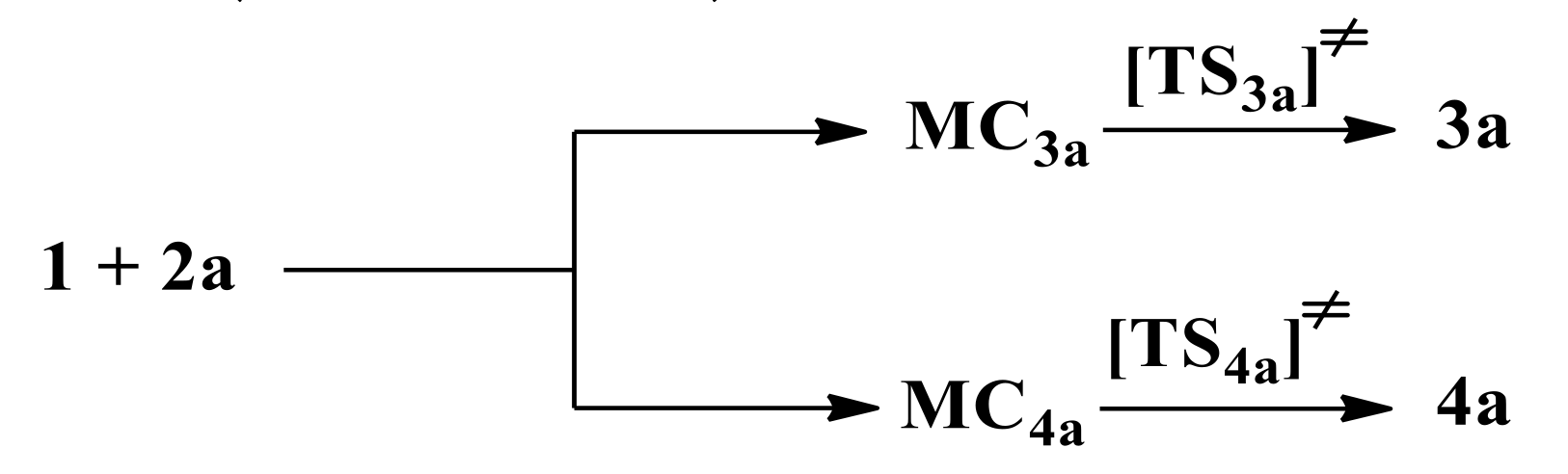
3.2. Reaction Profiles of 32CAbetween Benzonitrile N-Oxide and β-Phosphorylated Analogues of Nitroethenes
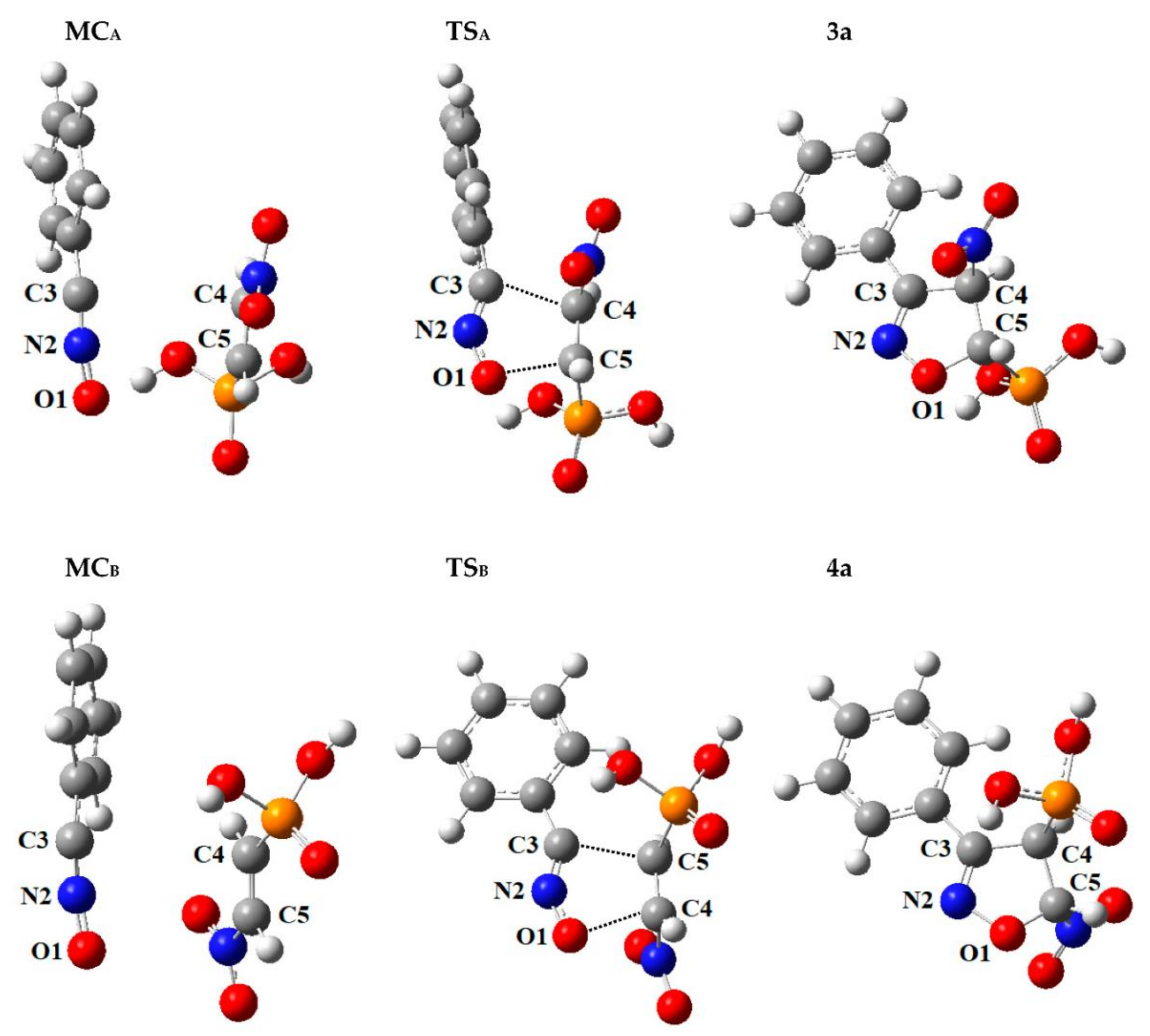
3.3. Critical Structures for Reaction of 32CA between Benzonitrile N-Oxide and β-Phosphorylated Nitroethenes
3.3.1. Pre-Reaction Molecular Complexes (MC)
3.3.2. Transition States (TS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huisgen, R. 1,3-Dipolare Cycloadditionen Rückschau und Ausblick. Angew. Chem. Int. Ed. 1963, 75, 604–637. [Google Scholar] [CrossRef]
- Jasiński, R.; Żmigrodzka, M.; Dresler, E.; Kula, K. A full regio- and stereoselective synthesis of 4-nitroisoxazolidines via stepwise [3+2] cycloaddition reactions between (Z)-C-(9-anthryl)-N-arylnitrones and (E)-3,3,3-trichloro-1-nitroprop-1-ene: Comprehensive experimental and theoretical study. J. Heterocyc. Chem. 2017, 54, 3314–3320. [Google Scholar] [CrossRef]
- Łapczuk-Krygier, A.; Kącka-Zych, A.; Kula, K. Recent progress in the field of cycloaddition reactions involving conjugated nitroalkenes. Curr. Chem. Lett. 2019, 8, 13–38. [Google Scholar] [CrossRef]
- Fryzlewicz, A.; Łapczuk-Krygier, A.; Kula, K.; Demchuk, O.M.; Dresler, E.; Jasiński, R. Regio- and stereoselective synthesis of nitro-functionalized analogs of nicotine. Chem. Heterocycl. Compd. 2020, 56, 120–122. [Google Scholar] [CrossRef]
- Martina, K.; Tagliapietra, S.; Veselov, V.V.; Cravotto, G. Green Protocols in Heterocycle Syntheses via 1,3-Dipolar Cycloadditions. Front. Chem. 2019, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.Z.; Li, X.L.; Chen, H.; Li, Y.N.; Wang, R. The synthesis and biological activity of novel spiro-isoxazoline C-disaccharides based on 1,3-dipolar cycloaddition of exo-glycals and sugar nitrile oxides. Tetrahedron Lett. 2007, 48, 7813–7816. [Google Scholar] [CrossRef]
- Jasinski, R.; Dresler, E. On the Question of Zwitterionic Intermediates in the [3+2] Cycloaddition Reactions: A Critical Review. Organics 2020, 7, 5. [Google Scholar] [CrossRef]
- Kula, K.; Łapczuk-Krygier, A. A DFT computational study on the [3+2] cycloaddition between parent thionitrone and nitroethene. Curr. Chem. Lett. 2018, 7, 27–34. [Google Scholar] [CrossRef]
- Kula, K.; Dobosz, J.; Jasiński, R.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Mirosław, B.; Demchuk, O.M. [3+2] Cycloaddition of diaryldiazomethanes with (E)-3,3,3-trichloro-1-nitroprop-1-ene: An experimental, theoretical and structural study. J. Mol. Struct. 2020, 1203, 127473. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Jeddeloh, M.R.; Holden, J.B.; Nouri, D.H.; Kurth, M.J. A Library of 3-aryl-4,5- dihydroisoxazole-5-carboxamides. J. Comb. Chem. 2007, 9, 1041–1045. [Google Scholar] [CrossRef]
- Quadrelli, P.; Martinez, N.V.; Scrocchi, R.; Corsaro, A.; Pistarà, V. Syntheses of Isoxazoline-Carbocyclic Nucleosides and Their Antiviral Evaluation: A Standard Protocol. Sci. World J. 2014, 2014, 492178. [Google Scholar] [CrossRef] [PubMed]
- Znati, M.; Debbabi, M.; Romdhane, A.; Ben Jannet, H.; Bouajila, J. Synthesis of new anticancer and anti-inflammatory isoxazolines and aziridines from the natural (-)-deltoin. J. Pharm. Pharmacol. 2018, 70, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Filial, I.; Bouajila, J.; Znati, M.; Bousejra-El Garah, F.; Ben Jannet, H. Synthesis of new isoxazoline derivatives from harmine and evaluation of their anti-Alzheimer, anti-cancer and anti-inflammatory activities. J. Enzyme Inhibit. Med. Chem. 2014, 30, 371–376. [Google Scholar] [CrossRef]
- Saravanan, G.; Alagarsamy, V.; Dineshkumar, P. Synthesis, analgesic, anti-inflammatory and in vitro antimicrobial activities of some novel isoxazole coupled quinazolin-4(3H)-one derivatives. Arch. Pharm. Res. 2013. [Google Scholar] [CrossRef]
- Sharifi, B.; Zade, B.G.; Zoladl, M.; Najafi, D.S.; Ghafarian, S.; Hamid, R.; Hashemi, M.; Abad, N. Side effects of risperidone. Life Sci. J. 2012, 9, 1463–1467. [Google Scholar] [CrossRef]
- Barceló, M.; Raviña, E.; Masaguer, C.F.; Domínguez, E.; Areias, F.M.; Brea, J.; Loza, M.I. Synthesis and binding affinity of new pyrazole and isoxazole derivatives as potential atypical antipsychotics. Bioorg. Med. Chem. Lett. 2007, 17, 4873–4877. [Google Scholar] [CrossRef]
- Pinho e Melo, T.M.V.D. Recent Advances on the Synthesis and Reactivity of Isoxazoles. Curr. Org. Chem. 2005, 9, 925–958. [Google Scholar] [CrossRef]
- Kanemasa, S.; Tsuge, O. Recent advances in synthetic applications of nitrile oxide cycloaddition. Heterocycles 1990, 30, 719–736. [Google Scholar] [CrossRef]
- Sewald, N. Synthetic Routes towards Enantiomerically Pure β-Amino Acids. Angew. Chem. Int. Ed. 2003, 42, 5794–5795. [Google Scholar] [CrossRef]
- Harada, K.; Kaji, E.; Takahashi, K.; Zen, S. Ring Transformation of 2-Isoxazoline 2-Oxides by Lewis Acids. Rev. Heteroatom Chem. 1997, 16, 171–195. [Google Scholar] [CrossRef]
- Ono, N. The Nitro Group in Organic Synthesis; Wiley-VSH: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Padwa, A.; Pearson, W.H. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; John Wiley & Sons: New York, NY, USA, 2002. [Google Scholar] [CrossRef]
- Boguszewska-Czubara, A.; Kula, K.; Wnorowski, A.; Biernasiuk, A.; Popiolek, P.; Miodowski, D.; Demchuk, O.M.; Jasiński, R. Novel functionalized β-nitrostyrenes: Promising candidates for new antibacterial drugs. Saudi Pharm. J. 2019, 27, 593–601. [Google Scholar] [CrossRef]
- Yan-Mei, L.; Ying-Wu, Y.; Yu-Fen, Z. Phosphoryl group participation leads to peptide formation from N-phosphorylamino acids. Int. J. Peptide Protein Res. 1992, 39, 375–381. [Google Scholar] [CrossRef]
- Nalwa, H.S. Handbook of Surfaces and Interfaces of Materials; Academic Press: Los Angeles, CA, USA, 2001. [Google Scholar]
- Ram, V.J.; Sethi, A.; Nath, M.; Pratap, R. The Chemistry of Heterocycles; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Mersbergen, D.; Wijnen, J.W.; Engberts, J.B.F.N. 1,3-Dipolar Cycloadditions of Benzonitrile Oxide with Various Dipolarophiles in Aqueous Solutions. A Kinetic Study. J. Org. Chem. 1998, 63, 8801–8805. [Google Scholar] [CrossRef]
- Jasiński, R.; Jasińska, E.; Dresler, E. A DFT computational study of the molecular mechanism of [3+2] cycloaddition reactions between nitroethene and benzonitrile N-oxides. J. Mol. Model. 2017, 23, 13. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Emamian, S.; Salami, M.; Ríos-Gutiérrez, M. Understanding the molecular mechanism of the [3+2] cycloaddition reaction of benzonitrile oxide toward electron-rich N-vinylpyrrole: A DFT study. J. Phys. Org. Chem. 2016, 29, 368–376. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos Gutiérrez, M.; Castellanos Soriano, J. Understanding the Origin of the Regioselectivity in Non-Polar [3+2] Cycloaddition Reactions through the Molecular Electron Density Theory. Organics 2020, 1, 3. [Google Scholar] [CrossRef]
- Jasiński, R.; Ziółkowska, M.; Demchuk, O.M.; Maziarka, A. Regio- and stereoselectivity of polar [2+3] cycloaddition reactions between (Z)-C-(3,4,5-trimethoxyphenyl)-N-methylnitrone and selected (E)-2-substituted nitroethenes. Cent. Eur. J. Chem. 2014, 12, 586–593. [Google Scholar] [CrossRef]
- Jasiński, R. Competition between one-step and two-step mechanism in polar [3+2] cycloadditions of (Z)-C-(3,4,5-trimethoxyphenyl)-N-methyl-nitrone with (Z)-2-EWG-1-bromo-1-nitroethenes. Comput. Theor. Chemia. 2018, 1125, 77–85. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Rev. A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jasiński, R. A new insight on the molecular mechanism of the reaction between (Z)-C,N-diphenylnitrone and 1,2-bismethylene-3,3,4,4,5,5-hexamethylcyclopentane. J. Mol. Graph. Model. 2020, 94, 107461. [Google Scholar] [CrossRef]
- Kącka-Zych, A. Participation of Phosphorylated Analogues of Nitroethene in Diels–Alder Reactions with Anthracene: A Molecular Electron Density Theory Study and Mechanistic Aspect. Organics 2020, 1, 4. [Google Scholar] [CrossRef]
- Kula, K.; Zawadzińska, K. Local nucleophile-electrophile interactions in [3+2] cycloaddition reactions between benzonitrile N-oxide and selected conjugated nitroalkenes in the light of MEDT computational study. Curr. Chem. Lett. 2021, 10, 9–16. [Google Scholar] [CrossRef]
- Stephens, P.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Domingo, L.R.; Kula, K.; Ríos-Gutiérrez, M. Unveiling the Reactivity of Cyclic Azomethine Ylides in [3+2] Cycloaddition Reactions within the Molecular Electron Density Theory. Eur. J. Org. Chem. 2020, 5938–5948. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Tapia, O. Solvent Effect Theories: Quantum and Classical Formalism and their Applications in Chemistry and Biochemistry. J. Math. Chem. 1992, 10, 131–181. [Google Scholar] [CrossRef]
- Tomasi, J.; Perisco, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2017–2094. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Chem. 1996, 225, 327–335. [Google Scholar] [CrossRef]
- Domingo, L.R. A New C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Mloston, G.; Jasiński, R.; Kula, K.; Heimgartner, H. A DFT Study on the Barton-Kellogg Reaction—The Molecular Mechanism of the Formation of Thiiranes in the Reaction between Diphenyldiazomethane and Diaryl Thioketones. Eur. J. Org. Chem. 2020, 176–182. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Duque-Noreña, M.; Chamorro, E. A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J. Mol. Struct. 2009, 895, 86–91. [Google Scholar] [CrossRef]
- Greelings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1,3-dipolar cycloaddition reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Saez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Domingo, L.R.; Saez, J.A. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. On the nature of organic electron density transfer complexes within molecular electron density theory. Org. Biomol. Chem. 2019, 17, 6478–6488. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| 1 | −3.83 | 5.02 | 1.46 | 2.78 |
| 2a | −5.54 | 5.15 | 2.98 | 1.00 |
| 2b | −6.01 | 4.90 | 3.68 | 0.66 |
| 2c | −6.09 | 4.79 | 3.87 | 0.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zawadzińska, K.; Kula, K. Application of β-Phosphorylated Nitroethenes in [3+2] Cycloaddition Reactions Involving Benzonitrile N-Oxide in the Light of a DFT Computational Study. Organics 2021, 2, 26-37. https://doi.org/10.3390/org2010003
Zawadzińska K, Kula K. Application of β-Phosphorylated Nitroethenes in [3+2] Cycloaddition Reactions Involving Benzonitrile N-Oxide in the Light of a DFT Computational Study. Organics. 2021; 2(1):26-37. https://doi.org/10.3390/org2010003
Chicago/Turabian StyleZawadzińska, Karolina, and Karolina Kula. 2021. "Application of β-Phosphorylated Nitroethenes in [3+2] Cycloaddition Reactions Involving Benzonitrile N-Oxide in the Light of a DFT Computational Study" Organics 2, no. 1: 26-37. https://doi.org/10.3390/org2010003
APA StyleZawadzińska, K., & Kula, K. (2021). Application of β-Phosphorylated Nitroethenes in [3+2] Cycloaddition Reactions Involving Benzonitrile N-Oxide in the Light of a DFT Computational Study. Organics, 2(1), 26-37. https://doi.org/10.3390/org2010003