
Activation of CO2 on the Surfaces of Bare, Ti-Adsorbed and Ti-Doped C60


Abstract
:1. Introduction
2. Computational Methods
3. Results
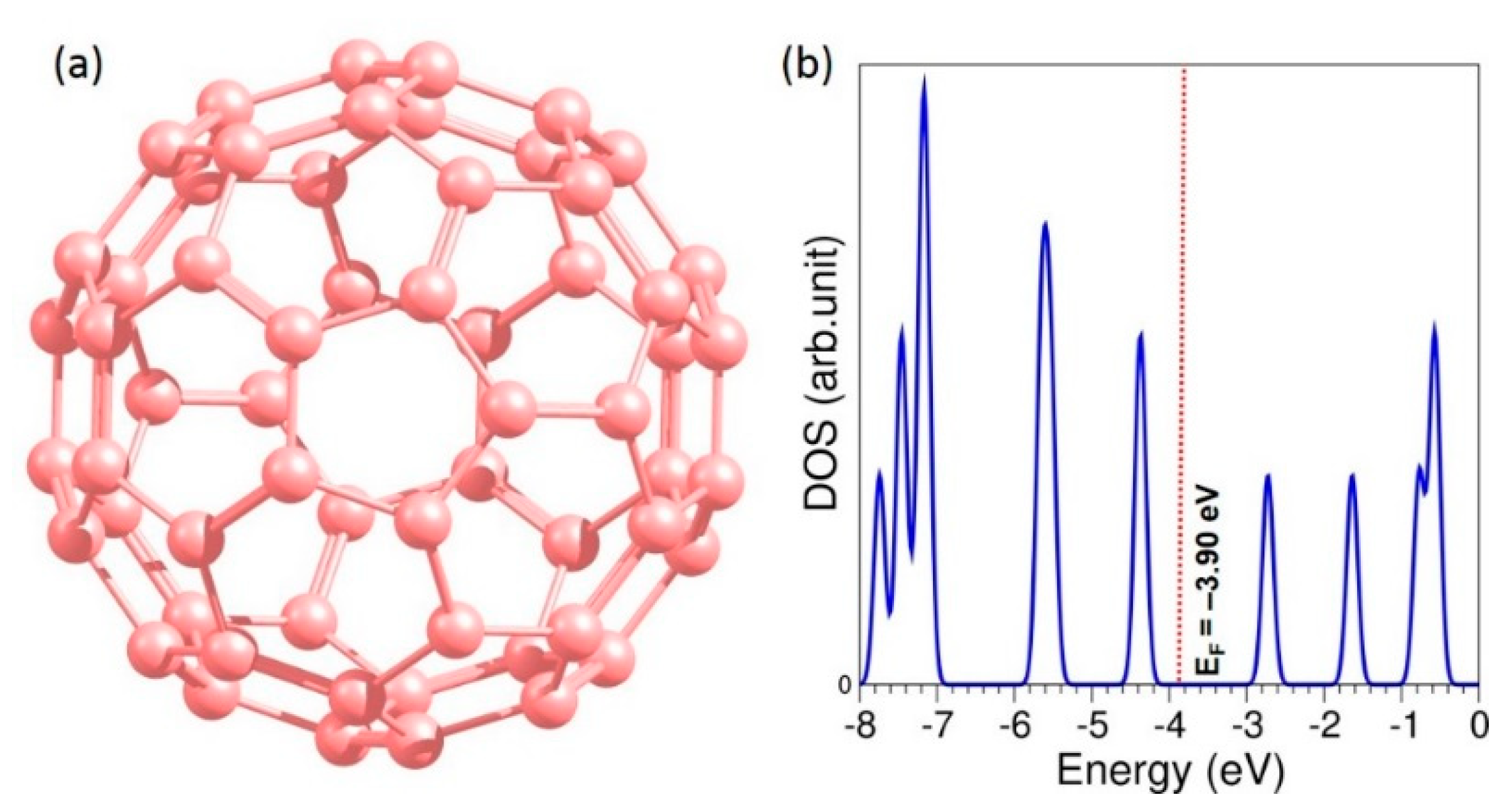
3.1. Strucure of C60
3.2. Encapsulation of CO2 Inside the Pure C60
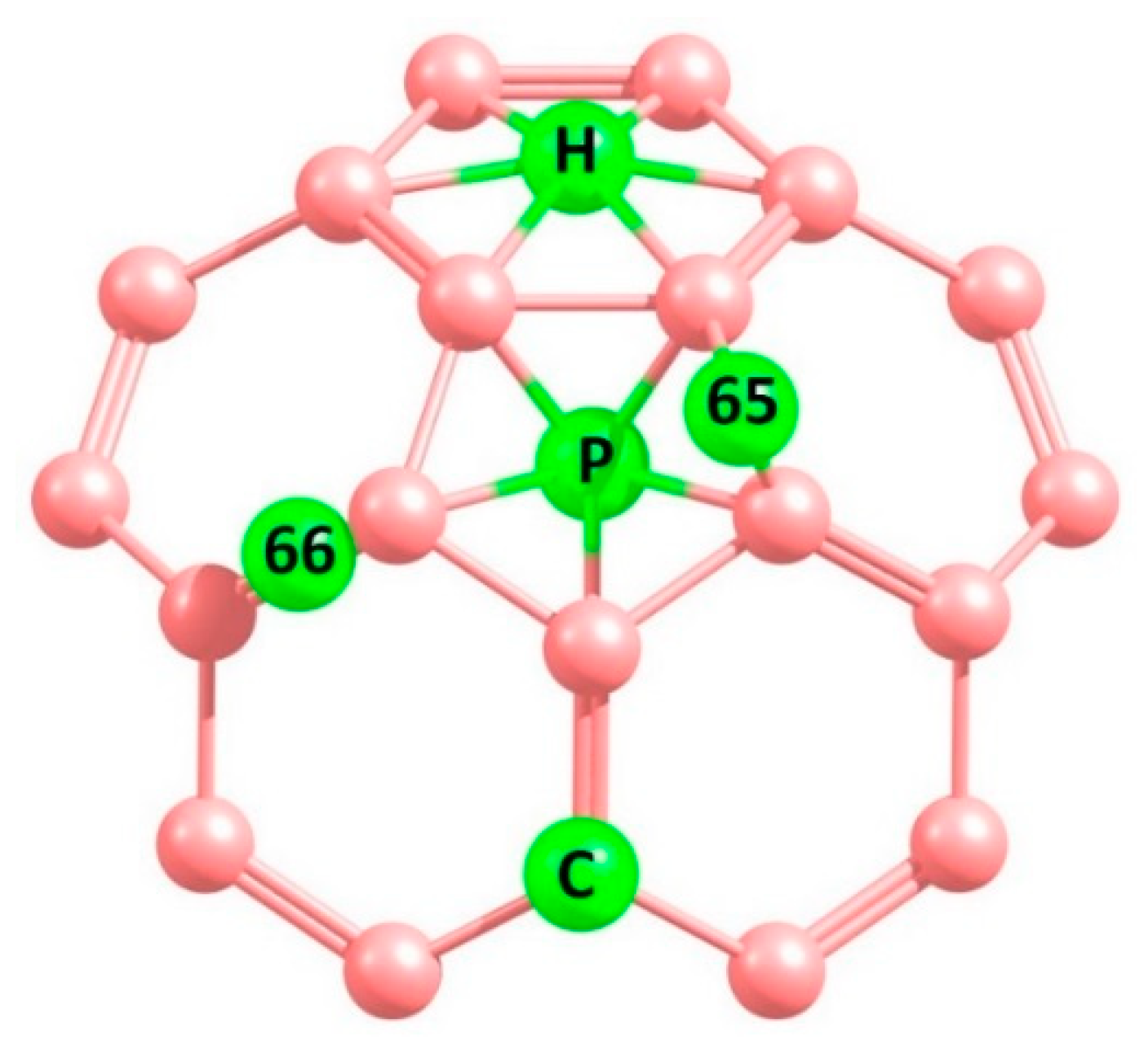
3.3. Adsorption of CO2 on the Surface of Pure C60
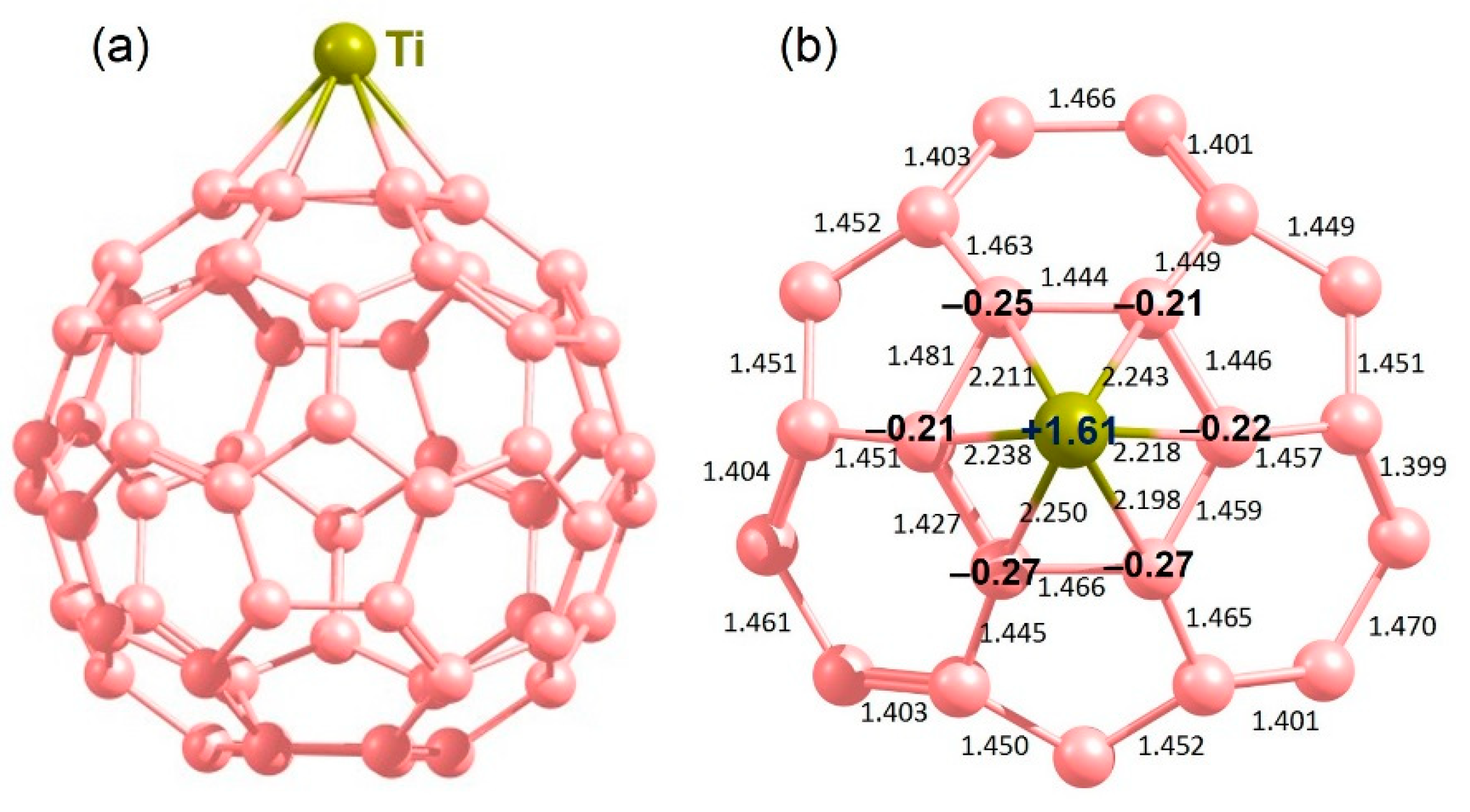
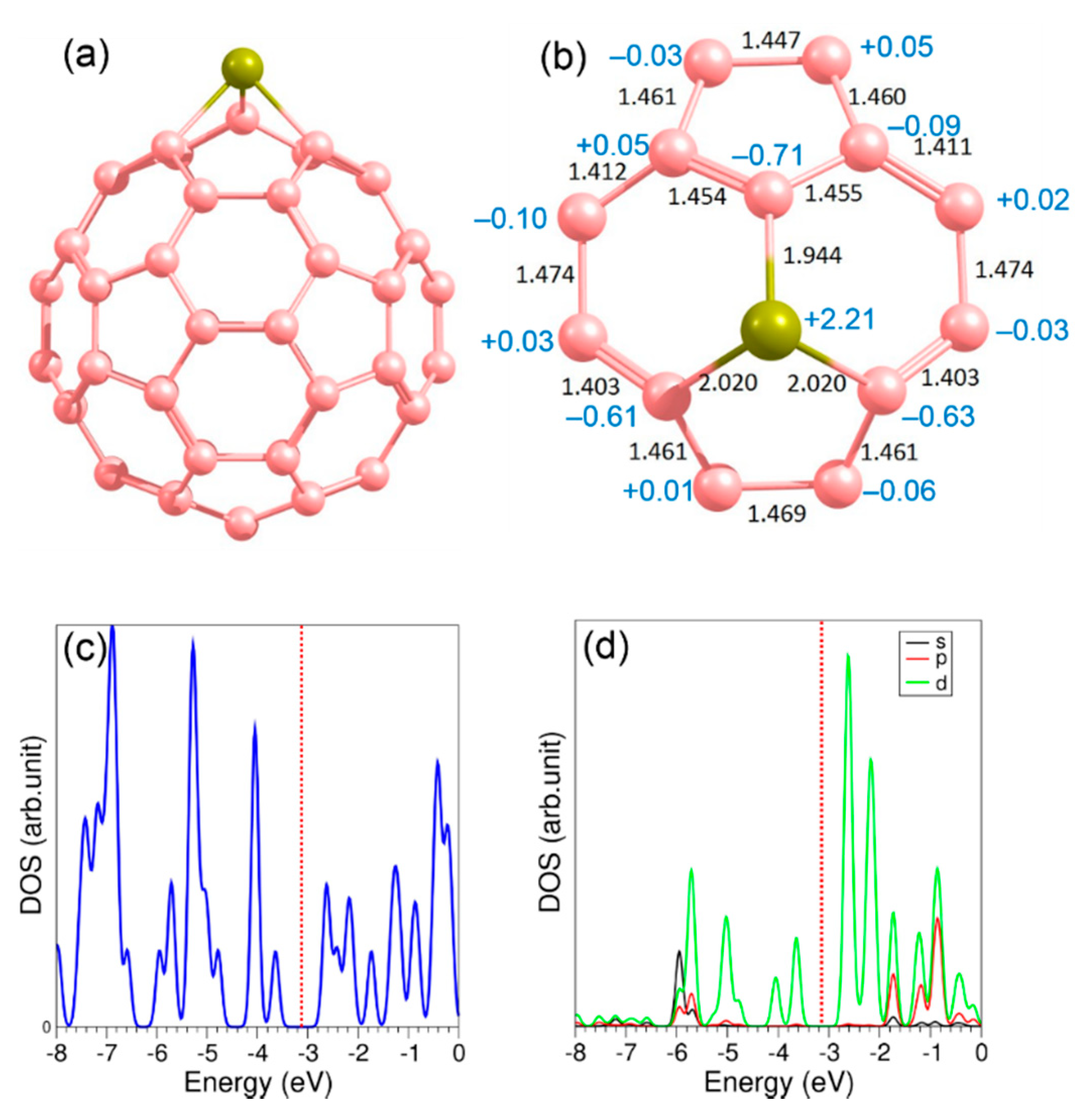
3.4. Adsorption of CO2 on the Surface of C60 Supported with Ti
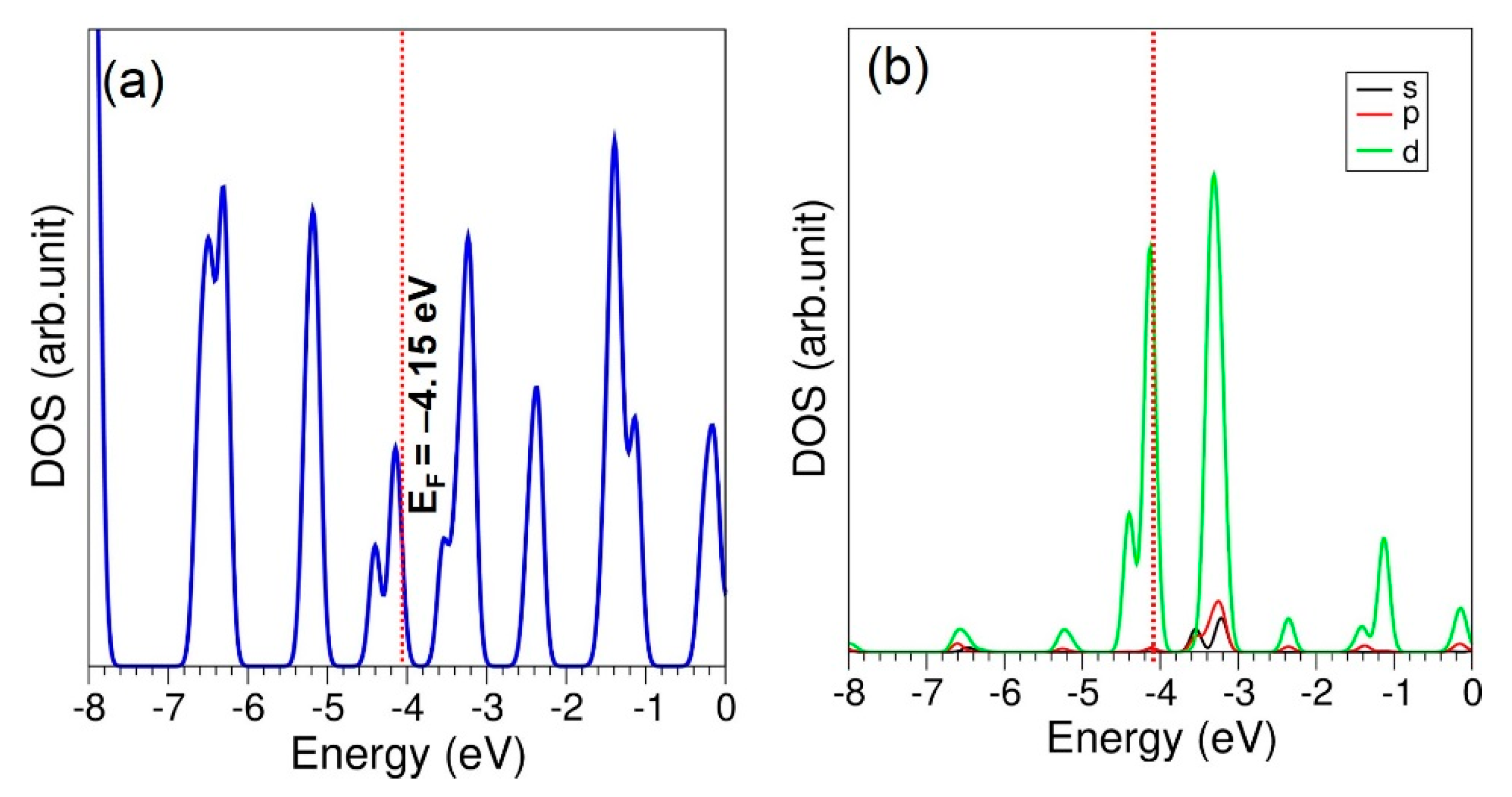
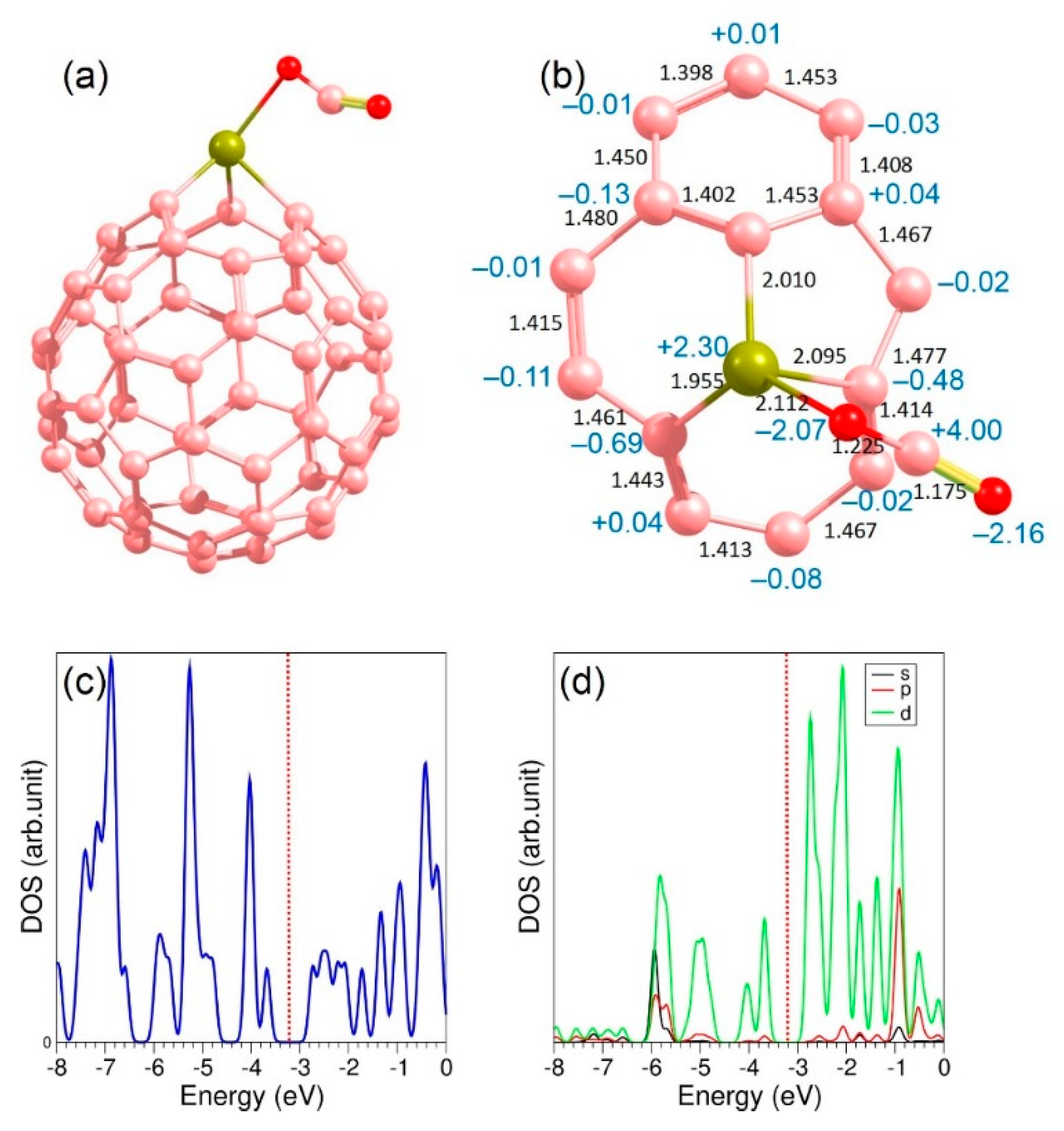
3.5. Adsorption of CO2 on the Surface Ti-Doped C60
4. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Liu, P.; Ou, C.; Dong, X. Porous Metal–Organic Frameworks for Carbon Dioxide Adsorption and Separation at Low Pressure. ACS Sustain. Chem. Eng. 2020, 8, 15378–15404. [Google Scholar] [CrossRef]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Gandara-Loe, J.; Pastor-Perez, L.; Bobadilla, L.F.; Odriozola, J.A.; Reina, T.R. Understanding the opportunities of metal–organic frameworks (MOFs) for CO2 capture and gas-phase CO2 conversion processes: A comprehensive overview. React. Chem. Eng. 2021, 6, 787–814. [Google Scholar] [CrossRef]
- Liu, J.; Chen, C.; Zhang, K.; Zhang, L. Applications of metal–organic framework composites in CO2 capture and conversion. Chin. Chem. Lett. 2021, 32, 649–659. [Google Scholar] [CrossRef]
- Mahdipoor, H.R.; Halladj, R.; Ganji Babakhani, E.; Amjad-Iranagh, S.; Sadeghzadeh Ahari, J. Synthesis, characterization, and CO2 adsorption properties of metal organic framework Fe-BDC. RSC Adv. 2021, 11, 5192–5203. [Google Scholar] [CrossRef]
- Megías-Sayago, C.; Bingre, R.; Huang, L.; Lutzweiler, G.; Wang, Q.; Louis, B. CO2 Adsorption Capacities in Zeolites and Layered Double Hydroxide Materials. Front. Chem. 2019, 7, 551. [Google Scholar] [CrossRef] [Green Version]
- Bakhtyari, A.; Mofarahi, M.; Lee, C.-H. Chapter 9—CO2 Adsorption by Conventional and Nanosized Zeolites. In Advances in Carbon Capture; Rahimpour, M.R., Farsi, M., Makarem, M.A., Eds.; Woodhead Publishing: Sawston, UK, 2020; pp. 193–228. [Google Scholar]
- Hauchhum, L.; Mahanta, P. Carbon dioxide adsorption on zeolites and activated carbon by pressure swing adsorption in a fixed bed. Int. J. Energy Environ. Eng. 2014, 5, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Seville, J.P.K.; Bell, S.D.; Ingram, A.; Zhang, H.; Sweygers, N.; Dewil, R.; Baeyens, J.; Appels, L. Reviewing Fundamental CO2 Adsorption Characteristics of Zeolite and Activated Carbon by In-situ Measurements With Radioactively Labelled CO2. Sep. Purif. Rev. 2021, 1, 12. [Google Scholar]
- Mutch, G.A.; Shulda, S.; McCue, A.J.; Menart, M.J.; Ciobanu, C.V.; Ngo, C.; Anderson, A.A.; Richards, M.R.; Vega-Maza, D. Carbon Capture by Metal Oxides: Unleashing the Potential of the (111) Facet. J. Am. Chem. Soc. 2018, 140, 4736–4742. [Google Scholar] [CrossRef] [Green Version]
- Othman, F.E.C.; Yusof, N.; Samitsu, S.; Abdullah, N.; Hamid, M.F.; Nagai, K.; Nizam Zainal Abidin, M.; Ariff Azali, M.; Fauzi Ismail, A.; Jaafar, J.; et al. Activated carbon nanofibers incorporated metal oxides for CO2 adsorption: Effects of different type of metal oxides. J. CO2 Util. 2021, 45, 101434. [Google Scholar] [CrossRef]
- Irani, V.; Khosh, A.G.; Tavasoli, A. Polyethyleneimine (PEI) Functionalized Metal Oxide Nanoparticles Recovered From the Catalytic Converters of Spent Automotive Exhaust Systems and Application for CO2 Adsorption. Front. Energy Res. 2020, 8, 196. [Google Scholar] [CrossRef]
- Xu, C.; Yu, G.; Yuan, J.; Strømme, M.; Hedin, N. Microporous organic polymers as CO2 adsorbents: Advances and challenges. Mater. Today Adv. 2020, 6, 100052. [Google Scholar] [CrossRef]
- Jiangfei, G.; Lizhi, W.; Zhang, D.; Huang, J. Amino-Functionalized Porphyrin-Based Porous Organic Polymers for CO2 Capture and Hg2+ Removal. Energy Fuels 2020, 34, 9771–9778. [Google Scholar] [CrossRef]
- Patel, H.A.; Hyun Je, S.; Park, J.; Chen, D.P.; Jung, Y.; Yavuz, C.T.; Coskun, A. Unprecedented high-temperature CO2 selectivity in N2-phobic nanoporous covalent organic polymers. Nat. Commun. 2013, 4, 1357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Aziz, B.; Hedin, N. Carbon dioxide adsorption on mesoporous silica surfaces containing amine-like motifs. Appl. Energy 2010, 87, 2907–2913. [Google Scholar] [CrossRef]
- Kishor, R.; Ghoshal, A.K. Amine-Modified Mesoporous Silica for CO2 Adsorption: The Role of Structural Parameters. Ind. Eng. Chem. Res. 2017, 56, 6078–6087. [Google Scholar] [CrossRef]
- Yu, J.; Le, Y.; Cheng, B. Fabrication and CO2 adsorption performance of bimodal porous silica hollow spheres with amine-modified surfaces. RSC Adv. 2012, 2, 6784–6791. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G.; Eklund, P.C. Science of Fullerenes and Carbon Nanotubes; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Wang, Y.; Tománek, D.; Ruoff, R.S. Stability of M@C60 endohedral complexes. Chem. Phys. Lett. 1993, 20, 79–85. [Google Scholar] [CrossRef]
- Brocławik, E.; Eilmes, A. Density functional study of endohedral complexes M@C60 (M=Li, Na, K, Be, Mg, Ca, La, B, Al): Electronic properties, ionization potentials, and electron affinities. J. Chem. Phys. 1998, 108, 3498–3503. [Google Scholar] [CrossRef]
- Kuganathan, N.; Green, J.C.; Himmel, H.-J. Dinitrogen fixation and activation by Ti and Zr atoms, clusters and complexes. New J. Chem. 2006, 30, 1253–1262. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Q.; Jena, P.; Kawazoe, Y. Clustering of Ti on a C60 Surface and Its Effect on Hydrogen Storage. J. Am. Chem. Soc. 2005, 127, 14582–14583. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, A.M.E. DFT study of hydrogen storage in Pd-decorated C60 fullerene. Mol. Phys. 2015, 113, 3531–3544. [Google Scholar] [CrossRef]
- Gao, B.; Zhao, J.-X.; Cai, Q.-H.; Wang, X.-G.; Wang, X.-Z. Doping of Calcium in C60 Fullerene for Enhancing CO2 Capture and N2O Transformation: A Theoretical Study. J. Phys. Chem. A 2011, 115, 9969–9976. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes. In C: The Art of Scientific Computing, 2nd ed.; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. The zero-flux surface and the topological and quantum definitions of an atom in a molecule. Theor. Chem. Acc. 2001, 105, 276–283. [Google Scholar] [CrossRef]
- Hawkins, J.M. Osmylation of C60: Proof and characterization of the soccer-ball framework. Acc. Chem. Res. 1992, 25, 150–156. [Google Scholar] [CrossRef]
- Goclon, J.; Winkler, K.; Margraf, J.T. Theoretical investigation of interactions between palladium and fullerene in polymer. RSC Adv. 2017, 7, 2202–2210. [Google Scholar] [CrossRef] [Green Version]
- Kuganathan, N.; Arya, A.K.; Rushton, M.J.D.; Grimes, R.W. Trapping of volatile fission products by C60. Carbon 2018, 132, 477–485. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Encapsulation Energy (eV) | Charge Transfer (e) | ||
|---|---|---|---|---|
| vdw-free | vdw | vdw-free | vdw | |
| CO2@C60 | 1.60 | 0.94 | –0.04 | –0.04 |
| Adsorption energy (eV) | Charge transfer (e) | |||
| CO2_C60 | 0.05 | –0.05 | –0.01 | –0.02 |
| Structure | Relative Energy (eV) | |
|---|---|---|
| vdw-free | vdw | |
| H | 0.00 | 0.00 |
| C | 0.42 | 0.42 |
| 66 | 0.42 | 0.42 |
| P | 0.58 | 0.59 |
| 65 | 0.84 | 0.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuganathan, N. Activation of CO2 on the Surfaces of Bare, Ti-Adsorbed and Ti-Doped C60. Fuels 2022, 3, 176-183. https://doi.org/10.3390/fuels3010011
Kuganathan N. Activation of CO2 on the Surfaces of Bare, Ti-Adsorbed and Ti-Doped C60. Fuels. 2022; 3(1):176-183. https://doi.org/10.3390/fuels3010011
Chicago/Turabian StyleKuganathan, Navaratnarajah. 2022. "Activation of CO2 on the Surfaces of Bare, Ti-Adsorbed and Ti-Doped C60" Fuels 3, no. 1: 176-183. https://doi.org/10.3390/fuels3010011
APA StyleKuganathan, N. (2022). Activation of CO2 on the Surfaces of Bare, Ti-Adsorbed and Ti-Doped C60. Fuels, 3(1), 176-183. https://doi.org/10.3390/fuels3010011