Frequency estimates of specific mutations appearing within CoV-2 genomes can be produced by utilizing the number of times a certain mutation is observed divided by total genomes analyzed (Equation (1)). The probability (
P) multiplication rule can then be employed in this scenario to infer the
P of a certain mutation appearing in multiple CoV-2 genomes selected at random [
7]. From our experience with molecular epidemiology, we observed that the CoV-2 ORF1a region contains a degree of nucleotide instability (i.e., more informative) when compared to the Spike region. This is true within the CoV-2 Delta strain, and similar observations were noted within Omicron’s ORF1a regions. Regions with relative genomic instabilities can be used to compare outbreak strains qualitatively in their mutational repertoire utilizing low frequency mutations as a first-tier choice. Here, were propose rather the quantitative utility of mutational frequencies incorporating the full mutational repertoire within ORF1a (i.e., both low and high frequency mutations). It is important to note that, due to temporal mutational dynamics, we should expect a degree of undulation within each nucleotide site [
8,
9]. For this reason, frequency calculations should be dependent on recent circulating variants, not based on the bulk of total CoV-2 viral sequences with strains that are no longer circulating. For example, applying frequency data from the Delta wave to samples collected during the Omicron wave can be substantially misleading. Date ranges of observed mutations can be set within COVIDCG online software.
We hence propose the use of the ORF1a genomic region to produce a combined, time-framed, frequency profile of the mutational repertoire integrated with the number of samples containing this profile (i.e.,
P product-rule). These calculations are then applied to the Essen-Möller equation initially produced to prove or disprove paternity [
10]. The merged equations are utilized to produce a Similarity Index as we show in the following derivations.
3.1. Similarity Index–Derivation of Base Equation
Mutational profiles, factored in as mutational frequencies, can be applied when comparing two or more strains that are part of a localized outbreak. Here, two variables are of most importance. The first is each nucleotide’s mutational frequency. The second is the number of outbreak samples carrying that same mutation.
Therefore, the
P (
Pm) of a certain mutation with Frequency (
F) to appear together in a pool of CoV-2 samples at random or by chance is:
where
F relates to the calculated mutational frequency (Equation (1)), and (
s1,
s2,
s3,
sn) relate to the individual outbreak samples carrying the exact mutation with frequency
F. Since all samples are carrying the same mutation with the same
F, the equation can be simplified to the following form:
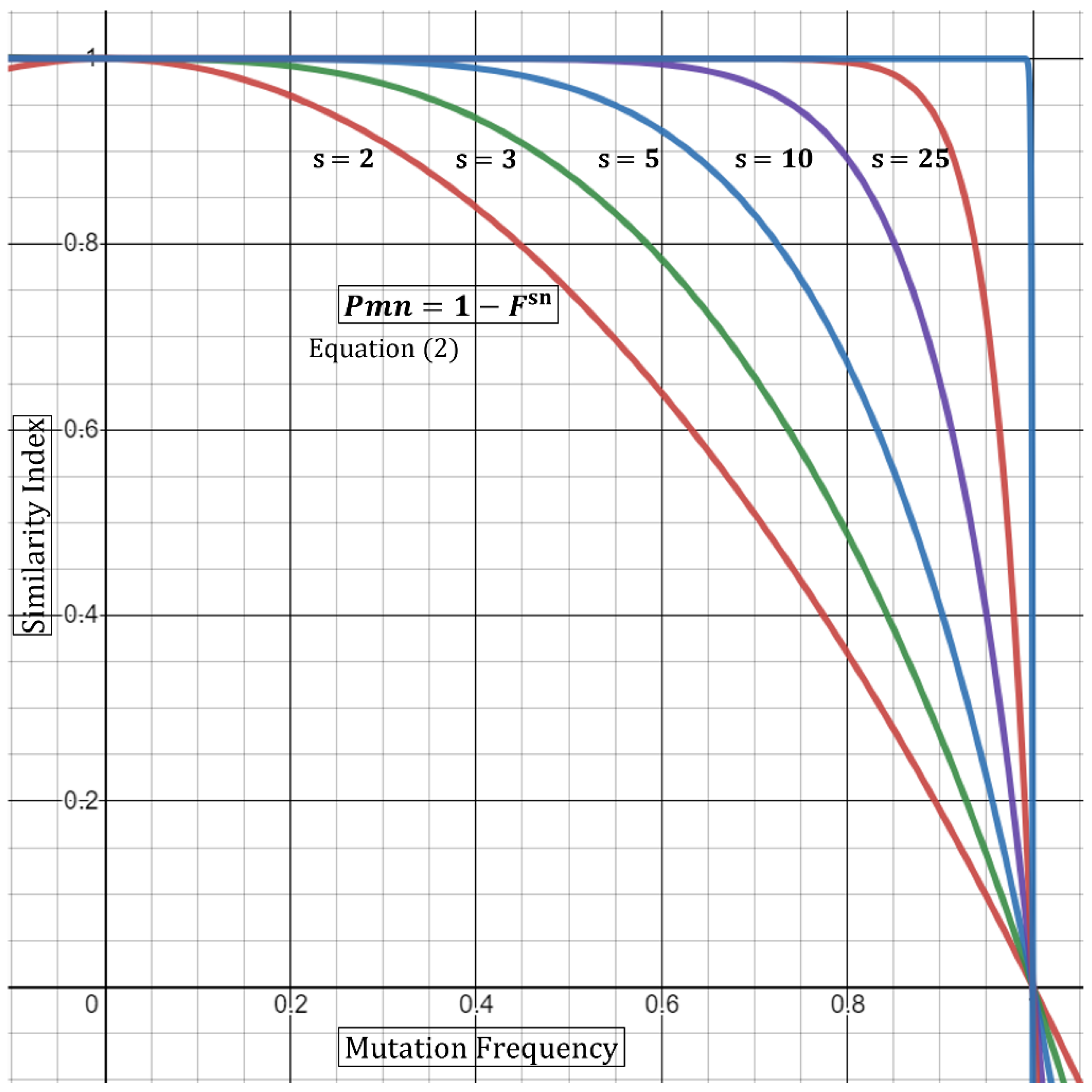
where
sn denotes the total number of samples carrying a mutation with frequency
F. Next, in an outbreak scenario, we are rather interested in defining the
P that a certain mutation will not appear together at random (
Pmn). Thus, the above equation can be transformed to the following form:
where
F relates to a single mutational frequency, and
sn relates to the total sample number where all samples are carrying the same mutation with frequency
F.
Thus, Equation (2) states that the
P of a certain mutation not appearing at random is dependent on the number of samples as well as the mutational frequency carried by
sn samples. We can also extrapolate from this base formula that incorporating a large outbreak sample number,
sn, will compensate for a higher frequency (e.g., 0.15–0.65). In other words, five outbreak samples carrying a mutation with 0.3 (30%) frequency produce a higher
P in samples that are related, compared to only two samples (
Table 1). Alternatively, a low or rare mutational frequency would also raise the
P in a smaller outbreak sample (
Table 1). Geometrical representation of this argument is displayed in
Figure 1. As the frequency (
F) of shared mutations approaches zero, the
P of association approaches 1 (100%). Similarly, as the number of samples,
sn, carrying a mutation with multiplied-frequency (
F) approaches infinity, the
P of association drifts towards one. One limitation in the utility of mutation frequencies in
P calculations is that mutations should be independent. Specifically, utilizing mutations that are co-dependent will produce misleading outputs utilizing the
P multiplication rule [
7]. To this end, Fang et al. utilized a concurrence-ratio whereby two single nucleotide variants can be assessed for the likelihood of coexisting in the same viral genomes [
11].
3.2. Incorporation of Mutational Differences
Heterogeneity of the mutational repertoire in outbreak samples is evidence towards non-similarity, depending on the degree of divergence. Therefore, it would be misleading to base the P model (Equation (2)) entirely on identical/unique mutations. We specifically observed in our epidemiological investigations that although strains within an outbreak can carry unique mutations, some nevertheless had non-identical mutational signatures. It would be important to estimate quantitatively the relatedness of these strains based on both similar and dissimilar mutations. The main question is, how confident are we that the strains on-hand are related? The answer to this can in part be effectuated by incorporating dissimilar mutational frequencies within the final P value in order to normalize for mutational differences between samples.
To this end, we propose the use of a modified Essen-Möller’s
W value as a base-equation:
In its original form,
W combines two hypotheses: X (paternity) and Y (non-paternity). Essen-Möller proposed this in order to include both possibilities wherein X + Y becomes a probability of 1 [
10].
In our case, we seek to calculate
W by summing two
Ps (i.e., similar or dissimilar strains based on mutational profiles) and then dividing the
P of interest (similar) by the sum, thence attaining an index on which we shall name here the Similarity Index (SI) [
10]. To this end, Equation (2) and Essen-Möller’s equation are coalesced as follows for comparing multiple (≥2) samples:
where
F1→
Fn and
Fd1→
Fdn relate to the shared and non-shared (d = different) mutations with a frequency of
F, respectively.
s1→
sn relates to the total sample number associated with the frequency
F1→
Fn. For example, if
F1 is (0.05), and
s1 relates to 5 samples, then
F1
s1 = 0.05
5. This would be followed by calculations and summing of all frequencies (
F2
s2,
F3
s3,
Fnsn) relating to each specific CoV-2 nucleotide mutation. In essence, (
F1
s1 +
F2
s2 +
Fnsn) present in both the numerator and denominator would be equivalent to “X” in Essen-Möller’s ratio. In contrast, (
F1
ds1+
F2
ds2+
Fndsn) is equivalent to “Y” in in Essen-Möller’s ratio.
This equation allows a multi-sample approach incorporating the differences in mutations, frequencies, and sample number which all contribute to the Similarity Index. The convolution of multi-sample comparison is evident when comparing samples carrying a host of identical mutations along with mutations only present within a certain subset of the group. It therefore becomes important to add in the factor of
s, where it can guide the statistical “swaying” power of each mutation present as shown geometrically in
Figure 1.
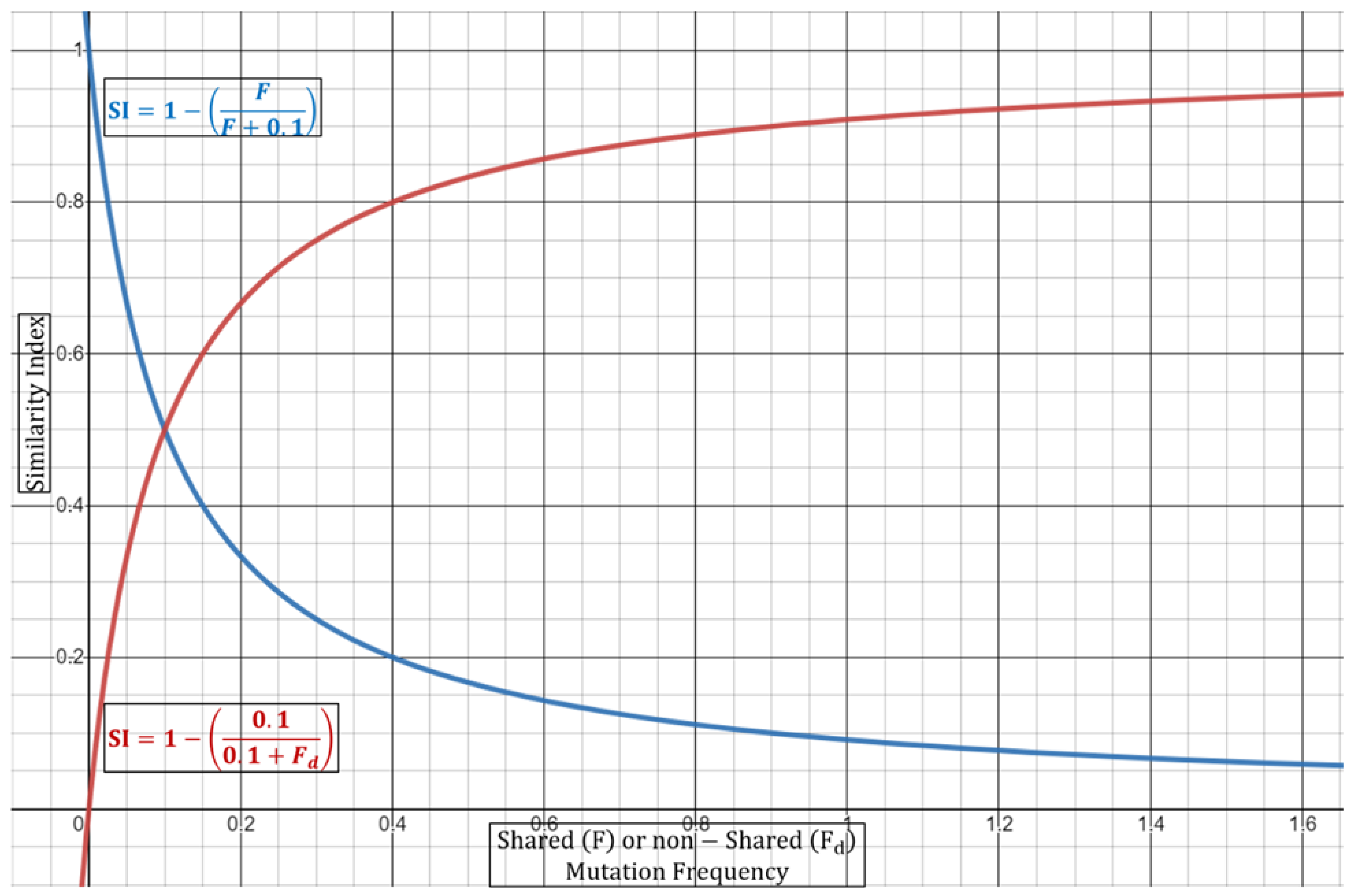
Based on this, the final equations can be summarized as follows:
where the product summation relates that every frequency of a shared mutation (
F) is summed until all total (
n) frequencies are incorporated. The same concept applies for non-shared mutational frequencies (
Fd). The ratio of the summation products is then subtracted from 1 to give a Similarity Index, or a confidence of relatedness, for the outbreak strains.
Equation (3) can be performed either in a dual fashion (two strains) or via a multi-comparison approach wherein all strains are weighted together. In the case of duality, the (
si) as the exponent will always be equivalent to 2, since by default there are only two samples. It is important to note here that if the ORF1a regions are identical, then Equation (3) cannot be used as it depends on shared vs. non-shared ratios. In this case, the full summation expression should be equivalent to 0 to give a Similarity Index of 1 (or 100% confidence in identity). In this case, it may be of use to include other genomic regions for inferring potential mutational differences. Next, as discussed above, the Similarity Index in Equation (3) will be dependent on the increase/decrease in the number of shared or different mutations. It will also depend on the number of samples carrying a certain mutation along with the mutational frequency.
Table 2 summarizes some aspects of the normalization incorporated in Equation (3) and their effects on the Similarity Index. With regards to the combined mutational frequencies, this can be observed geometrically wherein shared (
F) or non-shared (
Fd) combined frequencies oppositely shift the Similarity Index towards or away from 1 (100%) (
Figure 2).
3.3. Incorporation of Spatial Dynamics
Incorporation of distance into the overall Similarity Index is useful when comparing only two patients suspected of direct contact and with confirmed interactions such as a nurse physically assisting an infected patient or simply being in the same room. One may also estimate the general average distance observed between all outbreak patients (>2) and incorporate into the Similarity Index as such. The limitation here is that some infections occur indirectly through the action of touching mucous sites with un-sanitized hands where direct patient-to-patient contact was not a factor.
The CDC’s guidelines states that the risk of CoV-2 transmission is greatest within three to six feet of an infectious source. The risk is reduced post six feet but is not eliminated due to multiple variables, such as timing compounded with other factors. Specifically, even if one is distant from the infectious source by more than six feet, the chance of transmission increases if they are in the space for longer than 15 minutes (CDC). The chance of infection increases in enclosed spaces with inadequate ventilation or if the infected person is undergoing physical and vocal exertion (e.g., exercise, singing) due to increased dispersion of virions. Nonetheless, incorporation of temporal dynamics within the Similarity Index equation will require more studies to integrate most factors with optimal mathematical presumptions precisely.
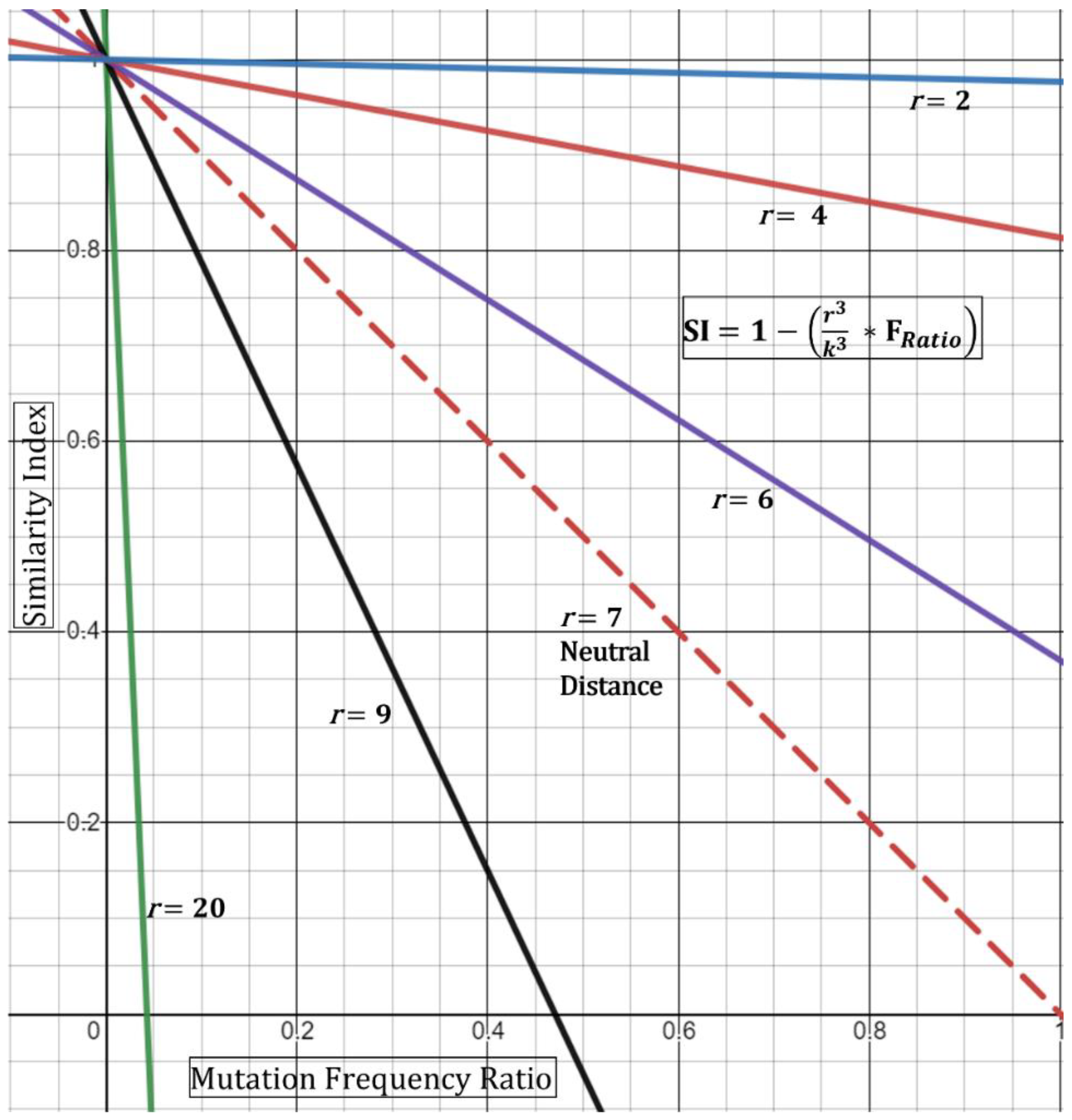
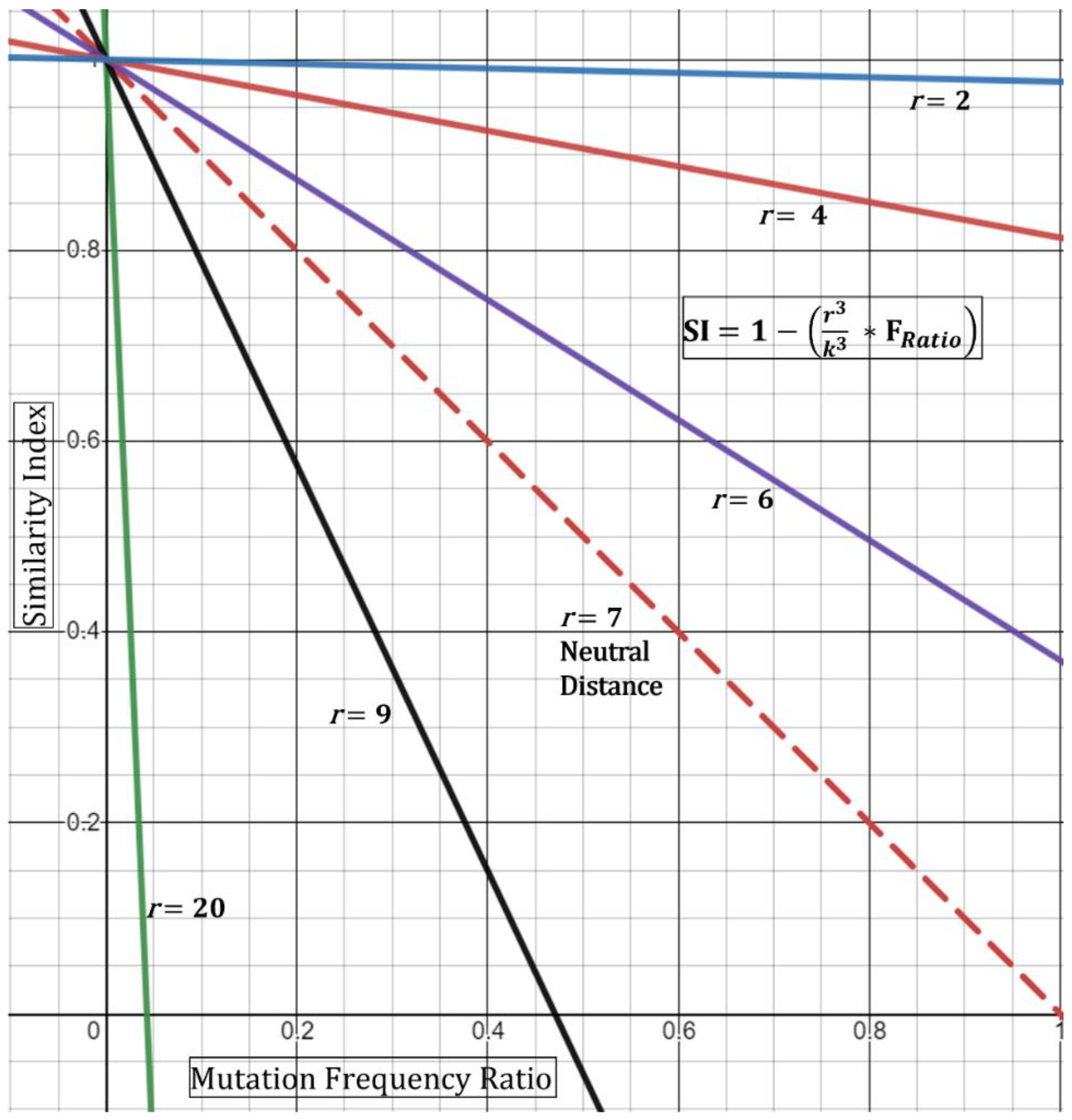
In order to incorporate patient-to-patient spatial dynamics at its simplest form, we propose utilizing a ratio of observed distance of two patients to the suggested safe distance. The physical space can be modeled as a sphere with volume,
. The geometry of the sphere (i.e., full vs. half) is not relevant since we are targeting a ratio. Therefore, the relationship can be written as such:
where
r is the minimum radius observed between patients;
k is the distance constant (7 feet—in alignment with recommendations of the CDC). The distance constant should be subject to amendment depending on observational/empirical studies.
From this, the new Similarity Index with simple spatial dynamics can be written as:
Based on this, if the minimum distance
r observed is 7 feet, then the
DR ratio collapses to “1” producing a “neutral” result (i.e., does not affect the index). If, however, the minimum distance is observed at 3.5 feet, then the ratio equates to 0.5, thence increasing the index—in other words, closer distances increase the chance that the strains are shared. This is geometrically displayed in (
Figure 3) in its simplest form utilizing the hypothetical Essen-Möller frequency ratio (F
Ratio). Here, as the radius
r approaches a distance of 0 feet, the Similarity Index approaches 1 (100%), regardless of the frequency ratio. This would be true, based on the model, even if the frequency ratio of mutations observed,
is equivalent to >0.8 (>80%). Conceptually, the fact that two patients were observed to interact at less than two feet while one of them was a known symptomatic CoV-2 positive substantially increases the Similarity Index even if there are no unique shared mutations. We must emphasize the assumptions in Equation (4)—that is, the patients should have confirmed or have been observed to be close together. Second, patient “zero” must have been confirmed to be infected around the time of the encounter to patient “one”. Third, patient “one” would need to test positive in a short period of time and/or show symptoms of infection. The latter point is already evident for many of the outbreak investigation samples.
A limitation with the spatial Similarity Index Equation (4), as seen in (
Figure 3), is that high
r values (above 7 feet) reduce the geometric curves ever towards the y-axis and limit the highest possible normalized frequency that can be used (i.e., resultant of
For example, at a
r = 9, a resultant frequency expression of ~0.47 would produce an index of 0 (
Figure 3) and with any frequency above 0.47 producing a negative index. Although this demonstrates the mathematical limits of this spatial equation, conceptually it means that similarity is unlikely—with an increased negative Similarity Index correlating with higher unlikeliness. Next, we do not foresee these limits to be reached incessantly given that, with our experience, most resultants of
show low frequency-ratio computations. Additionally, it would be seemingly rare to receive suspect localized outbreak samples with patients who were never observed less than 20–30 feet apart.
A second limitation for the spatial Similarity Index Equation (4) is that the curves generated by the distance ratio (
DR, and
Figure 3) are linear and would not fully represent real-time kinetics. A true representation of real-time spatial effects must include a temporal component where both factors are then guided by the physics of viral transmission, infectivity (Ct value), and length of exposure [
12]. Case-A analysis below demonstrates the use of Equation (4) where two of five specimens that are qualitatively expected to be similar had a reduced Similarity Index. Nonetheless, the equation should be used with full understanding of these limitations.
3.4. Analysis of Outbreak Samples via Similarity Index–Proof of Concept
To test our model, we utilized two cases (case-A and case-B) from our clinical reports on which we have concluded qualitatively as an outbreak case with similar and dissimilar strains, respectively. With regard to case-A, although it was qualitatively deemed to contain shared strains, there were samples that were more similar to each other when comparing nucleotide sequences within ORF1a. We utilized Equation (4) with a DR of one (i.e., neutral spatial dynamics).
Table 3 shows the mutations found within CoV-2 strains (extracted from suspected outbreak patients), while
Table 4 describes the Similarity Index between each pair of strains and the group. As expected, we can see that most samples have an index of >98%. The most dissimilarity was observed between p-1 and p-4 along with p-2 and p-4 showing a Similarity Index of 62.83% (
Table 4). This is logical as patients 1 and 2 are identical at ORF1a (
Table 4).
Interestingly, we observe here that p-1 and p-3 have a higher Similarity Index than p-1 and p-4. This seemed paradoxical since p-1 and p-4 have an extra shared mutation compared to p-1 and p-3 (
Table 4). The reason is that this extra shared mutation appears at a high frequency (68%). Albeit this seems paradoxical, it demonstrates that Equation (4) reduced the certainty of strain similarity since approximately 68% of circulating CoV-2 strains share this mutation and that the equation is based on random selection. It simply tells us that we have reduced confidence that p-1 and p-4 are similar given that they are chosen at random. This is one reason why it becomes important to include spatiotemporal components. These components integrate the important evidence, that the patients were close together in space and time during a known period of infection thence reducing the element of randomness with the Similarity Index. To this end, we shall hypothetically assume that p-1 and p-4 were observed at 5 feet, while one was symptomatic with a known positive low qRT-PCR Ct at the time of observation. In this case, using Equation (4) with a spatial component of 5 feet, the index is increased from 62.828% to 86.453%. Overall, these results show that lower frequency mutations are highly important in the differentiation process (i.e., excluding dissimilarity).
Using Equation (4), the combined Similarity Index was at 99.999%, but we also attempted to average the single indices for all five patients, producing an average of 92.135%. The discrepancy between Equation (4)-combined and the averaged single indices can be explained by the differential inclusion of all mutations with the total number of samples carrying shared vs. non-shared mutations, as detailed in the derivations section. Here, the combined Similarity Index by Equation (4) is heavily affected by low frequency mutations being shared, which increases the confidence that all samples are derived from the same source. In this case, the non-shared mutations had higher frequencies than the shared, which as we expect swayed the calculated values towards confidence of similarity (
Table 4).
Next, we analyzed case-B which we qualitatively reported clinically as a dissimilar group. We can observe that, although these three patients share plenty of mutations, they mostly nevertheless appear at high frequencies (
Table 5). Additionally, all three patients harbor non-shared low frequency mutations. As expected, the Similarity Index for all patients was overall lower than 5% (
Table 6). Thus, the equation demonstrates that even if all strains in question share several high frequency mutations, it still may not provide enough confidence to define similarity. Instead, high frequency mutations drive the index towards lower values due to reduced confidence stemming from the presence of high-prevalence mutations. The latter, occurring in the presence of non-shared low frequency mutations, substantially reduces the confidence in strain sharing as seen in this case. To this end, the combined Similarity Index by Equation (4) is calculated at 6.429% with averaged single indices at 3.031%.





{kind=link}
{kind=link}
{kind=link}