


The Etiological Diagnosis of Diabetes: Still a Challenge for the Clinician

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
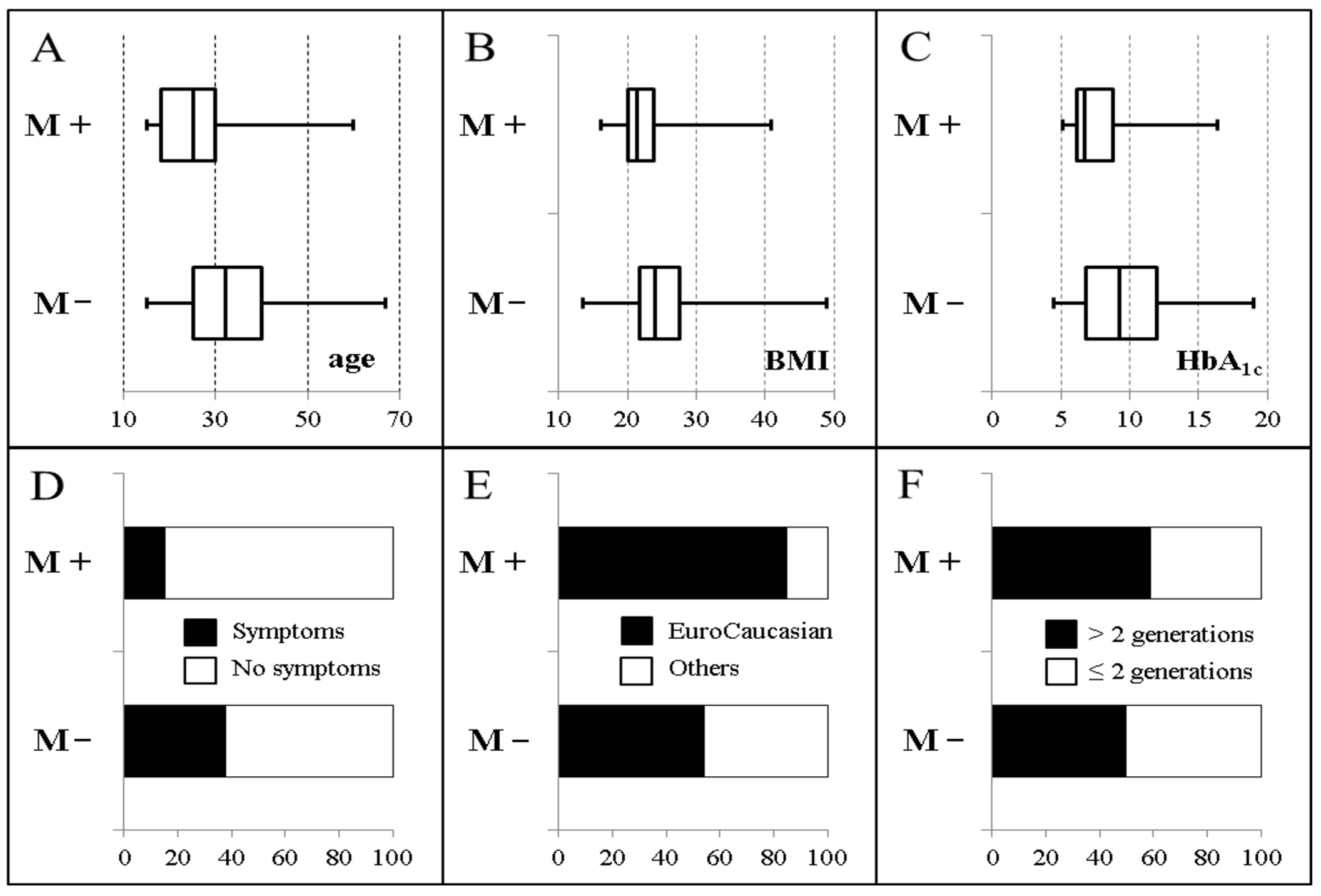
1. Introduction
- “Hybrid forms” of diabetes, including Slowly Evolving Immune-Mediated Diabetes (previously named Latent Autoimmune Diabetes in Adults, LADA) and Ketosis-Prone Type 2 Diabetes, the latter still being considered as a “non-autoimmune Type 1 diabetes” by the American Diabetes Association [5];
- “Unclassified Diabetes”, i.e., cases with no ascribable definite etiology, particularly at the time of diagnosis.
2. What Are the “Typical” Phenotypes of Type 1 and Type 2 Diabetes?
2.1. The Phenotype of Type 1 Diabetes Is Heterogenous
2.2. The Case of LADA/Slowly Evolving Immune-Mediated Diabetes
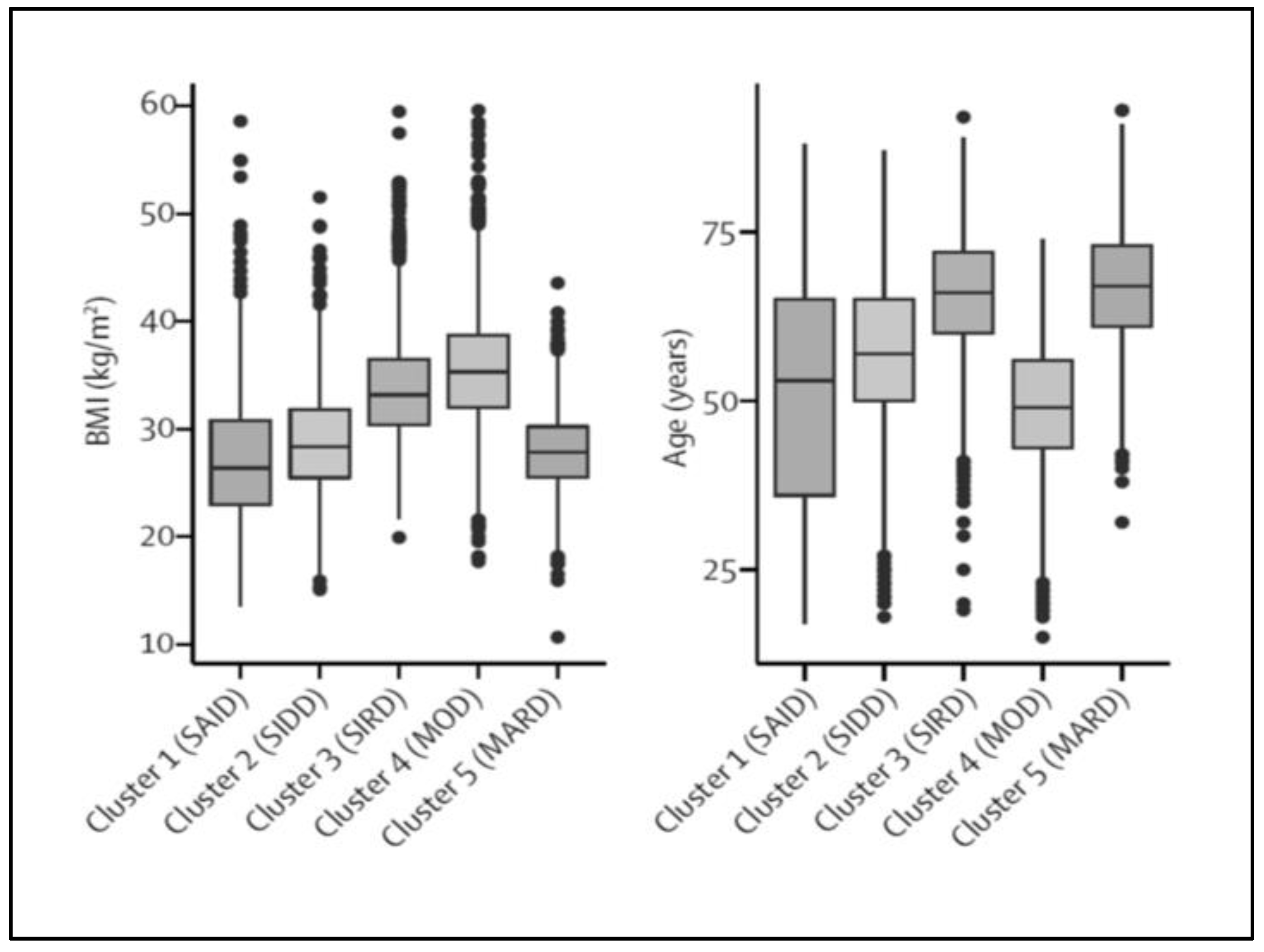
2.3. T2D Subtyping: Which Consequences for the Clinician?
2.4. The Case of “Ketosis-Prone Diabetes”
3. Monogenic Diabetes: A Multi-Faceted Diabetes Subtype
3.1. Epidemiology of MgD
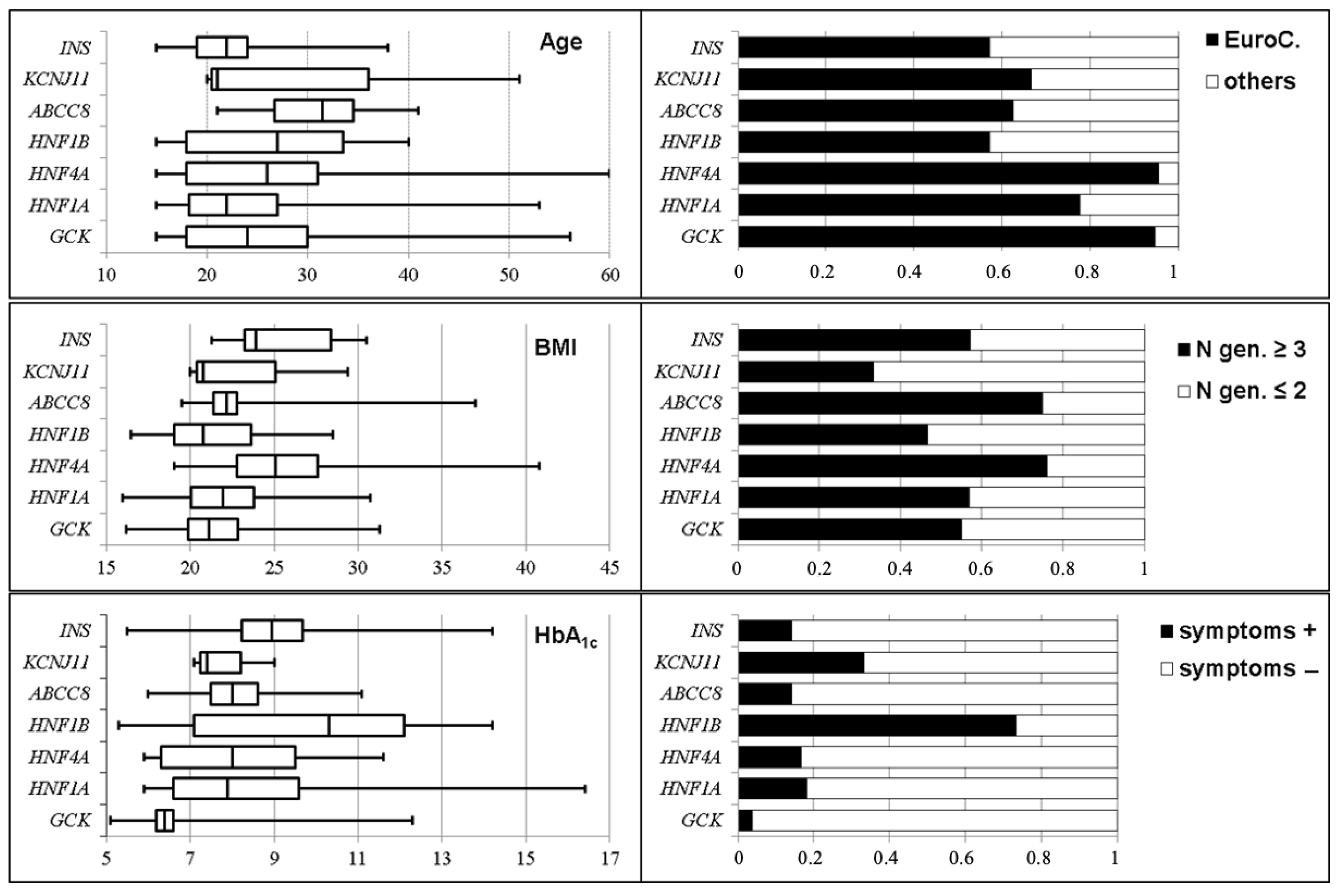
3.2. Genotype/Phenotype Correlations of the Most Frequent MgD Subtypes
3.2.1. GCK-MODY
3.2.2. HNF1A-MODY
3.2.3. HNF4A-MODY
3.2.4. HNF1B-Syndrome
3.2.5. Diabetes Associated with Pathogenic Variants of the Genes Encoding the K-ATP Channel Sub-Units
3.2.6. Maternally Inherited Diabetes and Deafness Syndrome
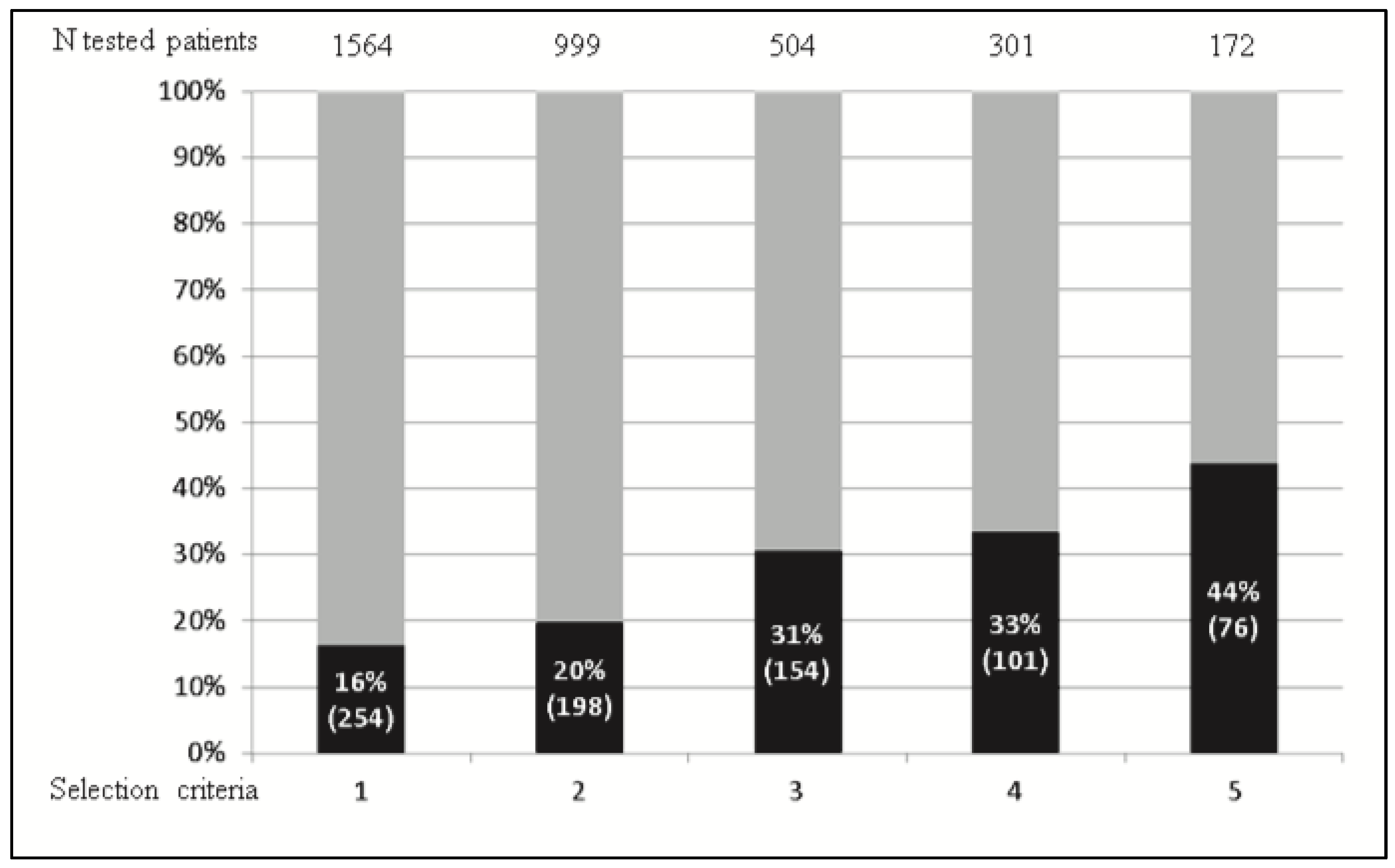
3.3. Monogenic Diabetes Often Remain Misdiagnosed
3.4. Differential Diagnosis with “Common” Diabetes Subtypes
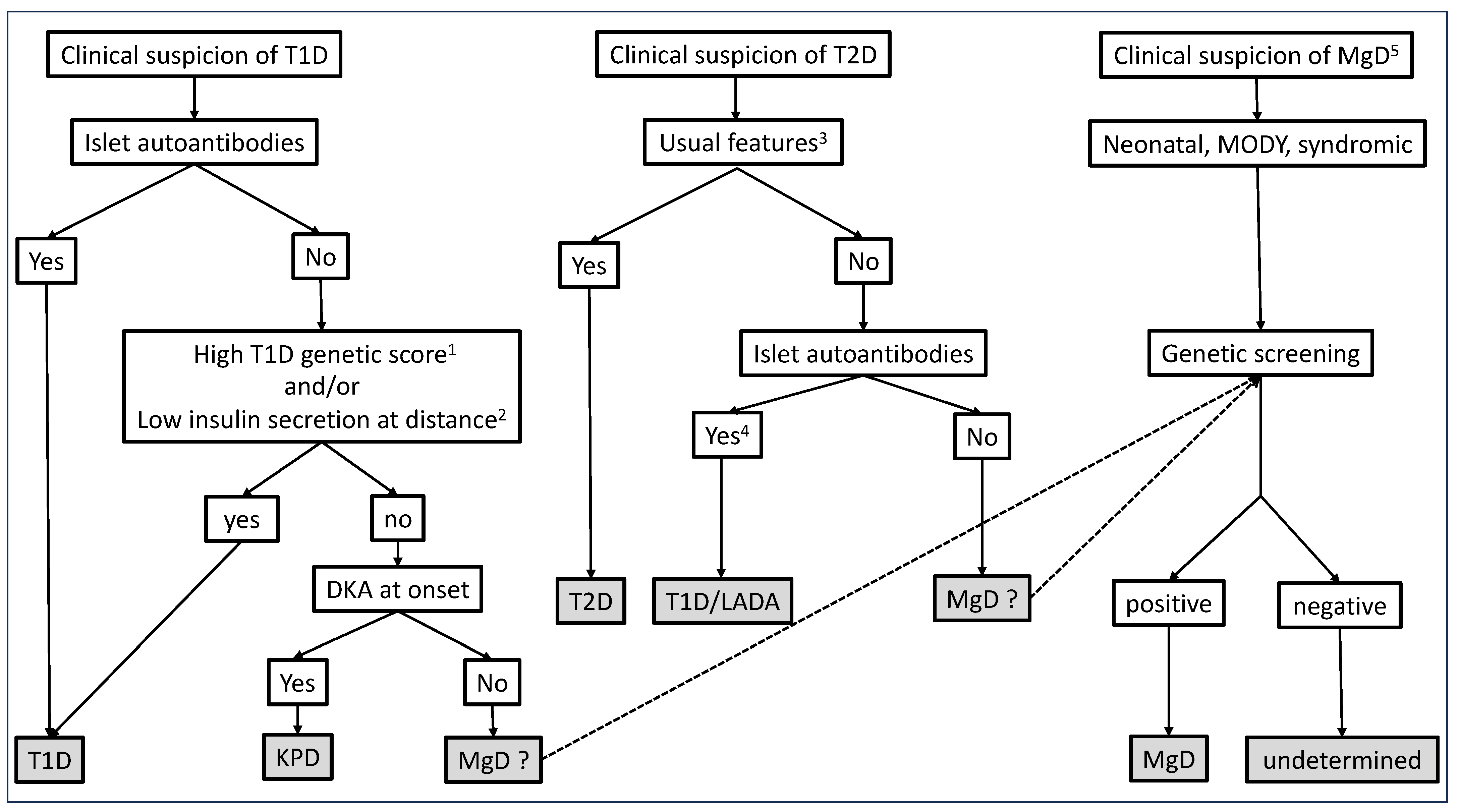
3.5. Which Strategy for the Genetic Diagnosis of MgD?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffin, S. Diabetes precision medicine: Plenty of potential, pitfalls and perils but not yet ready for prime time. Diabetologia 2022, 65, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, T.; Matsuda, A. Classification of diabetes on the basis of etiologies versus degree of insulin deficiency. Diabetes Care 1997, 20, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Classification of Diabetes Mellitus; World Health Organization: Geneva, Switzerland, 2019; Available online: https://apps.who.int/iris/handle/10665/325182 (accessed on 9 November 2022).
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes-2023. Diabetes Care 2023, 46 (Suppl. 1), S19–S40. [Google Scholar] [CrossRef] [PubMed]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Redondo, M.J.; Geyer, S.; Steck, A.K.; Sharp, S.; Wentworth, J.M.; Weedon, M.N.; Antinozzi, P.; Sosenko, J.; Atkinson, M.; Pugliese, A.; et al. A Type 1 Diabetes Genetic Risk Score Predicts Progression of Islet Autoimmunity and Development of Type 1 Diabetes in Individuals at Risk. Diabetes Care 2018, 41, 1887–1894. [Google Scholar] [CrossRef]
- Grubb, A.L.; McDonald, T.J.; Rutters, F.; Donnelly, L.A.; Hattersley, A.T.; Oram, R.A.; Palmer, C.N.A.; van der Heijden, A.A.; Carr, F.; Elders, P.J.M.; et al. A Type 1 Diabetes genetic risk score can identify patients with GAD65 autoantibody-positive type 2 diabetes who rapidly progress to insulin therapy. Diabetes Care. 2019, 42, 208–214. [Google Scholar] [CrossRef]
- Pugliese, A. Insulitis in the pathogenesis of type 1 diabetes. Pediatr. Diabetes 2016, 17 (Suppl. 22), 31–36. [Google Scholar] [CrossRef]
- Ziegler, A.G.; Rewers, M.; Simmel, O.; Simmel, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.I.; DeVries, J.H.; Hess-Fischl, A.; Hirsch, I.B.; Kirkman, M.S.; Klupa, T.; Ludwig, B.; Nørgaard, K.; Pettus, J.; Renard, E.; et al. The Management of Type 1 Diabetes in Adults. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2021, 44, 2589–2625. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Rogers, M.A.M.; Kim, C.; Banerjee, T.; Lee, J.M. Fluctuations in the incidence of type 1 diabetes in the United States from 2001 to 2015: A longitudinal study. BMC Med. 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Oram, R.A.; Patel, K.; Hill, A.; Shields, B.; McDonald, T.J.; Jones, A.; Hattersley, A.T.; Weedon, M.N. A type1 diabetes genetic risk score can aid discrimination between type 1 and type 2 diabetes in young adults. Diabetes Care 2016, 39, 337–344. [Google Scholar] [CrossRef]
- Thomas, N.J.; Jones, S.E.; Weedon, M.N.N.; Shields, B.M.; Oram, R.A.; Hattersley, A.T. Frequency and phenotype of type 1 diabetes in the first six decades of life: A crosssectional, genetically stratified survival analysis from US Biobank. Lancet Diabetes Endocrinol. 2018, 6, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.J.; Lynam, A.L.; Hill, A.V.; Weedon, M.N.; Shields, B.M.; Oram, R.A.; McDonald, T.J.; Hattersley, A.T.; Jones, A.G. Type 1 diabetes defined by severe insulin deficiency occurs after 30 years of age and is commonly treated as type 2 diabetes. Diabetologia 2019, 62, 1167–1172. [Google Scholar] [CrossRef]
- Eason, R.J.; Thomas, N.J.; Hill, A.V.; Knight, B.A.; Carr, A.; Hattersley, A.T.; McDonald, T.J.; Shields, B.M.; Jones, A.G.; StartRight Study Group. Routine islet autoantibody testing in clinically diagnosed adult-onset Type 1 Diabetes can help identify misclassification and the possibility of successful insulin cessation. Diabetes Care 2022, 45, 2844–2851. [Google Scholar] [CrossRef] [PubMed]
- Evertsen, J.; Alemzadeh, R.; Wang, X. Increasing incidence of pediatric type 1 diabetes mellitus in Southeastern Wisconsin: Relationship with body weight at diagnosis. PLoS ONE 2009, 4, e6873. [Google Scholar] [CrossRef] [PubMed]
- Minges, K.E.; Whittemore, R.; Weinzimer, S.A.; Irwin, M.L.; Redeker, N.S.; Grey, M. Correlates of overweight and obesity in 5529 adolescents with type 1 diabetes: The T1D Exchange Clinic Registry. Diabetes Res. Clin. Pract. 2017, 126, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Munoz, C.; Floreen, A.; Garey, C.; Karlya, T.; Jelley, D.; Alonso, G.T.; McAuliffe-Fogarty, A. Misdiagnosis and diabetic ketoacidosis at diagnosis of type 1 diabetes: Patient and caregiver perspectives. Clin. Diabetes 2019, 37, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E.; Banerji, M.A. Ketosis-prone diabetes (Flatbush diabetes): An emerging worldwide clinically important entity. Curr. Diab. Rep. 2018, 18, 120. [Google Scholar] [CrossRef] [PubMed]
- Usher-Smith, J.; Thompson, M.J.; Sharp, S.J.; Walter, F.M. Factors associated with the presence of diabetic ketoacidosis at diagnosis of diabetes in children and young adults: A systematic review. BMJ 2011, 343, d4092. [Google Scholar] [CrossRef]
- Lundgren, M.; Sahlin, A.; Svensson, C.; Carlsson, A.; Cedervall, E.; Jönsson, B.; Larsson, K.; Lernmark, A.; Neiderud, J.; Vigård, T.; et al. Reduced morbidity at diagnosis and improved glycemic control in children previously enrolled in DiPiS follow-up. Pediatr. Diabetes 2014, 15, 494–501. [Google Scholar] [CrossRef]
- Hekkala, A.M.; Ilonen, J.; Toppari, J.; Knip, M.; Veijola, R. Ketoacidosis at diagnosis of type 1 diabetes: Effect of prospective studies with newborn genetic screening and follow-up of risk children. Pediatr. Diabetes 2018, 19, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Ghalwash, M.; Anand, V.; Lou, O.; Martin, F.; Rewers, M.; Ziegler, A.G.; Toppari, J.; Hagopian, W.A.; Veijola, R.; Type 1 Diabetes Intelligence Study Group. Islet autoantibody screening in at-risk adolescents to predict type 1 diabetes until young adulthood: A prospective cohort study. Lancet Child Adolesc. Health 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Hodge, K.M.; Cousminer, D.L.; Leslie, R.D.; Grant, S.F.A. A global perspective of Latent Autoimmune Diabetes in Adults. Trend Endocrinol. Metab. 2018, 29, 638–650. [Google Scholar] [CrossRef]
- Writing Group for the SEARCH for Diabetes in Youth Study Group; Dabelea, D.; Bell, R.A.; D’Agostino, R.B., Jr.; Imperatore, G.; Johansen, J.M.; Linder, B.; Liu, L.L.; Loots, B.; Marcovina, S.; et al. Incidence of diabetes in youth in the United States. JAMA 2007, 297, 2716–2724. [Google Scholar] [CrossRef] [PubMed]
- Klingensmith, G.J.; Pyle, L.; Arslanian, S.; Copeland, K.C.; Cuttler, L.; Kaufman, F.; Laffel, L.; Marcovina, S.; Tollefsen, S.E.; Weinstock, R.S.; et al. The presence of GAD and IA-2 antibodies in youth with a type 2 diabetes phenotype. Results from the TODAY study. Diabetes Care 2010, 33, 1970–1975. [Google Scholar] [CrossRef]
- Cervin, C.; Lyssenko, V.; Bakhtadze, E.; Lindholm, E.; Nilsson, P.; Tuomi, T.; Corrado, M.C.; Groop, L. Genetic similarities between latent autoimmune diabetes in adults, type 1 diabetes, and type 2 diabetes. Diabetes 2008, 57, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Cousminer, D.L.; Ahlqvist, E.; Mishra, R.; Andersen, M.K.; Chesi, A.; Hawa, M.I.; Davis, A.; Hodge, K.M.; Bradfield, J.P.; Zhou, K.; et al. First genome-wide association study of Latent Autoimmune Diabetes in Adults reveals novel insights linking immune and metabolic diabetes. Diabetes Care 2018, 41, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Hjort, R.; Löfvenborg, J.E.; Ahlqvist, E.; Alfredsson, L.; Andersson, T.; Grill, V.; Groop, L.; Sørgjerd, E.P.; Tuomi, T.; Åsvold, B.O.; et al. Interaction between overweight and genotypes of HLA, TCF7L2, and FTO in relation to the risk of Latent Autoimmune Diabetes in Adults and type 2 diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 4815–4826. [Google Scholar] [CrossRef]
- Redondo, M.J.; Evans-Molina, C.; Steck, A.K.; Atkinson, M.A.; Sosenko, J. The influence of type 2 diabetes-associated factors on type 1 diabetes. Diabetes Care 2019, 42, 1357–1364. [Google Scholar] [CrossRef]
- Pieralice, S.; Pozzili, P. Latent Autoimmune Diabetes in Adults: A review on clinical implications and management. Diabetes Metab. J. 2018, 42, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, R.; Zampetti, S.; Maddaloni, E. Adult-onset autoimmune diabetes: Current knowledge and implications for management. Nat. Rev. Endocrinol. 2017, 13, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.G.; McDonald, T.J.; Shields, B.M.; Hagopian, W.; Hattersley, A.T. Latent Autoimmune Diabetes of Adults (LADA) is likely to represent a mixed population of autoimmune (Type 1) and nonautoimmune (Type 2) diabetes. Diabetes Care 2021, 44, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Ma, Y.; Knowler, W.C.; Marcovina, S.; Saudek, C.D.; Arakaki, R.; White, N.H.; Kahn, S.E.; Orchard, T.J.; Goldberg, R.; et al. Diabetes autoantibodies do not predict progression to diabetes in adults: The Diabetes Prevention Program. Diabet. Med. 2014, 31, 1064–1068. [Google Scholar] [CrossRef]
- Ziegler, A.G.; Kick, K.; Bonifacio, E.; Haupt, F.; Hippich, M.; Dunstheimer, D.; Lang, M.; Laub, O.; Warncke, K.; Lange, K.; et al. Yield of a Public Health Screening of Children for Islet Autoantibodies in Bavaria, Germany. JAMA 2020, 323, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, V.M.; Isomaa, B.; Lyssenko, V.; Laurila, E.; Korhonen, P.; Groop, L.C.; Tuomi, T.; Botnia Study Group. GAD antibody positivity predicts type 2 diabetes in an adult population. Diabetes 2010, 59, 416–422. [Google Scholar] [CrossRef]
- Vehik, K.; Lynch, K.F.; Schatz, D.A.; Akolkar, B.; Hagopian, W.; Rewers, M.; She, J.X.; Simell, O.; Toppari, J.; Ziegler, A.G.; et al. Reversion of β-Cell Autoimmunity Changes Risk of Type 1 Diabetes: TEDDY Study. Diabetes Care 2016, 39, 1535–1542. [Google Scholar] [CrossRef]
- Kawasaki, E.; Fukuyama, T.; Uchida, A.; Sagara, Y.; Tamai, H.; Nakano, Y.; Tojikubo, M.; Hiromatsu, Y.; Koga, N. Characterization of patients with diabetes who were incidentally found to be glutamic acid decarboxylase autoantibody-positive by bridging-type enzyme-linked immunosorbent assay. J. Diabetes Investig. 2020, 11, 1507–1510. [Google Scholar] [CrossRef]
- Wagner, R.; McNally, J.M.; Bonifacio, E.; Genovese, S.; Foulis, A.; McGill, M.; Christie, M.R.; Betterle, C.; Bosi, E.; Bottazzo, G.F. Lack of immunohistological changes in the islets of nondiabetic, autoimmune, polyendocrine patients with beta-selective GAD-specific islet cell antibodies. Diabetes 1994, 43, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Diedisheim, M.; Mallone, R.; Boitard, C.; Larger, E. β-cell Mass in Nondiabetic Autoantibody-Positive Subjects: An Analysis Based on the Network for Pancreatic Organ Donors Database. J. Clin. Endocrinol. Metab. 2016, 101, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Falorni, A.; Gambelunghe, G.; Forini, F.; Kassi, G.; Cosentino, A.; Candeloro, P.; Bolli, G.B.; Brunetti, P.; Calcinaro, F. Autoantibody recognition of COOH-terminal epitopes of GAD65 marks the risk for insulin requirement in adult-onset diabetes mellitus. J. Clin. Endocrinol. Metab. 2000, 85, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, P.; Hawa, M.I.; Krause, S.; Lampasona, V.; Jerram, S.T.; Williams, A.J.K.; Bonifacio, E.; Ziegler, A.G.; Leslie, R.D. Action LADA consortium. Autoantibodies to N-terminally truncated GAD improve clinical phenotyping of individuals with adult-onset diabetes: Action LADA 12. Diabetologia 2018, 61, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Pöllänen, P.M.; Härkönen, T.; Ilonen, J.; Toppari, J.; Veijola, R.; Siljander, H.; Knip, M. Autoantibodies to N-terminally Truncated GAD65(96-585): HLA Associations and Predictive Value for Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2022, 107, e935–e946. [Google Scholar] [CrossRef]
- Wucher, H.; Lepercq, J.; Carette, C.; Colas, C.; Dubois-Laforgue, D.; Gautier, J.F.; Lalej, D.; Larger, E.; Ledoux, S.; Mbemba, J.; et al. Poor prognosis of pregnancy in women with autoimmune type 1 diabetes mellitus masquerading as gestational diabetes. Diabetes Metab. 2011, 37, 47–51. [Google Scholar] [CrossRef]
- Incani, M.; Baroni, M.G.; Cossu, E. Testing for type 1 diabetes autoantibodies in gestational diabetes mellitus (GDM): Is it clinically useful? BMC Endoc. Disord. 2019, 19, 44. [Google Scholar] [CrossRef]
- Battaglia, M.; Ahmed, S.; Anderson, M.S.; Atkinson, M.A.; Becker, D.; Bingley, P.J.; Bosi, E.; Brusko, T.M.; DiMeglio, L.A.; Evans-Molina, C.; et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care 2020, 43, 5–12. [Google Scholar] [CrossRef]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An anti-CD3 antibody, Teplizumab, in relatives at risk for type 1 diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef]
- Tabak, A.G.; Jokela, M.; Akbaraly, T.N.; Brunner, E.J.; Kivimäki, M.; Witte, D.R. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: An analysis from the Whitehall II study. Lancet 2009, 373, 2215–2221. [Google Scholar] [CrossRef]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of diabetes by pathophysiology, natural history and prognosis. Diabetes 2017, 66, 241–255. [Google Scholar] [CrossRef]
- Del Prato, S. Heterogeneity of diabetes: Heralding the era of precision medicine. Lancet Diabetes Endocrinol. 2018, 7, 659–661. [Google Scholar] [CrossRef]
- Deutsch, A.J.; Ahlqvist, E.; Udler, M. Phenotpic and genetic classification of diabetes. Diabetologia 2022, 65, 1758–1769. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheng, W.Y.; Glicksberg, B.S.; Gottesman, O.; Tamler, R.; Chen, R.; Bottinger, E.P.; Dudley, J.T. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci. Transl. Med. 2015, 7, 311ra174. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 1844–1853. [Google Scholar] [CrossRef]
- Zaharia, O.P.; Strassburger, K.; Strom, A.; Bönhof, G.J.; Karusheva, Y.; Antoniou, S.; Bódis, K.; Markgraf, D.F.; Burkart, V.; Müssig, K.; et al. Risk of diabetes-associated diseases for patients with recent-onset diabetes: A 5-year follow-up study. Lancet Diabetes Endocrinol. 2019, 7, 684–694. [Google Scholar] [CrossRef]
- Mansour Aly, D.; Dwivedi, O.P.; Prasad, R.B.; Käräjämäki, A.; Hjort, R.; Thangam, M.; Åkerlund, M.; Mahajan, A.; Udler, M.S.; Florez, J.C.; et al. Genome-wide association analyses highlight etiological differences underlying newly defined subtypes of diabetes. Nat. Genet. 2021, 53, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Slieker, R.C.; Donnelly, L.A.; Fitipaldi, H.; Bouland, G.A.; Giordano, G.N.; Åkerlund, M.; Gerl, M.J.; Ahlqvist, E.; Ali, A.; Dragan, I.; et al. Distinct Molecular Signatures of Clinical Clusters in People With Type 2 Diabetes: An IMI-RHAPSODY Study. Diabetes 2021, 70, 2683–2693. [Google Scholar] [CrossRef]
- Zaghlool, S.B.; Halama, A.; Stephan, N.; Gudmundsdottir, V.; Gudnason, V.; Jennings, L.L.; Thangam, M.; Ahlqvist, E.; Malik, R.A.; Albagha, O.M.E.; et al. Metabolic and proteomic signatures of type 2 diabetes subtypes in an Arab population. Nat. Commun. 2022, 13, 7121. [Google Scholar] [CrossRef]
- Pigeyre, M.; Gerstein, H.; Ahlqvist, E.; Hess, S.; Paré, G. Identifying blood biomarkers for type 2 diabetes subtyping: A report from the ORIGIN trial. Diabetologia 2023, 66, 1045–1051. [Google Scholar] [CrossRef]
- Sattar, N.; Gill, J.M.R. Type 2 diabetes in migrant south Asians: Mechanisms, mitigation, and management. Lancet Diabetes Endocrinol. 2015, 3, 1004–1016. [Google Scholar] [CrossRef]
- Caleyachetty, R.; Barber, T.M.; Mohammed, N.I.; Cappuccio, F.P.; Hardy, R.; Mathur, R.; Banerjee, A.; Gill, P. Ethnicity-specific BMI cutoffs for obesity based on type 2 diabetes risk in England: A population-based cohort study. Lancet Diabetes Endocrinol. 2021, 9, 419–426. [Google Scholar] [CrossRef]
- Prasad, R.B.; Asplund, O.; Shukla, S.R.; Wagh, R.; Kunte, P.; Bhat, D.; Parekh, M.; Shah, M.; Phatak, S.; Käräjämäki, A.; et al. Subgroups of patients with young-onset type 2 diabetes in India reveal insulin deficiency as a major driver. Diabetologia 2022, 65, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Perng, W.; Conway, R.; Mayer-Davis, E.; Dabelea, D. Youth-Onset Type 2 Diabetes: The epidemiology of an awakening epidemic. Diabetes Care 2023, 46, 490–499. [Google Scholar] [CrossRef]
- Giannini, C.; Weiss, R.; Cali, A.; Bonadonna, R.; Santoro, N.; Pierpont, B.; Shaw, M.; Caprio, S. Evidence for early defects in insulin sensitivity and secretion before the onset of glucose dysregulation in obese youth. Diabetes 2012, 61, 606–614. [Google Scholar] [CrossRef]
- Dennis, J.M.; Shields, B.M.; Henley, W.E.; Jones, A.G.; Hattersley, A.T. Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: An analysis using clinical trial data. Lancet Diabetes Endocrinol. 2019, 7, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.T.N.; Wesolowska-Andersen, A.; Brorsson, C.; Rajendrakumar, A.L.; Hapca, S.; Gan, S.; Dawed, A.Y.; Donnelly, L.A.; McCrimmon, R.; Doney, A.S.F.; et al. Heterogeneity in phenotype, disease progression and drug response in type 2 diabetes. Nat. Med. 2022, 28, 982–988. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Brorsson, C.A.; Bizzotto, R.; Mari, A.; Tura, A.; Koivula, R.; Mahajan, A.; Vinuela, A.; Tajes, J.F.; Sharma, S.; et al. Four groups of type 2 diabetes contribute to the etiological and clinical heterogeneity in newly diagnosed individuals: An IMI DIRECT study. Cell Rep. Med. 2022, 3, 100477. [Google Scholar] [CrossRef]
- Winter, W.E.; Maclaren, N.K.; Riley, W.J.; Clarke, D.W.; Kappy, M.S.; Spillar, R.P. Maturity-onset diabetes of youth in black Americans. N. Engl. J. Med. 1987, 316, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Banerji, M.A.; Chaiken, R.L.; Huey, H.; Tuomi, T.; Norin, A.J.; Mackay, I.R.; Rowley, M.J.; Zimmet, P.Z.; Lebovitz, H.E. GAD antibody negative NIDDM in adult black subjects with diabetic ketoacidosis and increased frequency of human leukocyte antigen DR3 and DR4. Flatbush diabetes. Diabetes 1994, 43, 741–745. [Google Scholar] [CrossRef]
- Balasubramanyam, A. Syndromes of ketosis-prone diabetes. Trans. Am. Clin. Climatol. Assoc. 2019, 130, 145–155. [Google Scholar]
- Maldonado, M.; Hampe, C.S.; Gaur, L.K.; D’Amico, S.; Iyer, D.; Hammerle, L.P.; Bolgiano, D.; Rodriguez, L.; Rajan, A.; Lernmark, A.; et al. Ketosis-prone diabetes: Dissection of a heterogeneous syndrome using an immunogenetic and beta-cell functional classification, prospective analysis, and clinical outcomes. J. Clin. Endocrinol. Metab. 2003, 88, 5090–5098. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S.; Nalini, R.; Maldonado, M.R.; Hall, T.R.; Garza, G.; Iyer, D.; Balasubramanyam, A. Association of amino-terminal-specific antiglutamate decarboxylase (GAD65) autoantibodies with beta-cell functional reserve and a milder clinical phenotype in patients with GAD65 antibodies and ketosis-prone diabetes mellitus. J. Clin. Endocrinol. Metab. 2007, 92, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Nalini, R.; Ozer, K.; Maldonado, M.; Patel, S.G.; Hampe, C.S.; Guthikonda, A.; Villanueva, J.; O’Brian Smith, E.; Gaur, L.K.; Balasubramanyam, A. Presence or absence of a known diabetic ketoacidosis precipitant defines distinct syndromes of "A-β+" ketosis-prone diabetes based on long-term β-cell function, human leukocyte antigen class II alleles, and sex predilection. Metabolism 2010, 59, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Vellanki, P.; Umpierrez, G.E. Diabetic ketoacidosis: A common debut of diabetes among African American with Type 2 diabetes. Endocr. Pract. 2017, 23, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Hsu, J.W.; Jahoor, F.; Coraza, I.; Bain, J.R.; Stevens, R.D.; Iyer, D.; Nalini, R.; Ozer, K.; Hampe, C.S.; et al. Pathogenesis of A⁻β⁺ ketosis-prone diabetes. Diabetes 2013, 62, 12–22. [Google Scholar] [CrossRef]
- Flannick, J.; Johansson, S.; Njølstad, P.R. Common and rare forms of diabetes mellitus: Towards a continuum of diabetes subtypes. Nat. Rev. Endocrinol. 2016, 12, 394–406. [Google Scholar] [CrossRef]
- Murphy, R.; Ellard, S.; Hattersley, A.T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 200–213. [Google Scholar] [CrossRef]
- Donath, X.; Saint-Martin, C.; Dubois-Laforgue, D.; Rajasingham, R.; Mifsud, F.; Ciangura, C.; Timsit, J.; Bellanné-Chantelot, C.; Monogenic Diabetes Study Group of the Société Francophone du Diabète. Next-generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult-onset diabetes selected on a clinical basis: A cross-sectional analysis. BMC Med. 2019, 17, 132. [Google Scholar] [CrossRef]
- Bansal, V.; Gassenhuber, J.; Phillips, T.; Oliveira, G.; Harbaugh, R.; Villarasa, N.; Topol, E.J.; Seufferlein, T.; Boehm, B.O. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Med. 2017, 15, 213. [Google Scholar] [CrossRef]
- Colclough, K.; Ellard, S.; Hattersley, A.; Patel, K. Syndromic monogenic diabetes genes should be tested in patients with a clinical suspicion of Maturity-Onset Diabetes of the Young. Diabetes 2022, 71, 530–537. [Google Scholar] [CrossRef]
- Saint-Martin, C.; Bouvet, D.; Bastide, M.; Bellanné-Chantelot, C. Gene panel sequencing of patients with monogenic diabetes brings to Light genes typically associated with syndromic presentations. Diabetes 2022, 71, 578–584. [Google Scholar] [CrossRef]
- Johansson, B.B.; Irgens, H.U.; Molnes, J.; Sztromwasser, P.; Aukrust, I.; Juliusson, P.B.; Søvik, O.; Levy, S.; Skrivarhaug, T.; Joner, G.; et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia 2017, 60, 625–635. [Google Scholar] [CrossRef]
- Shields, B.M.; Shepherd, M.; Hudson, M.; McDonald, T.J.; Colclough, K.; Peters, J.; Knight, B.; Hyde, C.; Ellard, S.; Pearson, E.R.; et al. Population-based assessment of a biomarker-based screening pathway to aid diagnosis of monogenic diabetes in young-onset patients. Diabetes Care 2017, 40, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Chakera, A.J.; Steele, A.M.; Gloyn, A.L.; Shepherd, M.H.; Shields, B.; Ellard, S.; Hattersley, A.T. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 2015, 38, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Mirshahi, U.L.; Colclough, K.; Wright, C.F.; Wood, A.R.; Beaumont, R.N.; Tyrrell, J.; Laver, T.W.; Stahl, R.; Golden, A.; Goehringer, J.M.; et al. Reduced penetrance of MODY-associated HNF1A/HNF4A variants but not GCK variants in clinically unselected cohorts. Am. J. Hum. Genet. 2022, 109, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Stride, A.; Shields, B.; Gill-Carey, O.; Chakera, A.; Colclough, K.; Ellard, S.; Hattersley, A.T. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia 2014, 57, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Bellanné-Chantelot, C.; Lévy, D.J.; Carette, C.; Saint-Martin, C.; Riveline, J.P.; Larger, E.; Valéro, R.; Gautier, J.F.; Reznik, Y.; Sola, A.; et al. Clinical characteristics and diagnostic criteria of Maturity-Onset Diabetes of the Young (MODY) due to molecular anomalies of the HNF1A gene. J. Clin. Endocrinol. Metab. 2011, 96, E1346–E1351. [Google Scholar] [CrossRef]
- Shepherd, M.; Shields, B.; Ellard, S.; Rubio-Cabezas, O.; Hattersley, A.T. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabetic Med. 2009, 26, 437–441. [Google Scholar] [CrossRef]
- Bellanné-Chantelot, C.; Carette, C.; Riveline, J.P.; Valéro, R.; Gautier, J.F.; Larger, E.; Reznik, Y.; Ducluzeau, P.H.; Sola, A.; Hartemann-Heurtier, A.; et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with Maturity-Onset Diabetes of the Young (MODY)-3. Diabetes 2008, 57, 503–508. [Google Scholar] [CrossRef]
- Locke, J.M.; Saint-Martin, C.; Laver, T.W.; Patel, K.A.; Wood, A.R.; Sharp, S.A.; Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T.; Harries, L.W.; et al. The common HNF1A variant I27L is a modifier of age at diabetes diagnosis in individuals with HNF1A-MODY. Diabetes 2018, 67, 1903–1907. [Google Scholar] [CrossRef]
- Ludwig-Słomczyńska, A.; Seweryn, M.T.; Radkowski, P.; Kapusta, P.; Machlowska, J.; Pruhova, S.; Gasperikova, D.; Bellanne-Chantelot, C.; Hattersley, A.; Kandasamy, B.; et al. Variants influencing age at diagnosis of HNF1A-MODY. Mol. Med. 2022, 28, 113. [Google Scholar] [CrossRef] [PubMed]
- Stride, A.; Shepherd, M.; Frayling, T.M.; Bulman, M.P.; Ellard, S.; Hattersley, A.T. Intrauterine hyperglycemia is associated with an earlier diagnosis of diabetes in HNF-1alpha gene mutation carriers. Diabetes Care 2002, 25, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Klupa, T.; Warram, J.H.; Antonellis, A.; Pezzolesi, M.; Nam, M.; Malecki, M.T.; Doria, A.; Rich, S.S.; Krolewski, A.S. Determinants of the development of diabetes (maturity-onset diabetes of the young-3) in carriers of HNF-1alpha mutations: Evidence for parent-of-origin effect. Diabetes Care 2002, 25, 2292–2301. [Google Scholar] [CrossRef]
- Pearson, E.R.; Pruhova, S.; Tack, C.J.; Johansen, A.; Castleden, H.A.J.; Lumb, P.J.; Wierzbicki, A.S.; Clark, P.M.; Lebl, J.; Pedersen, O.; et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 2005, 48, 878–885. [Google Scholar] [CrossRef]
- Pearson, E.R.; Boj, S.F.; Steele, A.M.; Barrett, T.; Stals, K.; Shield, J.P.; Ellard, S.; Ferrer, J.; Hattersley, A.T. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLOS Med. 2007, 4, e118. [Google Scholar] [CrossRef] [PubMed]
- Clissold, R.L.; Hamilton, A.J.; Hattersley, A.T.; Ellard, S.; Bingham, C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat. Rev. Nephrol. 2015, 11, 102–112. [Google Scholar] [CrossRef]
- Dubois-Laforgue, D.; Cornu, E.; Saint-Martin, C.; Coste, J.; Bellanné-Chantelot, C.; Timsit, J.; Monogenic Diabetes Study Group of the Société Francophone du Diabète. Diabetes, associated clinical spectrum and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1B) molecular defects: A long-term study. Diabetes Care 2017, 40, 1436–1443. [Google Scholar] [CrossRef]
- Bellanné-Chantelot, C.; Clauin, S.; Chauveau, D.; Collin, P.; Daumont, M.; Douillard, C.; Dubois-Laforgue, D.; Dusselier, L.; Gautier, J.F.; Jadoul, M.; et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 2005, 54, 3126–3132. [Google Scholar] [CrossRef]
- Flanagan, S.; Patch, A.M.; Mackay, D.J.G.; Edghill, E.L.; Gloyn, A.K.; Robinson, D.; Shield, J.P.H.; Temple, K.; Ellard, S.; Hattersley, A.T. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007, 56, 1930–1937. [Google Scholar] [CrossRef]
- Babenko, A.P.; Polak, M.; Cavé, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006, 355, 456–466. [Google Scholar] [CrossRef]
- Timmers, M.; Dirinck, E.; Lauwers, P.; Wuyts, W.; De Block, C. ABCC8 variants in MODY12: Review of the literature and report of a case with severe complications. Diabetes Metab Res. Rev. 2021, 37, e3459. [Google Scholar] [CrossRef] [PubMed]
- Bowman, P.; Sulen, Å.; Barbetti, F.; Beltrand, J.; Svalastoga, P.; Codner, E.; Tessmann, E.H.; Juliusson, P.B.; Skrivarhaug, T.; Pearson, E.R.; et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: An international cohort study. Lancet Diabetes Endocrinol. 2018, 6, 637–646. [Google Scholar] [CrossRef]
- Bowman, P.; Mathews, F.; Barbetti, F.; Shepherd, M.H.; Sanchez, J.; Piccini, B.; Beltrand, J.; Letourneau-Freiberg, L.R.; Polak, M.; Greeley, S.A.W.; et al. Long-term Follow-up of glycemic and neurological outcomes in an international series of patients with sulfonylurea-treated ABCC8 permanent neonatal diabetes. Diabetes Care 2021, 44, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Guillausseau, P.J.; Dubois-Laforgue, D.; Massin, P.; Laloi-Michelin, M.; Bellanné-Chantelot, C.; Gin, H.; Bertin, E.; Blickle, J.F.; Bauduceau, B.; Bouhanick, B.; et al. Heterogeneity of diabetes phenotype in patients with 3243 bp mutation of mitochondrial DNA (Maternally Inherited Diabetes and Deafness or MIDD). Diabetes Metab. 2004, 30, 181–186. [Google Scholar] [CrossRef]
- Guillausseau, P.J.; Massin, P.; Dubois-LaForgue, D.; Timsit, J.; Virally, M.; Gin, H.; Bertin, E.; Blickle, J.F.; Bouhanick, B.; Cahen, J.; et al. Maternally inherited diabetes and deafness: A multicenter study. Ann. Intern. Med. 2001, 134, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Pihoker, C.; Gilliam, L.K.; Ellard, S.; Dabelea, D.; Davis, C.; Dolan, L.M.; Greenbaum, C.J.; Imperatore, G.; Lawrence, J.M.; Marcovina, S.M.; et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: Results from the SEARCH for Diabetes in Youth. J. Clin. Endocrinol. Metab. 2013, 98, 4055–4062. [Google Scholar] [CrossRef]
- Shepherd, M.; Shields, B.; Hammersley, S.; Hudson, M.; McDonald, T.J.; Colclough, K.; Oram, R.A.; Knight, B.; Hyde, C.; Cox, J.; et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5 % of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care 2016, 39, 1879–1888. [Google Scholar] [CrossRef]
- Shields, B.M.; Hicks, S.; Shepherd, M.H.; Colclough, K.; Hattersley, A.T.; Ellard, S. Maturity-onset diabetes of the young (MODY): How many cases are we missing? Diabetologia 2010, 53, 2504–2508. [Google Scholar] [CrossRef]
- van der Zwaag, A.M.; Weinreich, S.S.; Bosma, A.R.; Rigter, T.; Losekoot, M.; Henneman, L.; Cornel, M.C. Current and best practices of genetic testing for maturity onset diabetes of the young: Views of professional experts. Public Health Genomics. 2015, 18, 52–59. [Google Scholar] [CrossRef]
- Naylor, R. Economics of Genetic Testing for Diabetes. Curr. Diab. Rep. 2019, 19, 23. [Google Scholar] [CrossRef]
- Zhang, H.; Kleinberger, J.W.; Maloney, K.A.; Guan, Y.; Mathias, T.J.; Bisordi, K.; Streeten, E.A.; Blessing, K.; Snyder, M.N.; Bromberger, L.A.; et al. Model for integration of Monogenic Diabetes diagnosis into routine Care: The Personalized Diabetes Medicine Program. Diabetes Care 2022, 45, 1799–1806. [Google Scholar] [CrossRef]
- Carlsson, A.; Shepherd, M.; Ellard, S.; Weedon, M.; Lernmark, Å.; Forsander, G.; Colclough, K.; Brahimi, Q.; Valtonen-Andre, C.; Ivarsson, S.A.; et al. Absence of Islet Autoantibodies and Modestly Raised Glucose Values at Diabetes Diagnosis Should Lead to Testing for MODY: Lessons From a 5-Year Pediatric Swedish National Cohort Study. Diabetes Care 2020, 43, 82–89. [Google Scholar] [CrossRef]
- Harsunen, M.; Kettunen, J.L.T.; Härkönen, T.; Dwivedi, O.; Lehtovirta, M.; Vähäsalo, P.; Veijola, R.; Ilonen, J.; Miettinen, P.J.; Knip, M.; et al. Identification of monogenic variants in more than ten per cent of children without type 1 diabetes-related autoantibodies at diagnosis in the Finnish Pediatric Diabetes Register. Diabetologia 2023, 66, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Colclough, K.; Saint-Martin, C.; Timsit, J.; Ellard, S.; Bellanné-Chantelot, C. Clinical utility gene card for: Maturity-onset diabetes of the young. Eur. J. Hum. Genet. 2014, 22, 1153. [Google Scholar] [CrossRef] [PubMed]
- Wilmot-Roussel, H.; Lévy, D.J.; Carette, C.; Caillat-Zucman, S.; Boitard, C.; Timsit, J.; Dubois-Laforgue, D. Factors associated with the presence of glutamic acid decarboxylase and islet antigen-2 autoantibodies in patients with long-standing type 1 diabetes. Diabetes Metab. 2013, 39, 144–149. [Google Scholar] [CrossRef]
- Williams, C.L.; Fareed, R.; Mortimer, G.L.M.; Aitken, R.J.; Wilson, I.V.; George, G.; Gillespie, K.M.; Williams, A.J.K.; BOX Study Group; Long, A.E. The longitudinal loss of islet autoantibody responses from diagnosis of type 1 diabetes occurs progressively over follow-up and is determined by low autoantibody titres, early-onset, and genetic variants. Clin. Exp. Immunol. 2022, 210, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.A.; Rich, S.S.; Wood, A.R.; Jones, S.E.; Beaumont, R.N.; Harrison, J.W.; Schneider, D.A.; Locke, J.M.; Tyrrell, J.; Weedon, M.N.; et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care 2019, 42, 200–207. [Google Scholar] [CrossRef]
- Lynam, A.; McDonald, T.; Hill, A.; Dennis, J.; Oram, R.; Pearson, E.; Weedon, M.; Hattersley, A.; Owen, K.; Shields, B.; et al. Development and validation of multivariable clinical diagnostic models to identify type 1 diabetes requiring rapid insulin therapy in adults aged 18-50 years. BMJ Open 2019, 9, e031586. [Google Scholar] [CrossRef]
- Patel, K.A.; Oram, R.A.; Flanagan, S.E.; De Franco, E.; Colclough, K.; Shepherd, M.; Ellard, S.; Weedon, M.N.; Hattersley, A.T. Type 1 Diabetes Genetic Risk Score: A Novel Tool to Discriminate Monogenic and Type 1 Diabetes. Diabetes 2016, 65, 2094–2099. [Google Scholar] [CrossRef]
- Bonnefond, A.; Boissel, M.; Bolze, A.; Durand, E.; Toussaint, B.; Vaillant, E.; Gaget, S.; Graeve, F.; Dechaume, A.; Allegaert, F.; et al. Pathogenic variants in actionable MODY genes are associated with type 2 diabetes. Nat. Metabolism. 2020, 2, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- RADIANT Study Group. The Rare and Atypical Diabetes Network (RADIANT) Study: Design and Early Results. Diabetes Care 2023, 45, 1265–1270, Epub ahead of print. [Google Scholar] [CrossRef]
- Biesecker, L.G. Genomic screening for monogenic forms of diabetes. BMC Med. 2018, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Colclough, K.; Patel, K.A.; Hattersley, A.T. Prediction algorithms: Pitfalls in interpreting genetic variants of autosomal dominant monogenic diabetes. J. Clin. Investig. 2020, 130, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubois-Laforgue, D.; Timsit, J. The Etiological Diagnosis of Diabetes: Still a Challenge for the Clinician. Endocrines 2023, 4, 437-456. https://doi.org/10.3390/endocrines4020033
Dubois-Laforgue D, Timsit J. The Etiological Diagnosis of Diabetes: Still a Challenge for the Clinician. Endocrines. 2023; 4(2):437-456. https://doi.org/10.3390/endocrines4020033
Chicago/Turabian StyleDubois-Laforgue, Danièle, and José Timsit. 2023. "The Etiological Diagnosis of Diabetes: Still a Challenge for the Clinician" Endocrines 4, no. 2: 437-456. https://doi.org/10.3390/endocrines4020033
APA StyleDubois-Laforgue, D., & Timsit, J. (2023). The Etiological Diagnosis of Diabetes: Still a Challenge for the Clinician. Endocrines, 4(2), 437-456. https://doi.org/10.3390/endocrines4020033