Abstract
Finely tuned cartilage mineralization, endochondral ossification, and normal bone formation are necessary for normal bone growth. Hypertrophic chondrocytes in the epiphyseal cartilage secrete matrix vesicles, which are small extracellular vesicles initiating mineralization, into the intercolumnar septa but not the transverse partitions of the cartilage columns. Bone-specific blood vessels invade the unmineralized transverse septum, exposing the mineralized cartilage cores. Many osteoblast precursors migrate to the cartilage cores, where they synthesize abundant bone matrices, and mineralize them in a process of matrix vesicle-mediated bone mineralization. Matrix vesicle-mediated mineralization concentrates calcium (Ca) and inorganic phosphates (Pi), which are converted into hydroxyapatite crystals. These crystals grow radially and are eventually get out of the vesicles to form spherical mineralized nodules, leading to collagen mineralization. The influx of Ca and Pi into the matrix vesicle is regulated by several enzymes and transporters such as TNAP, ENPP1, PiT1, PHOSPHO1, annexins, and others. Such matrix vesicle-mediated mineralization is regulated by osteoblastic activities, synchronizing the synthesis of organic bone material. However, osteocytes reportedly regulate peripheral mineralization, e.g., osteocytic osteolysis. The interplay between cartilage mineralization and vascular invasion during endochondral ossification, as well as that of osteoblasts and osteocytes for normal mineralization, appears to be crucial for normal bone growth.
1. Introduction
The growth of long bone depends on endochondral ossification, which can be sequentially divided into cartilage matrix mineralization, vascular invasion into the epiphyseal cartilage to expose the mineralized cartilage matrix, osteoblastic migration into the mineralized cartilage cores, and bone deposition to form the primary trabeculae. Hypertrophic chondrocytes play a key role in normal cartilage mineralization, and subsequently in endochondral ossification. These hypertrophic chondrocytes secrete matrix vesicles, extracellular small vesicles that initiate mineralization, and also produce vascular endothelial growth factor (VEGF) allowing the vascular endothelial cells to invade the epiphyseal cartilage. Cartilage mineralization is involved in the modeling of long bones and their changes of shape and size, i.e., the development and growth of the metaphyseal trabeculae. Finely tuned interplays among chondrocytes, vascular endothelial cells, osteoclasts (chondroclasts), and osteoblasts is apparently necessary for adequate endochondral ossification [1].
Bone is a mineralized tissue composed of calcium phosphates and organic materials such as collagen and proteoglycans. There are two phases of bone mineralization: primary and secondary. Primary mineralization is achieved by osteoblasts. Osteoblasts also produce a large amount of matrix vesicles, which mineralize in nodules (globular assemblies of hydroxyapatite crystals) and then extend into the collagen fibrils secreted by the osteoblasts. In contrast to primary mineralization, secondary mineralization is the process whereby the mineral density of bone increases after primary mineralization. It is postulated that secondary mineralization is regulated through physical crystal maturation, and by the cellular activities of osteocytes embedded in the bone matrix. However, the exact mechanism of secondary mineralization is not yet fully understood.
Histological processes of primary mineralization in the bones can be divided into two phases: matrix vesicle-mediated mineralization and collagen mineralization. In matrix vesicle-mediated mineralization, osteoblasts appear to regulate the secretion speed and the amount of matrix vesicle according to the synthesis of bone matrix. The discovery of matrix vesicles was a breakthrough in the field of bone mineralization [2,3,4,5,6,7,8], and many membrane transporters and enzymes related to matrix vesicle-mediated mineralization have recently been discovered. In addition to matrix vesicle-mediated mineralization, recent reports have suggested that osteocytes putatively regulate the mineralization in the periphery. As osteoblasts and osteocytes are directly connected to each other by means of their cytoplasmic processes, bone mineralization may be regulated by the interplay of osteoblasts and osteocytes. Updated knowledge of the matrix vesicles and osteocytic network in bone mineralization may deepen the understanding of mineral metabolism in bones.
In this review, we present the ultrastructural and histological aspects of endochondral ossification, matrix vesicle-mediated mineralization, and osteocytic regulation of bone mineralization.
2. Histological Aspects on Endochondral Ossification
2.1. Cartilage Mineralization by Hypertrophic Chondrocytes
Epiphyseal cartilage can be divided into three distinctive zones: resting, proliferating, and hypertrophic zones. Chondrocytes form the longitudinal columns in the proliferative and hypertrophic zones, but the proliferative chondrocytes synchronously enlarge in the hypertrophic phenotype [1]. Parathyroid hormone (PTH)-related peptide (PTHrP) has been reported to regulate hypertrophic differentiation of chondrocytes by mediating the Indian hedgehog (IHH)/PTHrP negative feedback [9]. IHH expressed in the prehypertrophic zone (the upper region of the hypertrophic zone) stimulates PTHrP expression in the early differentiation stage of chondrocytes. PTHrP promotes the proliferation activity of chondrocytes by binding to the common receptor of PTH and PTHrP (PTH/PTHrP receptor) in the proliferative zone. PTHrP alternatively inhibits the hypertrophic phenotype of chondrocytes, and IHH expression is then turned off. In addition to IHH/PTHrP negative feedback, another important regulatory factor in chondrocyte proliferation is fibroblast growth factor receptor 3 (FGFR3). Point mutations in FGFR3 cause achondroplasia and thanatophoric dysplasia by continuous activation of the transcription factor Stat1 [10,11]. FGFR3 signaling has also been proposed to increase the pool of proliferating cells by stimulating chondrocytes in the resting zone and promoting their transit to the proliferative zone [12,13]. Thus, the action of IHH/PTHrP and FGFR3 may be essential for chondrocyte proliferation and differentiation [14]. Hypertrophic chondrocytes have large and translucent cell bodies and produce type I and X collagens, tissue nonspecific alkaline phosphatase (TNAP), proteoglycan, and osteopontin [15,16,17,18,19]. Hypertrophic chondrocytes do not proliferate but acquire mineralization ability in the cartilage matrix. Hypertrophic chondrocytes also reportedly secrete VEGF, an angiogenic molecule that has been implicated in matrix metabolism enabling vascular invasion of the epiphyseal cartilage [20]. Hypertrophic chondrocytes of the epiphyseal cartilage secrete matrix vesicles, in which crystalline calcium phosphates appear, forming hydroxyapatite crystals that grow and eventually break through the membrane to form mineralized nodules in the cartilage matrix. Hypertrophic chondrocytes deposit matrix vesicles in the intercolumnar septae but not in the transverse partitions, consequently forming mineralized longitudinal septae and unmineralized transverse partitions. The regular distribution of mineralized cartilage matrix in the longitudinal intercolumnar septum allows the vertical invasion of vascular endothelial cells, which infiltrate into the cartilage by penetrating the unmineralized transverse partitions. After the formation of these calcified cartilage cores exposed to bone tissues, many osteoblast precursors migrate and attach to the mineralized cartilage cores to deposit abundant organic bone matrices including type I collagen, osteocalcin, osteopontin, and so forth, thereby forming the primary trabeculae. Thus, the process of endochondral ossification involves a well-defined series of events which include the invasion of vascular endothelial cells, osteogenic cell migration, new bone deposition onto the cartilage core, and the formation of primary trabeculae.
2.2. Vascular Invasion at the Chondro-Osseous Junction
Vascular endothelial cells can invade the epiphyseal cartilage by piercing the incompletely calcified transverse partition of the columns. We demonstrated endomucin-reactive blood vessels invading the chondrocyte lacunae at the chondro-osseous junction [21]. Transmission electron microscopic (TEM) observation verified that the vascular endothelial cells, present in blood vessels close to the cartilaginous matrix, extend their fine cytoplasmic processes into the matrix. The tips of the extended cytoplasmic processes showed fine finger-like structures facing the cartilaginous matrix, suggesting that the apical region of the invading endothelial cells may be partially open. In some observations, cell debris was present inside the blood vessels facing the cartilaginous columns at the chondro-osseous junction, while erythrocytes were found outside the blood vessels. Since the apical region of invading blood vessels might be open, blood vessels could presumably invade the cartilaginous matrix and exclude unnecessary impeditive materials (mainly cellular debris) to avoid accumulation at the junction (Figure 1).
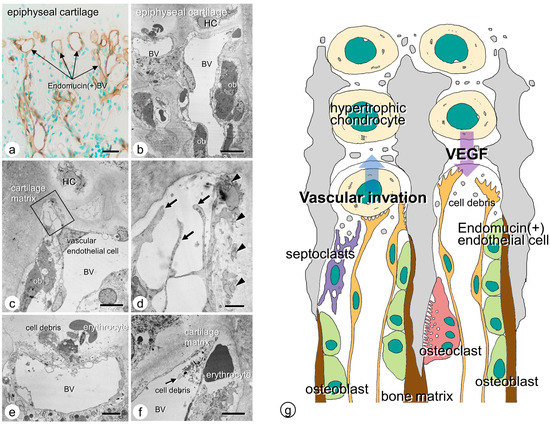
Figure 1.
Vascular endothelial cells at the chondro-osseous junction. (a) Endomucin-immunoreactive (brown color) blood vessels at the chondro-osseous junction under light microscope. (b–f) TEM images of blood vessels at the chondro-osseous junction. Invading blood vessels are seen beneath the chondrocytic lacunae. (c,d) When observed under higher magnification as shown in panel c, fine cytoplasmic processes (arrows) are seen extending from the vascular endothelial cell, with invaginations of the cell membranes in the superficial layer of the cartilaginous matrix. (e,f) Panel e demonstrates cell debris, including erythrocytes from the blood vessels, and panel f reveals an erythrocyte outside the vessel and cell debris in the vessels. (g) Schematic design of vascular invasion at the chondro-osseous junction. HP: hypertrophic chondrocyte; BV: blood vessel, ob: osteoblast. Bar, (a) 20 µm, (b) 10 µm, (c,e,f) 5 µm, (d) 1 µm.
2.3. Osteoclasts’ Function at the Chondro-Osseous Junction
It is well known that osteoclasts, also referred to as chondroclasts, accumulate in the chondro-osseous junction. Osteoclasts at the chondro-osseous junction show intense matrix metalloproteinase (MMP)-9 immunoreactivity [22]. Additionally, MMP-9 immunoreactivity is exhibited in the tips of the vascular endothelial cells facing the cartilaginous matrix, unlike the other areas distant from the chondro-osseous junction [20]. Therefore, osteoclasts and vascular endothelial cells apparently synthesize MMP-9, which dissolves the cartilaginous matrix [23,24]. Vascular invasion rather than osteoclastic resorption seems essential during endochondral ossification. Studies have found that op/op mice, c-fos−/− mice, and receptor activator of nuclear factor κβ ligand (Rankl)−/− mice preserve similar lengths of long bones to those seen in their wild-type counterparts in murine models that lack osteoclasts. However, without osteoclasts, the primary trabeculae form a disorganized but highly connected meshwork in the long bones. As described by Marks and Odgren [25], it seems likely that osteoclastic activity during endochondral ossification resorbs the excess mineralized cartilage matrices and scattered islets of mineralized cartilage in the chondro-osseous junction, enabling the longitudinal arrangement of primary trabeculae. Furthermore, another cell type, septoclasts, also referred to as perivascular cells, may also be involved in vascular invasion during endochondral ossification [26,27,28]. Septoclasts are positive for Dolichos biflorus agglutinin lectin histochemistry [26] and E-FABP [29,30], featuring well-developed Golgi apparatus and several cytoplasmic lysosomes filled with abundant cathepsin B [27]. We speculate that one major function of septoclasts is to remove excess extracellular organic (non-mineralized) debris that would otherwise interrupt the vascular invasion path into the cartilage, and it is unlikely that osteoclasts are designated to resorb the excess mineralized matrices in the cartilage.
3. Ultrastructural Aspects of Matrix Vesicle-Mediated Mineralization in Bone
3.1. Formation of Crystalline Calcium Phosphates in Matrix Vesicles
The primary trabeculae resulting from endochondral ossification can be mineralized by osteoblasts. Osteoblasts secrete matrix vesicles enveloped by a plasma membrane (ranging 30–1000 nm in diameter) into the osteoid (incompletely mineralized areas beneath the osteoblasts) [3]. Matrix vesicles are equipped with several enzymes and membrane transporters on the plasma membrane and inside the vesicles, enabling calcium phosphate nucleation and subsequent crystal growth. A crystalline calcium phosphate such as hydroxyapatite crystal [Ca10(PO4)6(OH)2] appears inside the matrix vesicles and grows radially, eventually breaking out of the vesicle membrane to form mineralized nodules in a globular assembly of radially oriented hydroxyapatite crystals with a small ribbon-like structure approximately 25 nm wide, 10 nm high, and 50 nm long [31,32].
It seems likely that crystal nucleation begins on the inner leaflet of the vesicle membrane, because the deposition of amorphous mineral crystals is initially observed on the inner leaflet. Acidic phospholipids such as phosphatidylserine and phosphatidylinositol, which have a high affinity for Ca2+, are abundantly present in the matrix vesicles and consequently form a stable calcium phosphate–phospholipid complex associated with the inner leaflet of the vesicle membrane [8]. Therefore, it is possible that such complexes may play important roles in crystal nucleation in the matrix vesicles.
3.2. Mineralized Nodules Develop from Matrix Vesicles
After crystal formation, matrix vesicles develop mineralized nodules in a globular assembly of needle-like hydroxyapatite crystals (Figure 2). The growth of mineralized nodules appears to be regulated by a large amount of extracellular Ca/Pi and organic materials in the osteoid. To allow the growth of mineralized nodules, many enzymes and transporters on the vesicle membrane may participate in the accumulation of Ca and Pi on the mineralized nodules. However, osteopontin and osteocalcin are suited to the function of regulating mineralization, because they effectively inhibit calcium phosphate nucleation and crystal growth [33,34]. Osteopontin is localized in the periphery of mineralized nodules, where it might block excessive mineralization [35]. Osteocalcin includes γ-carboxyglutamic acid, which binds to mineral crystals [36,37,38]. When warfarin, an inhibitor of glutamine residue γ-carboxylation, was administered in our previous study, numerous fragments of needle-shaped mineral crystals were dispersed throughout the osteoid [39] (Figure 3), and γ-carboxylase-deficient mice demonstrated similar abnormality, showing disassembled and scattered crystal minerals in the bones [40]. It seems feasible that γ-carboxylated osteocalcin may bind hydroxyapatite crystals to form and maintain the mineralized nodules.
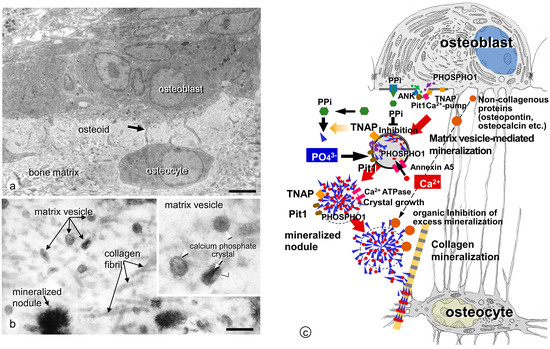
Figure 2.
Matrix vesicle-mediated bone mineralization by osteoblasts. (a,b) TEM observation of osteoblasts, osteocytes, and matrix vesicles. (a) Mature osteoblasts located on the bone surface (osteoid) connected to osteocytes with their cytoplasmic processes (black arrow). (b) At a higher magnification, many matrix vesicles and mineralized nodules are observed. Note the lipid bilayer of the vesicles (white arrowheads) and calcium phosphate crystals (white arrow) in the inset. (c) Schematic design of matrix vesicle-mediated bone mineralization. Bar, (a) 3 mm, (b) 400 nm. Panel C is modified from ref [41].
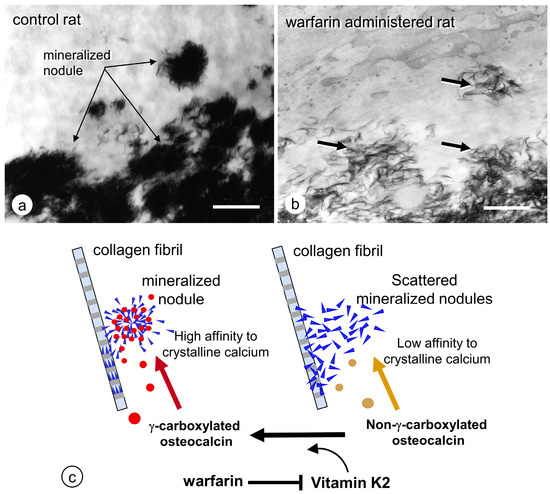
Figure 3.
Ultrastructure of dispersed mineral crystals in rats administered with warfarin. (a) TEM image of mineralized nodules with globular assembled mineral crystals in the control rats. (b) The rats administrated with warfarin demonstrate many dispersed mineral crystals (arrows) in the osteoid under TEM. (c) Schematic design of forming mineralized nodules by osteocalcin. Bar, 2 mm. Panel C is modified from ref [42].
3.3. Enzymes and Membrane Transporter Necessary for Matrix Vesicle-Mediated Mineralization in Bone
Matrix vesicles enable the influx of Ca2+ and phosphate ions (PO43−) by a variety of enzymes and membrane transporters such as tissue nonspecific alkaline phosphatase (TNAP) [6,43,44,45,46,47,48,49,50], ectonucleotide pyrophosphatase/phosphodiesterase 1(ENPP1) [51,52,53], ankylosis (ANK) [54,55], phosphoethanolamine/phosphocholine phosphatase 1 (PHOSPHO1) [54,55,56,57,58,59,60,61], and annexins [62]. TNAP, a glycosylphosphatidylinositol-anchored enzyme on the cell membrane, is one of the most important enzymes to initiate mineralization. In bones and cartilages, ENPP1 cleaves the extracellular ATPs into AMPs and pyrophosphate (PPi), and then TNAP hydrolyzes PPi, a phosphorus oxyanion with two phosphorus atoms in a P-O-P linkage, consequently producing PO43−. The resultant PO43− is transported into the matrix vesicles through sodium/phosphate co-transporter type III, also referred to as PiT1. Alternatively, Ca2+ can be delivered into the matrix vesicles by passage through annexins. TNAP is expressed not only by mature osteoblasts but also by preosteoblasts (osteoblast precursors), and therefore has been used as an osteoblastic lineage marker.
3.3.1. TNAP
TNAP is localized on the cell membranes of hypertrophic chondrocytes, mature osteoblasts, and preosteoblasts, as well as on the plasma membranes of matrix vesicles [43,44]. However, TNAP is not uniformly localized on the cell membranes of mature bone-synthesizing osteoblasts that possess cell polarity with distinct basolateral and secretory (osteoidal) domains. In one study, although Ca2+ transport ATPase was restricted to the osteoidal domain of the osteoblasts, TNAP was predominantly seen on the basolateral domain of the cell membranes [63]. Thus, the membranous domains in bone that feature an abundant TNAP are not matched to the region where TNAP actively serves for matrix vesicle-mediated mineralization. Tnap−/− mice have previously been generated [64,65] to mimic severe hypophosphatasia, with the implication that TNAP is involved in mineralization. Tnap−/− fetuses and neonatal mice have intact bones, but gradually show growth retardation and skeletal deformities. TNAP deficiency not only gives rise to hypomineralization in the skeleton, but also markedly disrupts the alignment of mineral crystals [66]. Thus, TNAP is necessary for normal mineralization and the ultrastructural arrangement of crystalline calcium phosphates in bone. In 2015, the development of the drug asfotase alfa (Strensiq) based on the long-lasting research on TNAP shed a ray of light on the treatment of hypophosphatasia caused by a hereditary mutation of Tnap gene [67,68].
3.3.2. ENPP1
ENPP1 cleaves the phosphodiester and pyrophosphate bonds of nucleotides and nucleotide sugars. Analysis of the crystalline structure of ENPP1 showed that nucleotides were accommodated in a pocket formed in the catalytic domain of this molecule, verifying that extracellular ATPs are a substrate for ENPP1 [69]. In bone and cartilage, the catalytic activity of ENPP1 generates PPi, which strongly inhibits mineralization by binding to hydroxyapatite crystals and disrupting their extension [51,52,53]. However, TNAP cleaves PPi into PO43−, which is a component of crystalline calcium phosphates in bone. Therefore, balanced interplay between ENPP1 and TNAP seems necessary for bone mineralization [70]. Alternatively, the lack of ENPP1 was proven to be related to the spontaneous mineralization of infantile arteries and periarticular regions [71,72]. In a normal state, therefore, PPi produced by ENPP1 may regulate the growth of hydroxyapatite crystals. In our observations, TNAP was mainly seen in mature osteoblasts and overlying preosteoblasts, while ENPP1 was detected in mature osteoblasts and osteocytes [73]. Genetic ENPP1 dysfunction leading to arterial mineralization may suggest that PPi deficiency or insufficiency can induce osteoblastic differentiation in vascular smooth muscle cells. Enpp1−/− mice, also known as tiptoe walking (ttw) mice, undergo ossification of the posterior longitudinal ligament of the spine (OPLL) including progressive ankylosing intervertebral and peripheral joint hyperostosis, as well as articular cartilage mineralization [74,75,76,77,78]. Despite the ectopic mineralization, Enpp1−/− mice show reduced serum concentrations of Ca2+ and PO43− as well as significantly elevated serum levels of fibroblast growth factor 23 (FGF23) [78,79]. FGF23 is an osteocyte-derived molecule that inhibits phosphate reabsorption and 1α-hydroxylase synthesis in the kidney [80,81,82]. Hence, in Enpp1−/− mice, the induction of Fgf23 mRNA expression, which increases the concentration of serum FGF23, may lead to reductions in the concentrations of Ca2+ and PO43−.
3.3.3. ANK
ENPP1 can be found not only on the cell surface but also in cytoplasmic regions, generating PPi in both locations. ANK reportedly transports intracellular PPi to the extracellular milieu, i.e., serves as a transmembrane PPi-channeling protein [54,55]. Therefore, it is feasible that ANK-mediated extracellular PPi levels may provide an equivalent balance by disallowing excessive or ectopic mineralization or hypomineralization in various tissues. In previous reports, infants with Ank gene mutations exhibited a three to five-fold decrease in extracellular PPi [54], while calcium pyrophosphate (CPP) crystal deposition (CPPD) was elevated in the synovial fluid by gain-of-function mutations in human ANK genes [83]. Thus, local PPi production naturally inhibits hydroxyapatite deposition, blocking undesirable mineralization in articular cartilage and other tissues. However, with the loss of ANK activity, extracellular PPi levels attenuate, intracellular PPi levels rise, and unregulated mineralization occurs.
3.3.4. PHOSPHO1
PHOSPHO1 is an enzyme abundantly present in bone-forming mature osteoblasts and hypertrophic chondrocytes [56]. Roberts et al. documented that PHOSPHO1 is restricted to the mineralizing regions of the bone and growth plate and plays a role in the initiation of matrix vesicle-mediated mineralization [57]. PHOSPHO1 is reportedly present not only in the cytoplasmic regions of bone-forming osteoblasts and hypertrophic chondrocytes but also in the matrix vesicles. PHOSPHO1 inside the matrix vesicles cleaves PO43− from phosphatidylcholine and phosphoethanolamine at the inner leaflet of the vesicles’ plasma membranes [56,57,58]. A recent report suggested that phospholipase A2 as well as ENPP6 are also included in matrix vesicles, acting in sequence to produce phosphocholine, which PHOSPHO1 subsequently hydrolyzes into PO43− [84]. Thus, PHOSPHO1 plays a pivotal role in the increased concentration of PO43− by cooperating with the PO43− supply by means of ENPP1/TNAP interplay. Neonatal Phospho1−/− mice demonstrated incomplete mineralization of the bone, often with spontaneous greenstick fractures [59,60]. Millán’s team demonstrated that PHOSPHO1 controls TNAP expression in mineralizing cells and is essential for mechanically competent mineralization [59,61]. Taken together, the PO43− supplementation necessary for matrix vesicle-mediated mineralization appears to be derived at least in part from TNAP/ENPP1 interaction outside the matrix vesicles as well as PHOSPHO1 activity inside the vesicles.
3.3.5. Annexins
Annexins are a group of proteins that show high affinity for Ca2+ and lipids, serving as ion channels for the transport of Ca2+ into the matrix vesicles. Three annexins, annexin A2, A5, and A6, that are abundantly present in vascular endothelial cells, heart, and skeletal muscles, have been discovered in matrix vesicles [62,85,86,87]. In the initial process of matrix vesicle-mediated mineralization, amorphous calcium phosphates are formed associated with the inner leaflet of the plasma membranes of the matrix vesicles. The annexin A5 might serve as a Ca2+ ion channel inside the matrix vesicles. Consequently, transported Ca2+ showed strong binding to phosphatidylserine in the inner leaflet of the membrane enclosing the matrix vesicle, which is enriched with anionic lipids [88,89]. Thus, it is feasible that annexin A5 might play an important role in Ca2+ transport and subsequent Ca2+-dependent phosphatidylserine binding in the matrix vesicles. It is a possibility that the Pi transported through PiT1 present in the membrane could also bind to Ca2+ trapped on the inner leaflet, to form amorphous calcium phosphates. Unexpectedly, Annexin a5−/− mice did not show skeletal deformity or reduced mineralization, suggesting that other annexins could compensate for the functions of annexin A5. However, further examination is necessary to clarify the precise role of annexins in bone mineralization.
4. Regulation of Bone Mineralization by Osteocyte
4.1. Erosion of Bone Minerals in the Vicinity of Osteocytes
Osteocytes are located in bone cavities known as osteocytic lacunae, and connect to neighboring osteocytes and osteoblasts on the bone surfaces via fine cytoplasmic processes that run through osteocytic canaliculi [90]. Osteocytes interconnect their cytoplasmic processes via gap junctions, thereby building functional syncytia referred to as the osteocytic lacunar canalicular system (OLCS) [41]. Mature well-mineralized bone develops an OLCS with an orderly arrangement, while immature bone has an irregular and disorganized OLCS [41]. The osteocytic network has been speculated to have roles in multiple processes including mechanical sensing, molecular transport, and regulation of peripheral mineralization [41].
Belanger proposed the concept of osteocytic osteolysis in the 1960s [91], suggesting that osteocytes have the potential not only to erode the peripheral bone minerals but also reversibly to remineralize the bone (Figure 4). This notion was not immediately accepted, however, many researchers have since observed that osteocytes and their canaliculi are involved in the mineral maintenance of the bone matrix [92,93,94,95,96,97,98,99]. The occurrence of osteocytic osteolysis has been reported in cases of PTH administration, including hyperparathyroidism [100,101], during lactation [96,102], in vitamin D receptor deficiency [103], and with sclerostin treatment [104]. During lactation, osteocytes reportedly erode the surrounding bone matrix by exhibiting a pattern of gene expression similar to that of osteoclasts during bone resorption, e.g., an elevation in tartrate-resistant acid phosphatase, cathepsin K, carbonic anhydrase, Na+/H+ exchanger, ATPase H+ transporting lysosomal subunits, and matrix metalloproteinase [96]. Using synchrotron X-ray microscopy, Nango et al. analyzed the degree of bone mineralization in mouse cortical bone around the lacunar canalicular network and reported the dissolution of bone mineral along the osteocyte canaliculi [105]. However, one criticism of the osteocytic osteolysis concept might be that the proteolytic enzymes and acids secreted from the bone-resorbing osteoclasts pass through the osteocytic canaliculi to reach distant osteocytes. Recently, using Rankl−/− mice, we have obtained microscopic findings that support the idea of osteocytic osteolysis [106]; osteocytic osteolysis is independent of osteoclastic activity and is discernible in mature cortical bone showing a regular distribution of the osteocytic network (Figure 5).
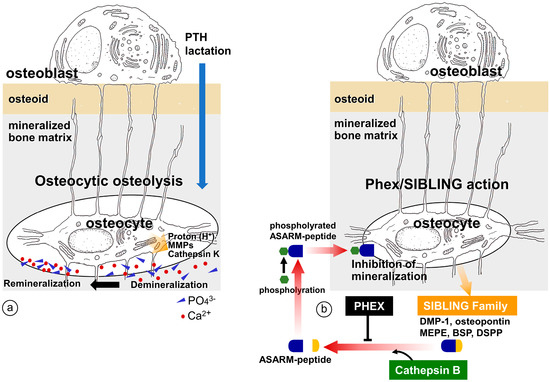
Figure 4.
Schematic representations of the two hypotheses: (a) osteocytic osteolysis and (b) regulation of bone mineralization by PHEX/SIBLINGs. During PTH administration or lactation, osteocytes secrete acids and proteolytic enzymes such as cathepsin K and MMPs to erode the surrounding bone. However, osteocytic osteolysis is reversible, so once-eroded bone can be remineralized. In contrast, SIBLINGs such as MEPE, DMP-1, and osteopontin are cleaved by cathepsin B to generate ASARM, which is then phosphorylated to inhibit mineralization. Alternatively, PHEX blocks the inhibition of mineralization.
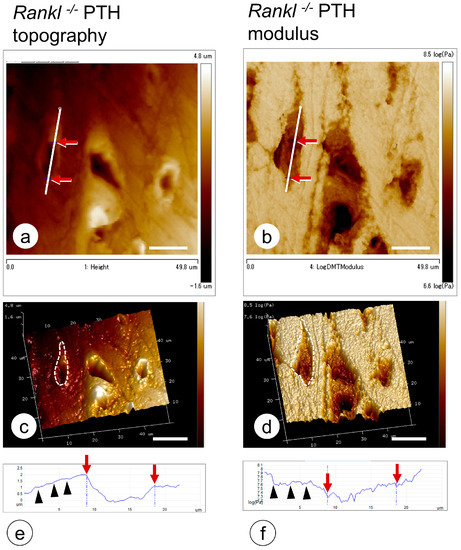
Figure 5.
Nano-indentation by atomic force microscopy on bone matrix surrounding osteocytic lacunae. (a,c) Topography of osteocytic lacunae in the femoral cortical bone of the PTH-administered Rankl−/− mice. (b,d) Elastic modulus of the osteocytic lacunae in the femoral cortical bone of the PTH-administered Rankl−/− mice. Red arrows along the white lines in a and b are matched with the red arrows in the graphs (e,f). Note the slightly expanded diameters in the three-dimensional images of the elastic modulus (compare the dotted circles in (c,d)), and that the index of the elastic modulus is lower than the topography in the graph (black arrowheads). Bar, 20 mm.
However, several reports have cautioned that (1) large osteocytic lacunae do not always represent the signs of osteocytic osteolysis [107], (2) the vitamin D receptor is not associated with osteocytic osteolysis [108], and (3) despite considerable research, osteocytic osteolysis has continued to be looked upon with skepticism [109]. Nevertheless, many researchers have attempted to elucidate whether osteocytic osteolysis would affect the mechanical properties of bone, and to extend the concept from including merely osteolysis to encompass a remodeling of the osteocytic network’s peripheral bone matrix. Recently, Kaya et al. reported that changes in bone mechanical properties induced by lactation and recovery appear to depend predominantly on the volume of osteocytic lacunae and canaliculi, suggesting that tissue-level mechanical properties of cortical bone are rapidly and reversibly modulated by osteocytes in response to physiological challenges [110]. Emami et al. have consistently reported notable canalicular changes following fracture that could affect mechanical properties of bone [111]. Vahidi et al. reported that femoral fracture in mice induced morphological changes of the canalicular network in the contralateral limb, suggesting decreased rates of bone formation and mineralization in the osteocytic lacunar canaliculi. They proposed that changes in canalicular remodeling by osteocytes involve utilization of the mineral from the bones for callus formation and bone repair after a fracture, but this process may also lessen bone quality and systemically elevate the fracture risk [112]. Osteocytes are the most abundant cells in bone, and the total area of osteocytes and their cytoplasmic processes is much larger than the areas of bone-forming osteoblasts or bone-resorbing osteoclasts. Therefore, osteocytes might be involved in the regulation of mineralization.
4.2. Regulation of Mineralization by Mediating SIBLING Family
Osteocytes are known to produce many important extracellular molecules, including fibroblast growth factor 23 (FGF23), small integrin-binding ligand N-linked glycoprotein (SIBLING) family proteins, and phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX). Through these molecules, osteocytes can regulate bone mineralization in two different manners: (1) systemic regulation of serum Pi by FGF23 in the kidney; and (2) local regulation of mineral crystal growth by PHEX/SIBLING family.
For systemic regulation of serum Pi, FGF23 secreted from osteocytes is circulated to reach the kidneys, where it binds to the receptor complex of fibroblast growth factor receptor Ic (FGFR1c) and αklotho expressed in the proximal renal tubules, to inhibit sodium/phosphate co-transporter type IIa/IIc (NaPi IIa/IIc). Since NaPi IIa/IIc reabsorb phosphate ions in the proximal renal tubules, FGF23 reduces the serum Pi concentration [80,81,82]. Human X-linked hypophosphatemia (XLH), one of the FGF23-related causes of hypophosphatemic rickets or osteomalacia in children and osteomalacia in adults, is caused by loss-of-function mutations in PHEX resulting in the elevated circulation of FGF23 and markedly decreased bone mineralization. This may indicate that the osteocyte-derived hormone FGF23, along with its function in the kidneys, may play a pivotal role in the systemic regulation of bone mineralization.
In contrast to systemic regulation of serum Pi and bone mineralization, osteocytes appear to regulate mineralization in the periphery of the osteocytic lacunae. Dentin matrix protein-1 (DMP-1), which is secreted by osteocytes, has high potential to bind Ca2+ and is postulated to play a role in the mineralization of the peripheral bone matrix of osteocytes [113]. The SIBLING family includes DMP-1, matrix extracellular phosphoglycoprotein (MEPE), osteopontin, bone sialoprotein (BSP), and dentin sialophosphoprotein (DSPP), which are encoded by a gene located on human chromosome 4q21 and mouse chromosome 5q21 [114,115]. We considered the possibility that the interaction between PHEX and the SIBLING family might regulate mineralization in the periphery of the osteocytic lacunae (Figure 4). For instance, MEPE secreted by osteocytes is cleaved by cathepsin B to release the carboxy terminal region, a novel functional domain referred to as the acidic serine-rich and aspirate-rich motif (ASARM) [116,117]. The resultant ASARM peptides are then phosphorylated to inhibit bone mineralization [117]. However, MEPE also binds to PHEX, forming the MEPE-PHEX complex. In this situation, cathepsin B is unable to cleave the MEPE-PHEX complex, which therefore blocks the synthesis of ASARM, so no phosphorylated ASARM inhibits mineralization, and normal mineralization is thereby attained [118]. It has been reported that the phosphorylated ASARM peptide of osteopontin inhibits mineralization in a phosphorylation-dependent manner, and PHEX disturbs the inhibition of mineralization [119]. These findings implicate the possibility that osteocyte-derived SIBLINGs may regulate peripheral bone mineralization by cooperating with PHEX. This idea is supported by the observation that the absence of DMP-1 results in rickets or osteomalacia in mice [120] and autosomal recessive hypophosphatemic rickets or osteomalacia (ARHR) in human patients [121]. However, PHEX/SIBLINGs are usually associated with congenital deformities, rickets, and osteomalacia, and therefore it is necessary to elucidate whether PHEX/SIBLINGs play an important role in the physiological regulation of bone mineralization in a healthy state.
5. Conclusions
During endochondral ossification, hypertrophic chondrocytes secrete matrix vesicles into the intercolumnar septa but not the transverse partitions of the cartilage columns; this allows vascular invasion into the epiphyseal cartilage and subsequent osteoblastic bone formation in the mineralized cartilage core. Thus, endochondral ossification is finely tuned by the cellular interplay at the chondro-osseous junction. To achieve matrix vesicle-mediated mineralization, many enzymes and membrane transporters including TNAP, ENPP1, PiT1, PHOSPHO1, annexins, and others are involved in the influx of Ca2+/Pi and the regulation of calcium phosphate crystal growth. In addition to their role in osteoblastic primary mineralization, osteocytes have recently been shown to regulate bone mineralization, presumably by controlling the synthesis of PHEX/SIBLING, as well as osteocytic osteolysis. Thus, normal mineralization is maintained by the orchestrated activities of bone cells.
Author Contributions
Conceptualization, writing—original draft preparation, T.H.; investigation; T.H., H.H., T.H., T.Y., T.M. and Y.M.; writing—review and editing, N.A.; supervision and funding acquisition, T.H. and N.A. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partially supported by grants from the Japanese Society for the Promotion of Science (JSPS, 22K09911 to T.H., 21K16928 to H.H., and 21H03103 to N.A.) and a grant-in-aid for young scientists provided by the Uehara Memorial Foundation (202110102 to T.H.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Amizuka, N.; Hasegawa, T.; Oda, K.; Freitas, P.H.L.; Hoshi, K.; Li, M.; Ozawa, H. Histology of epiphyseal cartilage calcification and endochondral ossification. Front. Biosci. 2012, 4, 2085–2100. [Google Scholar] [CrossRef]
- Ali, S.Y.; Sajdera, S.W.; Anderson, H.C. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc. Natl. Acad. Sci. USA 1970, 67, 1513–1520. [Google Scholar] [CrossRef]
- Anderson, H.C. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol. 1969, 41, 59–72. [Google Scholar] [CrossRef]
- Bonucci, E. Fine structure of early cartilage calcification. J. Ultrastruct. Res. 1967, 20, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, E. Fine structure and histochemistry of “calcifying globules” in epiphyseal cartilage. Z. Zellforsch. Mikrosk. Anat. 1970, 103, 192–217. [Google Scholar] [CrossRef]
- Ozawa, H.; Yamada, M.; Yajima, T. The ultrastructural and cytochemical aspects of matrix vesicles and calcification processes. In Formation and Calcification of Hard Tissues; Talmage, R.V., Ozawa, H., Eds.; Shakai Hoken Pub: Tokyo, Japan, 1978; pp. 9–57. [Google Scholar]
- Ozawa, H.; Yamada, M.; Yamamoto, T. Ultrastructural observations on the location of lead and calcium in the mineralizing dentine of rat incisor. In Matrix Vesicles; Ascenzi, A., Bonucci, E., de Bernard, B., Eds.; Wiching Editore srl: Milano, Italy, 1981; pp. 179–187. [Google Scholar]
- Wuthier, R.E. Lipid composition of isolated epiphyseal cartilage cells, membranes and matrix vesicles. Biochim. Biophys. Acta 1975, 409, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. Int. J. Mol. Sci. 2020, 21, 6665. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.L.; Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Wilkin, D.J.; Lachman, R.S.; Wilcox, W.R.; Rimoin, D.L.; Cohn, D.H.; Wasmuth, J.J. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995, 9, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Kitagawa, M.; Xue, N.; Xie, B.; Garofalo, S.; Cho, J.; Deng, C.; Horton, W.A.; Fu, X.Y. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997, 386, 288–292. [Google Scholar] [CrossRef]
- Peters, K.; Ornitz, D.; Werner, S.; Williams, L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev. Biol. 1993, 155, 423–430. [Google Scholar] [CrossRef]
- Deng, C.; Wynshaw-Boris, A.; Zhou, F.; Kuo, A.; Leder, P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 1996, 84, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Amizuka, N.; Davidson, D.; Liu, H.; Valverde-Franco, G.; Chai, S.; Maeda, T.; Ozawa, H.; Hammond, V.; Ornitz, D.M.; Goltzman, D.; et al. Signalling by fibroblast growth factor receptor 3 and parathyroid hormone-related peptide coordinate cartilage and bone development. Bone 2004, 34, 13–25. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Blackwood, H.J. Histochemical studies of chondrocyte function in the cartilage of the mandibular codyle of the rat. J. Anat. 1966, 100, 615–626. [Google Scholar] [PubMed]
- Ikeda, T.; Nomura, S.; Yamaguchi, A.; Suda, T.; Yoshiki, S. In situ hybridization of bone matrix proteins in undecalcified adult rat bone sections. J. Histochem. Cytochem. 1992, 40, 1079–1088. [Google Scholar] [CrossRef]
- Oshima, O.; Leboy, P.S.; McDonald, S.A.; Tuan, R.S.; Shapiro, I.M. Developmental expression of genes in chick growth cartilage detected by in situ hybridization. Calcif. Tissue Int. 1989, 45, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.R.; Pidoux, I.; Rosenberg, L. Role of proteoglycans in endochondral ossification: Immunofluorescent localization of link protein and proteoglycan monomer in bovine fetal epiphyseal growth plate. J. Cell Biol. 1982, 92, 249–260. [Google Scholar] [CrossRef]
- Schmid, T.M.; Linsenmayer, T.F. Immunohistochemical localization of short chain cartilage collagen (type X) in avian tissues. J. Cell Biol. 1985, 100, 598–605. [Google Scholar] [CrossRef]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef]
- Tsuchiya, E.; Hasegawa, T.; Hongo, H.; Yamamoto, T.; Abe, M.; Yoshida, T.; Zhao, S.; Tsuboi, K.; Udagawa, N.; Freitas, P.H.L.; et al. Histochemical assessment on the cellular interplay of vascular endothelial cells and septoclasts during endochondral ossification in mice. Microscopy 2021, 70, 201–214. [Google Scholar] [CrossRef]
- Kojima, T.; Hasegawa, T.; Freitas, P.H.L.; Yamamoto, T.; Sasaki, M.; Horiuchi, K.; Hongo, H.; Yamada, T.; Sakagami, N.; Saito, N.; et al. Histochemical aspects of the vascular invasion at the erosion zone of the epiphyseal cartilage in MMP-9-deficient mice. Biomed. Res. 2013, 34, 119–128. [Google Scholar] [CrossRef]
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Engsig, M.T.; Chen, Q.J.; Vu, T.H.; Pedersen, A.C.; Therkidsen, B.; Lund, L.R.; Henriksen, K.; Lenhard, T.; Foged, N.T.; Werb, Z.; et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. 2000, 151, 879–889. [Google Scholar] [CrossRef]
- Marks, S.C., Jr.; Odgren, P.R. The structure and development of the skeleton. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: New York, NY, USA, 2002; pp. 3–15. [Google Scholar]
- Nakamura, H.; Ozawa, H. Ultrastructural, enzyme-, lectin, and immunohistochemical studies of the erosion zone in rat tibiae. J. Bone Miner. Res. 1996, 11, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.R.; Lamplugh, L.; Shepard, N.L.; Mort, J.S. The septoclast, a cathepsin B-rich cell involved in the resorption of growth plate cartilage. J. Histochem. Cytochem. 1995, 43, 525–536. [Google Scholar] [CrossRef]
- Gartland, A.; Mason-Savas, A.; Yang, M.; MacKay, C.A.; Birnbaum, M.J.; Odgren, P.R. Septoclast deficiency accompanies postnatal growth plate chondrodysplasia in the toothless (tl) osteopetrotic, colony-stimulating factor-1 (CSF-1)-deficient rat and is partially responsive to CSF-1 injections. Am. J. Pathol. 2009, 175, 2668–2675. [Google Scholar] [CrossRef]
- Bando, Y.; Yamamoto, M.; Sakiyama, K.; Inoue, K.; Takizawa, S.; Owada, Y.; Iseki, S.; Kondo, H.; Amano, O. Expression of epidermal fatty acid binding protein (E-FABP) in septoclasts in the growth plate cartilage of mice. J. Mol. Histol. 2014, 45, 507–518. [Google Scholar] [CrossRef]
- Bando, Y.; Yamamoto, M.; Sakiyama, K.; Sakashita, H.; Taira, F.; Miyake, G.; Iseki, S.; Owada, Y.; Amano, O. Retinoic acid regulates cell-shape and -death of E-FABP (FABP5)-immunoreactive septoclasts in the growth plate cartilage of mice. Histochem. Cell Biol. 2017, 148, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S. Organization of extracellularly mineralized tissues: A comparative study of biological crystal growth. CRC Crit. Rev. Biochem. 1986, 20, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, H. Ultrastructural Concepts on Biological Calcification; Focused on Matrix Vesicles. J. Oral Biosci. 1985, 27, 751–774. [Google Scholar]
- Bosky, A.L.; Maresca, M.; Ullrich, W.; Doty, S.B.; Butler, W.T.; Prince, C.W. Osteopontin-hydroxyapatite interactions in vitro: Inhibition of hydroxyapatite formation and growth in a gelatin-gel. Bone Miner. 1993, 22, 147–159. [Google Scholar] [CrossRef]
- Hunter, G.K.; Hauschka, P.V.; Poole, A.R.; Rosenberg, L.C.; Goldberg, H.A. Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem. J. 1996, 317, 59–64. [Google Scholar] [CrossRef]
- Mark, M.P.; Butler, W.T.; Prince, C.W.; Finkleman, R.D.; Ruch, J.V. Developmental expression of 44-kDa phosphoprotein (osteopontin) and bone-carboxyglutamic acid (Gla)-containing protein (osteocalcin) in calcifying tissues of rat. Differentiation 1988, 37, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G.; Pauli, R.M.; Wilson, K.M. Maternal and fetal sequelae of anti-coagulation during pregnancy. Am. J. Med. 1980, 68, 122–140. [Google Scholar] [CrossRef]
- Hauschka, P.V.; Lian, J.B.; Gallop, P.M. Direct identification of the calcium-binding amino acid, gamma-carboxyglutamate, in mineralized tissue. Proc. Natl. Acad. Sci. USA 1975, 72, 3925–3929. [Google Scholar] [CrossRef]
- Price, P.A.; Otsuka, A.A.; Poser, J.W.; Kristaponis, J.; Raman, N. Characterization of a gamma-carboxyglutamic acid-containing protein from bone. Proc. Natl. Acad. Sci. USA 1976, 73, 1447–1451. [Google Scholar] [CrossRef]
- Amizuka, N.; Li, M.; Hara, K.; Kobayashi, M.; Freitas, P.H.L.; Ubaidus, S.; Oda, K.; Akiyama, Y. Warfarin administration disrupts the assembly of mineralized nodules in the osteoid. J. Electron. Microsc. 2009, 58, 55–65. [Google Scholar] [CrossRef]
- Azuma, K.; Shiba, S.; Hasegawa, T.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Ouchi, Y.; Amizuka, N.; Inoue, S. Osteoblast-specific γ-glutamyl carboxylase-deficient mice display enhanced bone formation with aberrant mineralization. J. Bone Miner. Res. 2015, 30, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Hongo, H.; Yamamoto, T.; Abe, M.; Yoshino, H.; Haraguchi-Kitakamae, M.; Ishizu, H.; Shimizu, T.; Iwasaki, N.; Amizuka, N. Matrix vesicle-mediated mineralization and osteocytic regulation of bone mineralization. Int. J. Mol. Sci. 2022, 23, 9941. [Google Scholar] [CrossRef]
- Hasegawa, T. Ultrastructure and biological function of matrix vesicles in bone mineralization. Histochem. Cell Biol. 2018, 149, 289–304. [Google Scholar] [CrossRef] [PubMed]
- de Bernard, B.; Bianco, P.; Bonucci, E.; Costantini, M.; Lunazzi, G.C.; Martinuzzi, P.; Modricky, C.; Moro, L.; Panfili, E.; Pollesello, P. Biochemical and immunohistochemical evidence that in cartilage an alkaline phosphatase is a Ca2+-binding glycoprotein. J. Cell Biol. 1986, 103, 1615–1623. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Anderson, H.C. Phosphatases of epiphyseal cartilage studied by electron microscopic cytochemical methods. J. Histochem. Cytochem. 1971, 19, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. Ultrastractural and cytochemical studies on the calcification of the tendon-bone joint. Arch. Histol. Jap. 1976, 39, 347–378. [Google Scholar] [CrossRef]
- Hoshi, K.; Ejiri, S.; Ozawa, H. Localizational alterations of calcium, phosphorus, and calcification-related organics such as proteoglycans and alkaline phosphatase during bone calcification. J. Bone Miner. Res. 2001, 16, 289–298. [Google Scholar] [CrossRef]
- Schmitz, J.P.; Schwartz, Z.; Sylvia, V.L.; Dean, D.D.; Calderon, F.; Boyan, B.D. Vitamin D3 regulation of stromelysin-1 (MMP-3) in chondrocyte cultures is mediated by protein kinase C. J. Cell Physiol. 1996, 168, 570–579. [Google Scholar] [CrossRef]
- Fleish, H.; Neuman, W.F. Mechanisms of calcification: Role of collagen, polyphosphates, and phosphatase. Am. J. Physiol. 1961, 200, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Fleish, H.; Neuman, W. The role of phosphatase and polyphosphates in calcification of collagen. Helv. Physiol. Pharmacol. Acta 1961, 19, C17–C18. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Ozawa, H.; Crenshaw, M.A. Ca-ATPase and ALPase activities at the initial calcification sites of dentine and enamel in the rat incisor. Cell Tissue Res. 1986, 243, 91–99. [Google Scholar] [CrossRef]
- Terkeltaub, R.; Rosenbach, M.; Fong, F.; Goding, J. Causal link between nucleotide pyrophosphohydrolase overactivity and increased intracellular inorganic pyrophosphate generation demonstrated by transfection of cultured fibroblasts and osteoblasts with plasma cell membrane glycoprotein-1. Arthritis Rheum. 1994, 37, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Vaingankar, S.; Chen, Y.; Moffa, A.; Goldring, M.B.; Sano, K.; Jin-Hua, P.; Sali, A.; Goding, J.; Terkeltaub, R. Differential mechanisms of inorganic pyrophosphate production by plasma cell membrane glycoprotein-1 and B10 in chondrocytes. Arthritis Rheum. 1999, 42, 1986–1997. [Google Scholar] [CrossRef]
- Johnson, K.; Moffa, A.; Chen, Y.; Pritzker, K.; Goding, J.; Terkeltaub, R. Matrix vesicle plasma membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J. Bone Miner. Res. 1999, 14, 883–892. [Google Scholar] [CrossRef]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef]
- Szeri, F.; Niaziorimi, F.; Donnelly, S.; Fariha, N.; Tertyshnaia, M.; Patel, D.; Lundkvist, S.; van de Wetering, K. The Mineralization Regulator ANKH Mediates Cellular Efflux of ATP, Not Pyrophosphate. J. Bone Miner. Res. 2022, 37, 1024–1031. [Google Scholar] [CrossRef]
- Houston, B.; Stewart, A.J.; Farquharson, C. PHOSPHO1-A novel phosphatase specifically expressed at sites of mineralisation in bone and cartilage. Bone 2004, 34, 629–637. [Google Scholar] [CrossRef]
- Roberts, S.; Narisawa, S.; Harmey, D.; Millán, J.L.; Farquharson, C. Functional involvement of PHOSPHO1 in matrix vesicle–mediated skeletal mineralization. J. Bone. Miner. Res. 2007, 22, 617–627. [Google Scholar] [CrossRef]
- Ciancaglini, P.; Yadav, M.C.; Simão, A.M.; Narisawa, S.; Pizauro, J.M.; Farquharson, C.; Hoylaerts, M.F.; Millán, J.L. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOSPHO1-deficient matrix vesicles. J. Bone. Miner. Res. 2010, 25, 716–723. [Google Scholar] [CrossRef]
- Huesa, C.; Yadav, M.C.; Finnilä, M.A.; Goodyear, S.R.; Robins, S.P.; Tanner, K.E.; Aspden, R.M.; Millán, J.L.; Farquharson, C. PHOSPHO1 is essential for mechanically competent mineralization and the avoidance of spontaneous fractures. Bone 2011, 48, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Boyde, A.; Staines, K.A.; Javaheri, B.; Millan, J.L.; Pitsillides, A.A.; Farquharson, C. A distinctive patchy osteomalacia characterizes PHOSPHO1-deficient mice. J. Anat. 2017, 231, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.C.; Simão, A.M.S.; Narisawa, S.; Huesa, C.; McKee, M.D.; Farquharson, C.; Millán, J.L. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function—A unified model of the mechanisms of initiation of skeletal calcification. J. Bone Miner. Res. 2011, 26, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T.; Nah, H.D.; Shapiro, I.M.; Pacifici, M. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J. Cell Biol. 1997, 137, 1149–1160. [Google Scholar] [CrossRef]
- Nakano, Y.; Beertsen, W.; van den Bos, T.; Kawamoto, T.; Oda, K.; Takano, Y. Site-specific localization of two distinct phosphatases along the osteoblast plasma membrane: Tissue non-specific alkaline phosphatase and plasma membrane calcium ATPase. Bone 2004, 35, 1077–1085. [Google Scholar] [CrossRef]
- Narisawa, S.; Fröhlander, N.; Millian, J.L. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn. 1997, 208, 432–446. [Google Scholar] [CrossRef]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Tesch, W.; Vandenbos, T.; Roschgr, P.; Fratzl-Zelman, N.; Klaushofer, K.; Beertsen, W.; Fratzl, P. Orientation of mineral crystallites and mineral density during skeletal development in mice deficient in tissue nonspecific alkaline phosphatase. J. Bone Miner. Res. 2003, 18, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Jakob, F.; Seefried, L.; Mentrup, B.; Graser, S.; Plotkin, H.; Girschick, H.J.; Liese, J. Recombinant enzyme replacement therapy in hypophosphatasia. Subcell. Biochem. 2015, 76, 323–341. [Google Scholar] [PubMed]
- Whyte, M.P.; Rockman-Greenberg, C.; Ozono, K.; Riese, R.; Moseley, S.; Melian, A.; Thompson, D.D.; Bishop, N.; Hofmann, C. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J. Clin. Endocrinol. Metab. 2016, 101, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Nishimasu, H.; Okudaira, S.; Mihara, E.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 16876–16881. [Google Scholar] [CrossRef]
- Andrilli, L.H.S.; Sebinelli, H.G.; Favarin, B.Z.; Cruz, M.A.E.; Ramos, A.P.; Bolean, M.; Millán, J.L.; Bottini, M.; Ciancaglini, P. NPP1 and TNAP hydrolyze ATP synergistically during biomineralization. Purinergic. Signal. 2022; in press. [Google Scholar] [CrossRef]
- Rutsch, F.; Vaingankar, S.; Johnson, K.; Goldfine, I.; Maddux, B.; Schauerte, P.; Kalhoff, H.; Sano, K.; Boisvert, W.A.; Superti-Furga, A.; et al. PC-1 Nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am. J. Pathol. 2001, 158, 543–554. [Google Scholar] [CrossRef]
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Höhne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 2003, 34, 379–381. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hasegawa, T.; Mae, T.; Hongo, H.; Yamamoto, T.; Abe, M.; Nasoori, A.; Morimoto, Y.; Maruoka, H.; Kubota, K.; et al. Comparative immunolocalization of tissue nonspecific alkaline phosphatase and ectonucleotide pyrophosphatase/phosphodiesterase 1 in murine bone. J. Oral Biosci. 2021, 63, 259–264. [Google Scholar] [CrossRef]
- Okawa, A.; Nakamura, I.; Goto, S.; Moriya, H.; Nakamura, Y.; Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 1998, 19, 271–273. [Google Scholar] [CrossRef]
- Johnson, K.; Pritzker, K.; Goding, J.; Terkeltaub, R. The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J. Rheumatol. 2001, 28, 2681–2691. [Google Scholar] [PubMed]
- Johnson, K.; Hashimoto, S.; Lotz, M.; Pritzker, K.; Goding, J.; Terkeltaub, R. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum. 2001, 44, 1071–1081. [Google Scholar] [CrossRef]
- Johnson, K.; Goding, J.; Van Etten, D.; Sali, A.; Hu, S.I.; Farley, D.; Krug, H.; Hessle, L.; Millán, J.L.; Terkeltaub, R. Linked deficiencies in extracellular PP(i) and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression. J. Bone Miner. Res. 2003, 18, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, N.C.; Zhu, D.; Milne, E.M.; van ‘t Hof, R.; Martin, A.; Darryl Quarles, L.; Millán, J.L.; Farquharson, C.; MacRae, V.E. Altered bone development and an increase in FGF-23 expression in Enpp1(-/-) mice. PLoS ONE 2012, 7, e32177. [Google Scholar] [CrossRef]
- Nam, H.K.; Emmanouil, E.; Hatch, N.E. Deletion of the pyrophosphate generating enzyme ENPP1 rescues craniofacial abnormalities in the TNAP-/- mouse model of Hypophosphatasia and reveals FGF23 as a marker of phenotype severity. Front. Dent. Med. 2022, 3, 846962. [Google Scholar] [CrossRef]
- Bergwitz, C.; Juppner, H. FGF23 and syndromes of abnormal renal phosphate handling. Adv. Exp. Med. Biol. 2012, 728, 41–64. [Google Scholar]
- Ho, B.B.; Bergwitz, C. FGF23 signalling and physiology. J. Mol. Endocrinol. 2021, 66, R23–R32. [Google Scholar] [CrossRef]
- Lederer, E. Regulation of serum phosphate. J. Physiol. 2014, 592, 3985–3995. [Google Scholar] [CrossRef]
- Abhishek, A.; Doherty, M. Pathophysiology of articular chondrocalcinosis--role of ANKH. Nat. Rev. Rheumatol. 2011, 7, 96–104. [Google Scholar] [PubMed]
- Stewart, A.J.; Leong, D.T.K.; Farquharson, C. PLA 2 and ENPP6 may act in concert to generate phosphocholine from the matrix vesicle membrane during skeletal mineralization. FASEB J. 2018, 32, 20–25. [Google Scholar] [CrossRef]
- Cao, X.; Genge, B.R.; Wu, L.N.; Buzzi, W.R.; Showman, R.M.; Wuthier, R.E. Characterization, cloning and expression of the 67-kDA annexin from chicken growth plate cartilage matrix vesicles. Biochem. Biophys. Res. Commun. 1993, 197, 556–561. [Google Scholar] [CrossRef]
- Kirsch, T.; Nah, H.D.; Demuth, D.R.; Harrison, G.; Golub, E.E.; Adams, S.L.; Pacifici, M. Annexin V-mediated calcium flux across membranes is dependent on the lipid composition: Implications for cartilage mineralization. Biochemistry 1997, 36, 3359–3367. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T.; Claassen, H. Matrix vesicles mediate mineralization of human thyroid cartilage. Calcif. Tissue Int. 2000, 66, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Majeska, R.J.; Holwerda, D.L.; Wuthier, R.E. Localization of phosphatidylserine in isolated chick epiphyseal cartilage matrix vesicles with trinitrobenzenesulfonate. Calcif. Tissue Int. 1979, 27, 41–46. [Google Scholar] [CrossRef]
- Taylor, M.G.; Simkiss, K.; Simmons, J.; Wu, L.N.; Wuthier, R.E. Structural studies of a phosphatidyl serine-amorphous calcium phosphate complex. Cell. Mol. Life Sci. 1998, 54, 196–202. [Google Scholar] [CrossRef]
- Hasegawa, T.; Yamamoto, T.; Hongo, H.; Qiu, Z.; Abe, M.; Kanesaki, T.; Tanaka, K.; Endo, T.; Freitas, P.H.L.; Li, M. Three-dimensional ultrastructure of osteocytes assessed by focused ion beam-scanning electron microscopy (FIB-SEM). Histochem. Cell Biol. 2018, 149, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, L.F. Osteocytic osteolysis. Calcif. Tissue. Res. 1969, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Collins, J.F.; Xu, H.; Xu, L.; Ghishan, F.K. Molecular cloning of a murine type III sodium-dependent phosphate cotransporter (Pit-2) gene promoter. Biochim. Biophys. Acta 2001, 1522, 42–45. [Google Scholar] [CrossRef]
- Qing, H.; Bonewald, L.F. Osteocyte remodeling of perilacunar and pericanalicular matrix. Int. J. Oral Sci. 2009, 1, 59–65. [Google Scholar] [CrossRef]
- Teit, A.; Zallone, A. Do osteocytes contribute to bone mikneral homeostasis? Osteocytic osteolysis revisited. Bone 2009, 44, 11–16. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Qing, H.; Ardeshirpour, L.; Pajevic, P.D.; Dusevich, V.; Jähn, K.; Kato, S.; Wysolmerski, J.; Bonewald, L.F. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res. 2012, 27, 1018–1029. [Google Scholar] [CrossRef]
- Whysolmerski, J.J. Osteocytic osteolysis: Time for a second look? BoneKEy Rep. 2012, 1, 229. [Google Scholar] [CrossRef]
- Whysolmerski, J.J. Osteocytes remove and replace perilacunar minewral during reproductive cycles. Bone 2013, 54, 230–236. [Google Scholar] [CrossRef]
- Sano, H.; Kikuta, J.; Furuya, M.; Kondo, N.; Endo, N.; Ishii, M. Intravital bone imaging by two-photon excitation microscopy to identify osteocytic osteolysis in vivo. Bone 2015, 74, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, E.; Gherardi, G.; Faraggiana, T. Bone changes in hemodialyzed uremic subjects. Comparative light and electron microscope investigations. Virchows Arch. A Pathol. Anat. Histol. 1976, 371, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Mosekilde, L.; Melsen, F. A tetracycline-based histomorphometric evaluation of bone resorption and bone turnover in hyperthyroidism and hyperparathyroidism. Acta Med. Scand. 1978, 204, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Jähn, K.; Kelkar, S.; Zhao, H.; Xie, Y.; Tiede-Lewis, L.M.; Dusevich, V.; Dallas, S.L.; Bonewald, L.F. Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo. J. Bone Miner. Res. 2017, 32, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Rolvien, T.; Krause, M.; Jeschke, A.; Yorgan, T.; Püschel, K.; Schinke, T.; Busse, B.; Demay, M.B.; Amling, M. Vitamin D regulates osteocyte survival and perilacunar remodeling in human and murine bone. Bone 2017, 103, 78–87. [Google Scholar] [CrossRef]
- Kogawa, M.; Wijenayaka, A.R.; Ormsby, R.T.; Thomas, G.P.; Anderson, P.H.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin regulates release of bone mineral by osteocytes by induction of carbonic anhydrase 2. J. Bone Miner. Res. 2013, 28, 2436–2448. [Google Scholar] [CrossRef]
- Nango, N.; Kubota, S.; Hasegawa, T.; Yashiro, W.; Momose, A.; Matsuo, K. Osteocyte-directed bone demineralization along canaliculi. Bone 2016, 84, 279–288. [Google Scholar] [CrossRef]
- Hongo, H.; Hasegawa, T.; Saito, M.; Tsuboi, K.; Yamamoto, T.; Sasaki, M.; Abe, M.; de Freitas, P.H.L.; Yurimoto, H.; Udagawa, N.; et al. Osteocytic osteolysis in PTH-treated wild-type and Rankl-/- mice examined by transmission electron microscopy, atomic force microscopy, and isotope microscopy. J. Histochem. Cytochem. 2020, 68, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Jandl, N.M.; von Kroge, S.; Stürznickel, J.; Baranowsky, A.; Stockhausen, K.E.; Mushumba, H.; Beil, F.T.; Püschel, K.; Amling, M.; Rolvien, T. Large osteocyte lacunae in iliac crest infantile bone are not associated with impaired mineral distribution or signs of osteocytic osteolysis. Bone 2020, 135, 115324. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.M.; Blouin, S.; Hofstaetter, J.G.; Roschger, P.; Zwerina, J.; Erben, R.G. No role of osteocytic osteolysis in the development and recovery of the bone phenotype induced by severe secondary hyperparathyroidism in vitamin D receptor deficient mice. Int. J. Mol. Sci. 2020, 21, 7989. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.A.; Kovacs, C.S. The puzzle of lactational bone physiology: Osteocytes masquerade as osteoclasts and osteoblasts. J. Clin. Investig. 2019, 129, 3041–3044. [Google Scholar] [CrossRef] [PubMed]
- Kaya, S.; Basta-Pljakic, J.; Seref-Ferlengez, Z.; Majeska, R.J.; Cardoso, L.; Bromage, T.G.; Zhang, Q.; Flach, C.R.; Mendelsohn, R.; Yakar, S.; et al. Lactation-induced changes in the volume of osteocyte lacunar-canalicular space alter mechanical properties in cortical bone tissue. J. Bone Miner. Res. 2017, 32, 688–697. [Google Scholar] [CrossRef]
- Emami, A.J.; Sebastian, A.; Lin, Y.Y.; Yee, C.S.; Osipov, B.; Loots, G.G.; Alliston, T.; Christiansen, B.A. Altered canalicular remodeling associated with femur fracture in mice. J. Orthop. Res. 2022, 40, 891–900. [Google Scholar] [CrossRef]
- Vahidi, G.; Rux, C.; Sherk, V.D.; Heveran, C.M. Lacunar-canalicular bone remodeling: Impacts on bone quality and tools for assessment. Bone 2021, 143, 115663. [Google Scholar]
- Sasaki, M.; Hasegawa, T.; Yamada, T.; Hongo, H.; de Freitas, P.H.; Suzuki, R.; Yamamoto, T.; Tabata, C.; Toyosawa, S.; Yamamoto, T.; et al. Altered distribution of bone matrix proteins and defective bone mineralization in klotho-deficient mice. Bone 2013, 57, 206–219. [Google Scholar] [CrossRef]
- Liu, S.; Rowe, P.S.; Vierthaler, L.; Zhou, J.; Quarles, L.D. Phosphorylated acidic serine-aspartate-rich MEPE-associated motif peptide from matrix extracellular phosphoglycoprotein inhibits phosphate regulating gene with homologies to endopeptidases on the X-chromosome enzyme activity. J. Endocrinol. 2007, 192, 261–267. [Google Scholar] [CrossRef]
- Staines, K.A.; MacRae, V.E.; Farquharson, C. The importance of the SIBLING family of proteins. J. Endocrinol. 2012, 214, 241–255. [Google Scholar] [CrossRef]
- Rowe, P.S.; de Zoysa, P.A.; Dong, R.; Wang, H.R.; White, K.E.; Econs, M.J.; Oudet, C.L. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics 2000, 67, 54–68. [Google Scholar] [CrossRef]
- Rowe, P.S.; Kumagai, Y.; Gutierrez, G.; Garrett, I.R.; Blacher, R.; Rosen, D.; Cundy, J.; Navvab, S.; Chen, D.; Drezner, M.K.; et al. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 2004, 34, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.S.; Garrett, I.R.; Schwarz, P.M.; Carnes, D.L.; Lafer, E.M.; Mundy, G.R.; Gutierrez, G.E. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: A model for impaired mineralization in X-linked rickets (HYP). Bone 2005, 36, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Masica, D.L.; Gray, J.J.; McKee, M.D. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J. Bone Miner. Res. 2010, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Mäkitie, O.; Pereira, R.C.; Kaitila, I.; Turan, S.; Bastepe, M.; Laine, T.; Kröger, H.; Cole, W.G.; Jüppner, H. Long-term clinical outcome and carrier phenotype in autosomal recessive hypophosphatemia caused by a novel DMP1 mutation. J. Bone Miner. Res. 2010, 25, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).