
Insights into the Molecular and Hormonal Regulation of Complications of X-Linked Hypophosphatemia




{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Phosphate Regulating Hormones: 1,25 Dihydroxyvitamin D3 (1,25D) and FGF23
1.2. Treatment of XLH
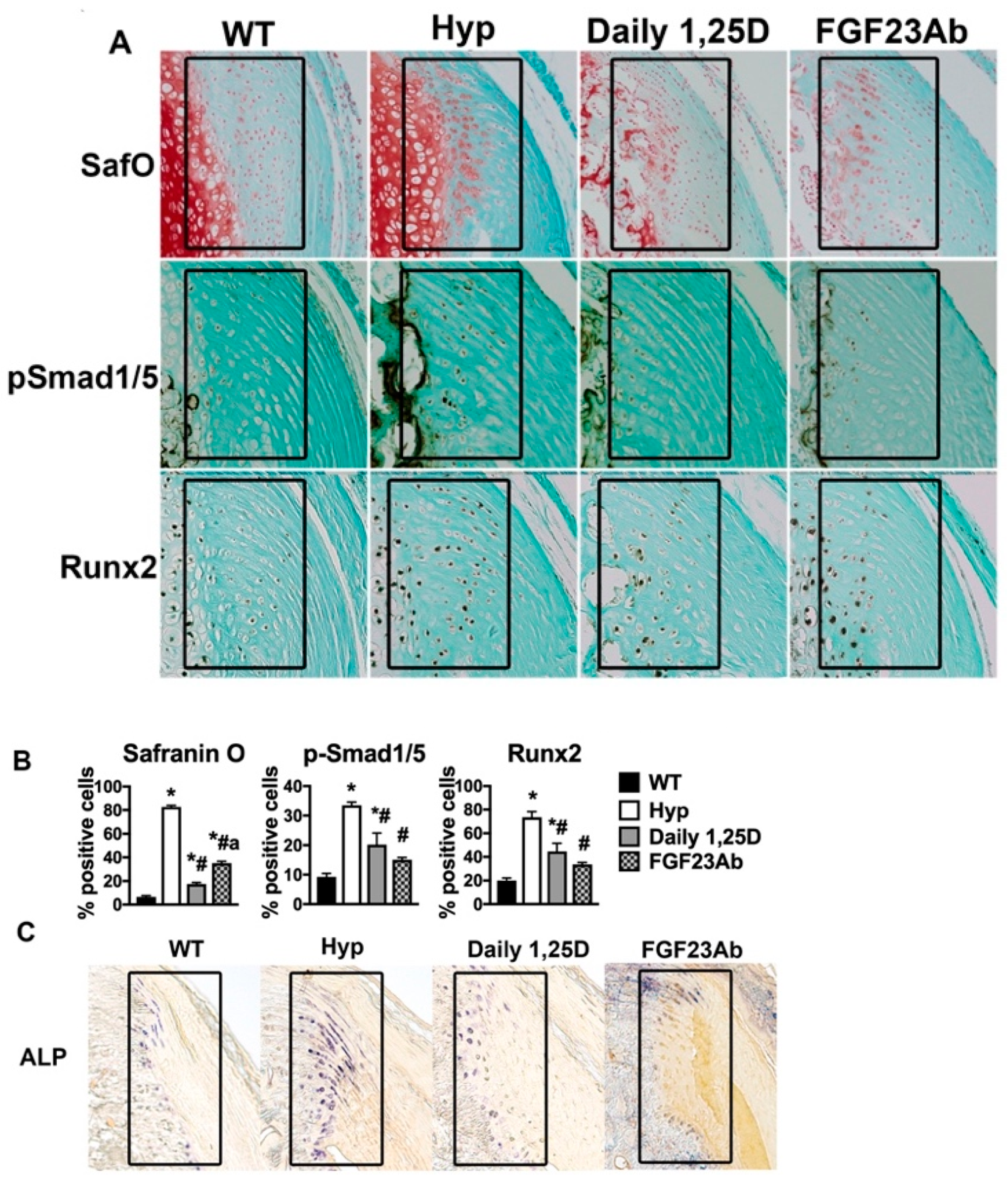
2. Impairment of Growth and Growth Plate Maturation
2.1. Growth in XLH
2.2. Growth Plate Abnormalities in XLH
2.3. Molecular Mechanisms of Impaired Growth in XLH
3. Skeletal Mineralization, Microarchitecture, and Biomechanics in XLH
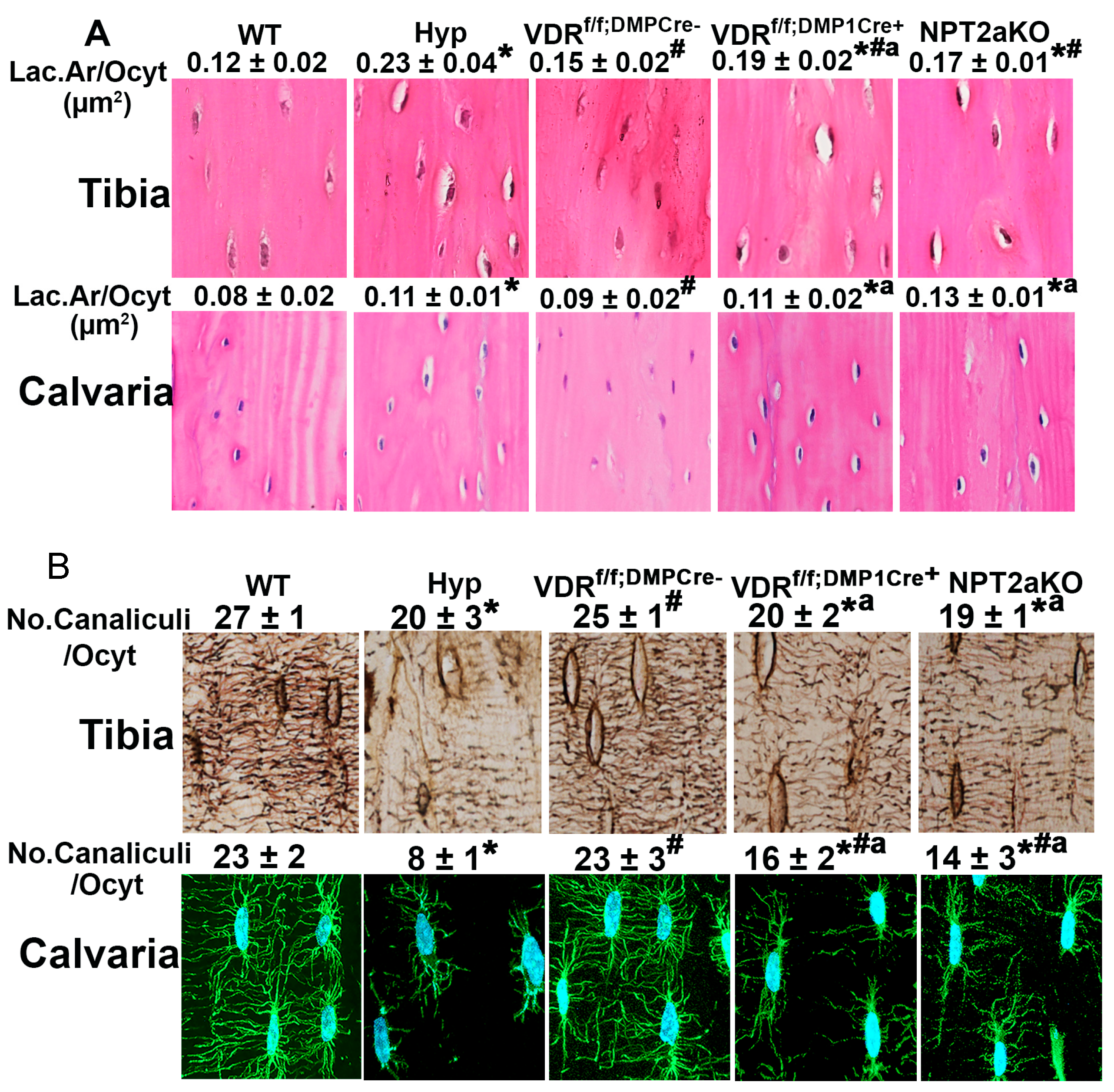
4. Osteocyte Perilacunar and Canalicular Organization
4.1. Osteocytes
4.2. Osteocyte LCN Remodeling
4.3. Regulation of Osteocyte LCN Organization in Hyp Mice
5. Enthesopathy
5.1. The Enthesis
5.2. Enthesopathy in XLH
5.3. Molecular Pathogenesis of XLH Enthesopathy
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jonsson, K.B.; Zahradnik, R.; Larsson, T.; White, K.E.; Sugimoto, T.; Imanishi, Y.; Yamamoto, T.; Hampson, G.; Koshiyama, H.; Ljunggren, Ö.; et al. Fibroblast Growth Factor 23 in Oncogenic Osteomalacia and X-Linked Hypophosphatemia. N. Engl. J. Med. 2003, 348, 1656–1663. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E38–E49. [Google Scholar] [CrossRef]
- Holm, I.A.; Nelson, A.E.; Robinson, B.G.; Mason, R.S.; Marsh, D.J.; Cowell, C.T.; Carpenter, T.O. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 2001, 86, 3889–3899. [Google Scholar] [CrossRef]
- Liu, E.S.; Carpenter, T.O.; Gundberg, C.M.; Simpson, C.A.; Insogna, K.L. Calcitonin Administration in X-Linked Hypophosphatemia. N. Engl. J. Med. 2011, 364, 1678–1680. [Google Scholar] [CrossRef]
- Farrow, E.G.; White, K.E. Recent advances in renal phosphate handling. Nat. Rev. Nephrol. 2010, 6, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Imel, E.A.; Holm, I.A.; de Beur, S.M.J.; Insogna, K.L. A clinician’s guide to X-linked hypophosphatemia. J. Bone Miner. Res. 2011, 26, 1381–1388. [Google Scholar] [CrossRef]
- Sarafrazi, S.; Daugherty, S.C.; Miller, N.; Boada, P.; Carpenter, T.O.; Chunn, L.; Dill, K.; Econs, M.J.; Eisenbeis, S.; Imel, E.A.; et al. Novel PHEX gene locus-specific database: Comprehensive characterization of vast number of variants associated with X-linked hypophosphatemia (XLH). Hum. Mutat. 2021, 43, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Schranck, F.W.; Armamento-Villareal, R. X-linked hypophosphatemia: A search for gender, race, anticipation, or parent of origin effects on disease expression in children. J. Clin. Endocrinol. Metab. 1996, 81, 4075–4080. [Google Scholar] [CrossRef] [PubMed]
- Dahir, K.M.; Black, M.; Gottesman, G.S.; Imel, E.A.; Mumm, S.; Nichols, C.M.; Whyte, M.P. X-Linked Hypophosphatemia Caused by the Prevailing North American PHEX Variant c.*231A>G; Exon 13–15 Duplication Is Often Misdiagnosed as Ankylosing Spondylitis and Manifests in Both Men and Women. JBMR Plus 2022, 6, e10692. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-R.; Park, J.-W.; Cho, D.-Y.; Yang, J.H.; Yoon, H.-R.; Jung, S.-C. PHEX Gene Mutations and Genotype-Phenotype Analysis of Korean Patients with Hypophosphatemic Rickets. J. Korean Med. Sci. 2007, 22, 981–986. [Google Scholar] [CrossRef]
- Eicher, E.M.; Southard, J.L.; Scriver, C.R.; Glorieux, F.H. Hypophosphatemia: Mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc. Natl. Acad. Sci. USA 1976, 73, 4667–4671. [Google Scholar] [CrossRef] [PubMed]
- Strom, T.M.; Francis, F.; Lorenz, B.; Böddrich, A.; Lehrach, H.; Econs, M.J.; Meitinger, T. Pex Gene Deletions in Gy and Hyp Mice Provide Mouse Models for X-Linked Hypophosphatemia. Hum. Mol. Genet. 1997, 6, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.S.; Martins, J.S.; Zhang, W.; Demay, M.B. Molecular analysis of enthesopathy in a mouse model of hypophosphatemic rickets. Development 2018, 145, dev163519. [Google Scholar] [CrossRef]
- Aono, Y.; Yamazaki, Y.; Yasutake, J.; Kawata, T.; Hasegawa, H.; Urakawa, I.; Fujita, T.; Wada, M.; Yamashita, T.; Fukumoto, S.; et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J. Bone Miner. Res. 2009, 24, 1879–1888. [Google Scholar] [CrossRef]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef]
- Bikle, D.; Christakos, S. New aspects of vitamin D metabolism and action—Addressing the skin as source and target. Nat. Rev. Endocrinol. 2020, 16, 234–252. [Google Scholar] [CrossRef]
- Sone, T.; Kerner, S.; Pike, J. Vitamin D receptor interaction with specific DNA. Association as a 1,25-dihydroxyvitamin D3-modulated heterodimer. J. Biol. Chem. 1991, 266, 23296–23305. [Google Scholar] [CrossRef]
- Sone, T.; Ozono, K.; Pike, J.W. A 55-Kilodalton Accessory Factor Facilitates Vitamin D Receptor DNA Binding. Mol. Endocrinol. 1991, 5, 1578–1586. [Google Scholar] [CrossRef]
- Okazaki, T.; Igarashi, T.; Kronenberg, H.M. 5’-flanking region of the parathyroid hormone gene mediates negative regulation by 1,25-(OH)2 vitamin D3. J. Biol. Chem. 1988, 263, 2203–2208. [Google Scholar] [CrossRef]
- Canalejo, A.; Almadén, Y.; Torregrosa, V.; Gomez-Villamandos, J.C.; Ramos, B.; Campistol, J.M.; Felsenfeld, A.J.; Rodríguez, M. The In Vitro Effect of Calcitriol on Parathyroid Cell Proliferation and Apoptosis. J. Am. Soc. Nephrol. 2000, 11, 1865–1872. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Wacker, M.J. FGF23 production by osteocytes. Pediatr. Nephrol. 2013, 28, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Kusano, K.; Kinosaki, M.; Ito, H.; Hirata, M.; Segawa, H.; Miyamoto, K.; Fukushima, N. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J. Biol. Chem. 2003, 278, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.-Y.; Miao, D.; Goltzman, D.; Karaplis, A.C. The Autosomal Dominant Hypophosphatemic Rickets R176Q Mutation in Fibroblast Growth Factor 23 Resists Proteolytic Cleavage and Enhances in Vivo Biological Potency. J. Biol. Chem. 2003, 278, 9843–9849. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Urakawa, I.; Yamazaki, Y.; Hasegawa, H.; Hino, R.; Yoneya, T.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem. Biophys. Res. Commun. 2004, 314, 409–414. [Google Scholar] [CrossRef]
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, O.; Tenenhouse, H.S.; Juppner, H.; Jonsson, K.B. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004, 145, 3087–3094. [Google Scholar] [CrossRef]
- Chanakul, A.; Zhang, M.Y.H.; Louw, A.; Armbrecht, H.J.; Miller, W.L.; Portale, A.A.; Perwad, F. FGF-23 Regulates CYP27B1 Transcription in the Kidney and in Extra-Renal Tissues. PLoS ONE 2013, 8, e72816. [Google Scholar] [CrossRef]
- Bai, X.; Miao, D.; Xiao, S.; Qiu, D.; St-Arnaud, R.; Petkovich, M.; Gupta, A.; Goltzman, D.; Karaplis, A.C. CYP24 inhibition as a therapeutic target in FGF23-mediated renal phosphate wasting disorders. J. Clin. Investig. 2016, 126, 667–680. [Google Scholar] [CrossRef]
- Insogna, K.L.; Briot, K.; Imel, E.A.; Kamenický, P.; Ruppe, M.D.; Portale, A.A.; Weber, T.; Pitukcheewanont, P.; Cheong, H.I.; de Beur, S.J.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J. Bone Miner. Res. 2018, 33, 1383–1393. [Google Scholar] [CrossRef]
- Carpenter, T.O.; Imel, E.A.; Ruppe, M.D.; Weber, T.J.; Klausner, M.A.; Wooddell, M.M.; Kawakami, T.; Ito, T.; Zhang, X.; Humphrey, J.; et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J. Clin. Investig. 2014, 124, 1587–1597. [Google Scholar] [CrossRef]
- Portale, A.A.; Carpenter, T.O.; Brandi, M.L.; Briot, K.; Cheong, H.I.; Cohen-Solal, M.; Crowley, R.; De Beur, S.J.; Eastell, R.; Imanishi, Y.; et al. Continued Beneficial Effects of Burosumab in Adults with X-Linked Hypophosphatemia: Results from a 24-Week Treatment Continuation Period After a 24-Week Double-Blind Placebo-Controlled Period. Calcif. Tissue Int. 2019, 105, 271–284. [Google Scholar] [CrossRef]
- Imel, E.A.; Zhang, X.; Ruppe, M.D.; Weber, T.J.; Klausner, M.A.; Ito, T.; Vergeire, M.; Humphrey, J.S.; Glorieux, F.H.; Portale, A.A.; et al. Prolonged Correction of Serum Phosphorus in Adults With X-Linked Hypophosphatemia Using Monthly Doses of KRN23. J. Clin. Endocrinol. Metab. 2015, 100, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Imel, E.A.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Ward, L.M.; Nilsson, O.; Simmons, J.; Padidela, R.; Namba, N.; Cheong, H.I.; et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: A randomised, active-controlled, open-label, phase 3 trial. Lancet 2019, 393, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Insogna, K.L.; Rauch, F.; Kamenický, P.; Ito, N.; Kubota, T.; Nakamura, A.; Zhang, L.; Mealiffe, M.; Martin, J.S.; Portale, A.A. Burosumab Improved Histomorphometric Measures of Osteomalacia in Adults with X-Linked Hypophosphatemia: A Phase 3, Single-Arm, International Trial. J. Bone Miner. Res. 2019, 34, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Beck-Nielsen, S.S.; Brusgaard, K.; Rasmussen, L.M.; Brixen, K.; Brock-Jacobsen, B.; Poulsen, M.R.; Vestergaard, P.; Ralston, S.H.; Albagha, O.M.E.; Poulsen, S.; et al. Phenotype Presentation of Hypophosphatemic Rickets in Adults. Calcif. Tissue Int. 2010, 87, 108–119. [Google Scholar] [CrossRef]
- Živičnjak, M.; Schnabel, D.; Billing, H.; Staude, H.; Filler, G.; Querfeld, U.; Schumacher, M.; Pyper, A.; Schröder, C.; Bramswig, J.; et al. Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr. Nephrol. 2010, 26, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, E.; Gil-Peña, H.; Chocron, S.; Madariaga, L.; de la Cerda-Ojeda, F.; Fernández-Fernández, M.; de Lucas-Collantes, C.; Gil, M.; Luis-Yanes, M.I.; Vergara, I.; et al. Phenotypic characterization of X-linked hypophosphatemia in pediatric Spanish population. Orphanet J. Rare Dis. 2021, 16, 104. [Google Scholar] [CrossRef] [PubMed]
- Schutt, S.; Schumacher, M.; Holterhus, P.; Felgenhauer, S.; Hiort, O. Effect of GH replacement therapy in two male siblings with combined X-linked hypophosphatemia and partial GH deficiency. Eur. J. Endocrinol. 2003, 149, 317–321. [Google Scholar] [CrossRef]
- Cagnoli, M.; Richter, R.; Bohm, P.; Knye, K.; Empting, S.; Mohnike, K. Spontaneous Growth and Effect of Early Therapy with Calcitriol and Phosphate in X-linked Hypophosphatemic Rickets. Pediatr. Endocrinol. Rev. 2017, 15 (Suppl. S1), 119–122. [Google Scholar] [CrossRef]
- Mao, M.; Carpenter, T.O.; Whyte, M.P.; Skrinar, A.; Chen, C.-Y.; Martin, J.S.; Rogol, A.D. Growth Curves for Children with X-linked Hypophosphatemia. J. Clin. Endocrinol. Metab. 2020, 105, 3243–3249. [Google Scholar] [CrossRef]
- Thacher, T.D.; Pettifor, J.M.; Tebben, P.J.; Creo, A.L.; Skrinar, A.; Mao, M.; Chen, C.-Y.; Chang, T.; Martin, J.S.; Carpenter, T.O. Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: Utility of the radiographic Rickets Severity Score. Bone 2019, 122, 76–81. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Street, J.; Bao, M.; DeGuzman, L.; Bunting, S.; Peale, F.V., Jr.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S.E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef]
- Liu, E.S.; Martins, J.S.; Raimann, A.; Chae, B.T.; Brooks, D.J.; Jorgetti, V.; Bouxsein, M.L.; Demay, M.B. 1,25-Dihydroxyvitamin D Alone Improves Skeletal Growth, Microarchitecture and Strength in a Murine Model of XLH, Despite Enhanced FGF23 Expression. J. Bone Miner. Res. 2016, 31, 929–939. [Google Scholar] [CrossRef]
- Sabbagh, Y.; Carpenter, T.O.; Demay, M.B. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 9637–9642. [Google Scholar] [CrossRef]
- Fuente, R.; Garcia-Bengoa, M.; Fernandez-Iglesias, A.; Gil-Pena, H.; Santos, F.; Lopez, J.M. Cellular and Molecular Alterations Underlying Abnormal Bone Growth in X-Linked Hypophosphatemia. Int. J. Mol. Sci. 2022, 23, 934. [Google Scholar] [CrossRef]
- Fuente, R.; Gil-Peña, H.; Claramunt-Taberner, D.; Hernández-Frías, O.; Fernández-Iglesias, Á.; Hermida-Prado, F.; Anes-González, G.; Rubio-Aliaga, I.; Lopez, J.M.; Santos, F. Marked alterations in the structure, dynamics and maturation of growth plate likely explain growth retardation and bone deformities of young Hyp mice. Bone 2018, 116, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Donohue, M.M.; Demay, M.B. Rickets in VDR Null Mice Is Secondary to Decreased Apoptosis of Hypertrophic Chondrocytes. Endocrinology 2002, 143, 3691–3694. [Google Scholar] [CrossRef]
- Bergwitz, C.; Roslin, N.M.; Tieder, M.; Loredo-Osti, J.C.; Bastepe, M.; Abu-Zahra, H.; Frappier, D.; Burkett, K.; Carpenter, T.O.; Anderson, D.; et al. SLC34A3 Mutations in Patients with Hereditary Hypophosphatemic Rickets with Hypercalciuria Predict a Key Role for the Sodium-Phosphate Cotransporter NaPi-IIc in Maintaining Phosphate Homeostasis. Am. J. Hum. Genet. 2006, 78, 179–192. [Google Scholar] [CrossRef]
- Miedlich, S.U.; Zhu, E.D.; Sabbagh, Y.; DeMay, M.B. The Receptor-Dependent Actions of 1,25-Dihydroxyvitamin D Are Required for Normal Growth Plate Maturation in NPt2a Knockout Mice. Endocrinology 2010, 151, 4607–4612. [Google Scholar] [CrossRef]
- Miedlich, S.U.; Zalutskaya, A.; Zhu, E.D.; Demay, M.B. Phosphate-induced Apoptosis of Hypertrophic Chondrocytes Is Associated with a Decrease in Mitochondrial Membrane Potential and Is Dependent upon Erk1/2 Phosphorylation. J. Biol. Chem. 2010, 285, 18270–18275. [Google Scholar] [CrossRef] [PubMed]
- Provot, S.; Nachtrab, G.; Paruch, J.; Chen, A.P.; Silva, A.; Kronenberg, H.M. A-Raf and B-Raf Are Dispensable for Normal Endochondral Bone Development, and Parathyroid Hormone-Related Peptide Suppresses Extracellular Signal-Regulated Kinase Activation in Hypertrophic Chondrocytes. Mol. Cell. Biol. 2008, 28, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Tanzawa, H.; Sato, K. The proto-oncogene C-raf-1 is highly expressed only in the hypertrophic zone of the growth plate. Calcif. Tissue Int. 1994, 54, 426–430. [Google Scholar] [CrossRef]
- Liu, E.S.; Raimann, A.; Chae, B.T.; Martins, J.S.; Baccarini, M.; Demay, M.B. C-Raf promotes Angiogenesis during Normal Growth Plate Maturation. Development 2015, 143, 348–355. [Google Scholar] [CrossRef]
- Papaioannou, G.; Petit, E.T.; Liu, E.S.; Baccarini, M.; Pritchard, C.; Demay, M.B. Raf Kinases Are Essential for Phosphate Induction of ERK1/2 Phosphorylation in Hypertrophic Chondrocytes and Normal Endochondral Bone Development. J. Biol. Chem. 2017, 292, 3164–3171. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.K.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP Receptor in Early Development and Indian Hedgehog—Regulated Bone Growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Schipani, E.; Lanske, B.; Hunzelman, J.; Luz, A.; Kovacs, C.S.; Lee, K.; Pirro, A.; Kronenberg, H.M.; Jüppner, H. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc. Natl. Acad. Sci. USA 1997, 94, 13689–13694. [Google Scholar] [CrossRef]
- Liu, E.S.; Zalutskaya, A.; Chae, B.T.; Zhu, E.D.; Gori, F.; Demay, M.B. Phosphate Interacts With PTHrP to Regulate Endochondral Bone Formation. Endocrinology 2014, 155, 3750–3756. [Google Scholar] [CrossRef]
- Beck, L.; Karaplis, A.C.; Amizuka, N.; Hewson, A.S.; Ozawa, H.; Tenenhouse, H.S. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc. Natl. Acad. Sci. USA 1998, 95, 5372–5377. [Google Scholar] [CrossRef]
- Kaneko, I.; Segawa, H.; Ikuta, K.; Hanazaki, A.; Fujii, T.; Tatsumi, S.; Kido, S.; Hasegawa, T.; Amizuka, N.; Saito, H.; et al. Eldecalcitol Causes FGF23 Resistance for Pi Reabsorption and Improves Rachitic Bone Phenotypes in the Male Hyp Mouse. Endocrinology 2018, 159, 2741–2758. [Google Scholar] [CrossRef]
- Jehan, F.; Gaucher, C.; Nguyen, T.M.; Walrant-Debray, O.; Lahlou, N.; Sinding, C.; Déchaux, M.; Garabédian, M. Vitamin D Receptor Genotype in Hypophosphatemic Rickets as a Predictor of Growth and Response to Treatment. J. Clin. Endocrinol. Metab. 2008, 93, 4672–4682. [Google Scholar] [CrossRef] [PubMed]
- Linglart, A.; Duplan, M.B.; Briot, K.; Chaussain, C.; Esterle, L.; Guillaume-Czitrom, S.; Kamenicky, P.; Nevoux, J.; Prié, D.; Rothenbuhler, A.; et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr. Connect. 2014, 3, R13–R30. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, O.; Doria, A.; Kooh, S.W.; Cole, W.G.; Daneman, A.; Sochett, E. Early Treatment Improves Growth and Biochemical and Radiographic Outcome in X-Linked Hypophosphatemic Rickets. J. Clin. Endocrinol. Metab. 2003, 88, 3591–3597. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.; Zhang, J.; Stillman, A.; Ellis, B.; Troiano, N.; Adams, D.J.; Gundberg, C.M.; Lifton, R.P.; Carpenter, T.O. Increased Bone Volume and Correction of HYP Mouse Hypophosphatemia in the Klotho/HYP Mouse. Endocrinology 2010, 151, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jagga, S.; Martins, J.S.; Rana, R.; Pajevic, P.D.; Liu, E.S. Impaired 1,25 dihydroxyvitamin D3 action and hypophosphatemia underlie the altered lacuno-canalicular remodeling observed in the Hyp mouse model of XLH. PLoS ONE 2021, 16, e0252348. [Google Scholar] [CrossRef]
- Barratt, K.R.; Sawyer, R.K.; Atkins, G.J.; St-Arnaud, R.; Anderson, P.H. Vitamin D supplementation improves bone mineralisation independent of dietary phosphate in male X-linked hypophosphatemic (Hyp) mice. Bone 2021, 143, 115767. [Google Scholar] [CrossRef]
- Marie, P.J.; Travers, R.; Glorieux, F.H. Healing of Bone Lesions with 1,25-Dihydroxyvitamin D 3 in the Young X-Linked Hypophosphatemic Male Mouse. Endocrinology 1982, 111, 904–911. [Google Scholar] [CrossRef]
- Xiao, Z.; Liu, J.; Liu, S.-H.; Petridis, L.; Cai, C.; Cao, L.; Wang, G.; Chin, A.L.; Cleveland, J.W.; Ikedionwu, M.O.; et al. Novel Small Molecule Fibroblast Growth Factor 23 Inhibitors Increase Serum Phosphate and Improve Skeletal Abnormalities in Hyp Mice. Mol. Pharmacol. 2022, 101, 408–421. [Google Scholar] [CrossRef]
- Ecarot, B.; Glorieux, F.; Desbarats, M.; Travers, R.; Labelle, L. Effect of 1,25-dihydroxyvitamin D3 treatment on bone formation by transplanted cells from normal and X-linked hypophosphatemic mice. J. Bone Miner. Res. 1995, 10, 424–431. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ecarot, B.; Glorieux, F. Abnormal response of osteoblasts from Hyp mice to 1,25-dihydroxyvitamin D3. Bone 1992, 13, 209–215. [Google Scholar] [CrossRef]
- Carpenter, K.A.; Ross, R.D. Sclerostin Antibody Treatment Increases Bone Mass and Normalizes Circulating Phosphate Levels in Growing Hyp Mice. J. Bone Miner. Res. 2020, 35, 596–607. [Google Scholar] [CrossRef]
- Dos Santos, E.L.; Chavez, M.; Tan, M.; Mohamed, F.; Kolli, T.; Foster, B.; Liu, E. Effects of Active Vitamin D or FGF23 Antibody on Hyp Mice Dentoalveolar Tissues. J. Dent. Res. 2021, 100, 1482–1491. [Google Scholar] [CrossRef]
- Ward, L.M.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Portale, A.A.; Högler, W.; Simmons, J.H.; Gottesman, G.S.; Padidela, R.; Namba, N.; et al. Effect of Burosumab Compared with Conventional Therapy on Younger vs Older Children with X-linked Hypophosphatemia. J. Clin. Endocrinol. Metab. 2022, 107, e3241–e3253. [Google Scholar] [CrossRef] [PubMed]
- Gadion, M.; Hervé, A.; Herrou, J.; Rothenbuhler, A.; Smail-Faugeron, V.; Courson, F.; Linglart, A.; Chaussain, C.; Duplan, M.B. Burosumab and Dental Abscesses in Children with X-Linked Hypophosphatemia. JBMR Plus 2022, 6, e10672. [Google Scholar] [CrossRef]
- Murali, S.K.; Andrukhova, O.; Clinkenbeard, E.L.; White, K.E.; Erben, R.G. Excessive Osteocytic Fgf23 Secretion Contributes to Pyrophosphate Accumulation and Mineralization Defect in Hyp Mice. PLoS Biol. 2016, 14, e1002427. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.; Prade, V.M.; Kunzke, T.; Erben, R.G.; Walch, A. Spatial metabolomics reveals upregulation of several pyrophosphate-producing pathways in cortical bone of Hyp mice. J. Clin. Investig. 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Nakano, Y.; Loisel, T.; Crine, P.; McKee, M.D. MEPE-ASARM Peptides Control Extracellular Matrix Mineralization by Binding to Hydroxyapatite: An Inhibition Regulated by PHEX Cleavage of ASARM. J. Bone Miner. Res. 2008, 23, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Masica, D.L.; Gray, J.J.; McKee, M.D. Phosphorylation-Dependent Inhibition of Mineralization by Osteopontin ASARM Peptides is Regulated by PHEX Cleavage. J. Bone Miner. Res. 2010, 25, 695–705. [Google Scholar] [CrossRef]
- Hoac, B.; Østergaard, M.; Wittig, N.K.; Boukpessi, T.; Buss, D.J.; Chaussain, C.; Birkedal, H.; Murshed, M.; McKee, M.D. Genetic Ablation of Osteopontin in Osteomalacic Hyp Mice Partially Rescues the Deficient Mineralization Without Correcting Hypophosphatemia. J. Bone Miner. Res. 2020, 35, 2032–2048. [Google Scholar] [CrossRef]
- Parfitt, A.M. The cellular basis of bone turnover and bone loss: A rebuttal of the osteocytic resorption—Bone flow theory. Clin. Orthop. Relat. Res. 1977, 127, 236–247. [Google Scholar] [CrossRef]
- Klein-Nulend, J.; Van Der Plas, A.; Semeins, C.M.; Ajubi, N.E.; Erangos, J.A.; Nijweide, P.J.; Burger, E.H. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. 1995, 9, 441–445. [Google Scholar] [CrossRef]
- McGarry, J.G.; Klein-Nulend, J.; Mullender, M.G.; Prendergast, P.J. A comparison of strain and fluid shear stress in stimulating bone cell responses—A computational and experimental study. FASEB J. 2005, 19, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; Ni, P.; Noonan, M.L.; White, K.E. Osteocytic FGF23 and Its Kidney Function. Front. Endocrinol. 2020, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Beck-Nielsen, S.S.; Mughal, Z.; Haffner, D.; Nilsson, O.; Levtchenko, E.; Ariceta, G.; de Lucas Collantes, C.; Schnabel, D.; Jandhyala, R.; Mäkitie, O. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J. Rare Dis. 2019, 14, 58. [Google Scholar] [CrossRef]
- Beno, T.; Yoon, Y.-J.; Cowin, S.C.; Fritton, S.P. Estimation of bone permeability using accurate microstructural measurements. J. Biomech. 2006, 39, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Hemmatian, H.; Bakker, A.D.; Klein-Nulend, J.; Van Lenthe, G.H. Aging, Osteocytes, and Mechanotransduction. Curr. Osteoporos. Rep. 2017, 15, 401–411. [Google Scholar] [CrossRef]
- Wittig, N.K.; Laugesen, M.; Birkbak, M.E.; Bach-Gansmo, F.L.; Pacureanu, A.; Bruns, S.; Wendelboe, M.H.; Brüel, A.; Sørensen, H.O.; Thomsen, J.S.; et al. Canalicular Junctions in the Osteocyte Lacuno-Canalicular Network of Cortical Bone. ACS Nano 2019, 13, 6421–6430. [Google Scholar] [CrossRef]
- Tokarz, D.; Martins, J.S.; Petit, E.T.; Lin, C.P.; Demay, M.B.; Liu, E.S. Hormonal Regulation of Osteocyte Perilacunar and Canalicular Remodeling in the Hyp Mouse Model of X-Linked Hypophosphatemia. J. Bone Miner. Res. 2017, 33, 499–509. [Google Scholar] [CrossRef]
- Qing, H.; Ardeshirpour, L.; Pajevic, P.D.; Dusevich, V.; Jähn, K.; Kato, S.; Wysolmerski, J.; Bonewald, L.F. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res. 2012, 27, 1018–1029. [Google Scholar] [CrossRef]
- Tazawa, K.; Hoshi, K.; Kawamoto, S.; Tanaka, M.; Ejiri, S.; Ozawa, H. Osteocytic osteolysis observed in rats to which parathyroid hormone was continuously administered. J. Bone Miner. Metab. 2004, 22, 524–529. [Google Scholar] [CrossRef]
- Tang, S.Y.; Herber, R.-P.; Ho, S.P.; Alliston, T. Matrix metalloproteinase-13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. J. Bone Miner. Res. 2012, 27, 1936–1950. [Google Scholar] [CrossRef] [PubMed]
- Holmbeck, K.; Bianco, P.; Pidoux, I.; Inoue, S.; Billinghurst, R.C.; Wu, W.; Chrysovergis, K.; Yamada, S.; Birkedal-Hansen, H.; Poole, A.R. The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. J. Cell Sci. 2005, 118, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Mikuni-Takagaki, Y.; Oikawa, K.; Itoh, T.; Inada, M.; Noguchi, T.; Park, J.-S.; Onodera, T.; Krane, S.M.; Noda, M.; et al. A Crucial Role for Matrix Metalloproteinase 2 in Osteocytic Canalicular Formation and Bone Metabolism. J. Biol. Chem. 2006, 281, 33814–33824. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J.; Glorieux, F.H. Relation between hypomineralized periosteocytic lesions and bone mineralization in vitamin D-resistant rickets. Calcif. Tissue Int. 1983, 35, 443–448. [Google Scholar] [CrossRef]
- Busse, B.; Bale, H.A.; Zimmermann, E.A.; Panganiban, B.; Barth, H.D.; Carriero, A.; Vettorazzi, E.; Zustin, J.; Hahn, M.; Ager, J.W., 3rd; et al. Vitamin D Deficiency Induces Early Signs of Aging in Human Bone, Increasing the Risk of Fracture. Sci. Transl. Med. 2013, 5, 193ra188. [Google Scholar] [CrossRef]
- Rolvien, T.; Krause, M.; Jeschke, A.; Yorgan, T.; Püschel, K.; Schinke, T.; Busse, B.; Demay, M.B.; Amling, M. Vitamin D regulates osteocyte survival and perilacunar remodeling in human and murine bone. Bone 2017, 103, 78–87. [Google Scholar] [CrossRef]
- Benjamin, M.; Ralphs, J. Entheses--the bony attachments of tendons and ligaments. Ital. J. Anat. Embryol. 2001, 106 (Suppl. S1), 151–157. [Google Scholar]
- Lu, H.H.; Thomopoulos, S. Functional Attachment of Soft Tissues to Bone: Development, Healing, and Tissue Engineering. Annu. Rev. Biomed. Eng. 2013, 15, 201–226. [Google Scholar] [CrossRef]
- Shaw, H.M.; Benjamin, M. Structure-function relationships of entheses in relation to mechanical load and exercise. Scand. J. Med. Sci. Sports 2007, 17, 303–315. [Google Scholar] [CrossRef]
- Woo, S.-Y. Ligament, tendon, and joint capsule insertions to bone. Inj. Repair Musculoskelet. Soft Tissues 1988, 133–166. [Google Scholar]
- Benjamin, M.; Toumi, H.; Ralphs, J.; Bydder, G.; Best, T.M.; Milz, S. Where tendons and ligaments meet bone: Attachment sites (‘entheses’) in relation to exercise and/or mechanical load. J. Anat. 2006, 208, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Genin, G.M.; Kent, A.; Birman, V.; Wopenka, B.; Pasteris, J.D.; Marquez, P.J.; Thomopoulos, S. Functional Grading of Mineral and Collagen in the Attachment of Tendon to Bone. Biophys. J. 2009, 97, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.G.; Pasteris, J.; Genin, G.M.; Daulton, T.L.; Thomopoulos, S. Mineral Distributions at the Developing Tendon Enthesis. PLoS ONE 2012, 7, e48630. [Google Scholar] [CrossRef] [PubMed]
- Killian, M.L.; Thomopoulos, S. Scleraxis is required for the development of a functional tendon enthesis. FASEB J. 2015, 30, 301–311. [Google Scholar] [CrossRef]
- Minina, E.; Wenzel, H.M.; Kreschel, C.; Karp, S.; Gaffield, W.; McMahon, A.P.; Vortkamp, A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001, 128, 4523–4534. [Google Scholar] [CrossRef]
- Blitz, E.; Viukov, S.; Sharir, A.; Shwartz, Y.; Galloway, J.L.; Pryce, B.A.; Johnson, R.L.; Tabin, C.J.; Schweitzer, R.; Zelzer, E. Bone Ridge Patterning during Musculoskeletal Assembly Is Mediated through SCX Regulation of Bmp4 at the Tendon-Skeleton Junction. Dev. Cell 2009, 17, 861–873. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Takimoto, A.; Akiyama, H.; Kist, R.; Scherer, G.; Nakamura, T.; Hiraki, Y.; Shukunami, C. Scx+/Sox9+ progenitors contribute to the establishment of the junction between cartilage and tendon/ligament. Development 2013, 140, 2280–2288. [Google Scholar] [CrossRef]
- Bi, W.M.; Deng, J.M.; Zhang, Z.P.; Behringer, R.R.; De Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef]
- Blitz, E.; Sharir, A.; Akiyama, H.; Zelzer, E. Tendon-bone attachment unit is formed modularly by a distinct pool of Scx- and Sox9-positive progenitors. Development 2013, 140, 2680–2690. [Google Scholar] [CrossRef]
- Felsenthal, N.; Rubin, S.; Stern, T.; Krief, S.; Pal, D.; Pryce, B.A.; Schweitzer, R.; Zelzer, E. Development of migrating tendon-bone attachments involves replacement of progenitor populations. Development 2018, 145, dev165381. [Google Scholar] [CrossRef]
- Chen, X.; Macica, C.M.; Dreyer, B.E.; Hammond, V.E.; Hens, J.R.; Philbrick, W.M.; Broadus, A.E. Initial Characterization of PTH-Related Protein Gene-Driven lacZ Expression in the Mouse. J. Bone Miner. Res. 2005, 21, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.G.; Long, F.; Thomopoulos, S. Enthesis fibrocartilage cells originate from a population of Hedgehog-responsive cells modulated by the loading environment. Development 2015, 142, 196–206. [Google Scholar] [CrossRef]
- Liang, G.; Katz, L.D.; Insogna, K.L.; Carpenter, T.O.; Macica, C.M. Survey of the Enthesopathy of X-Linked Hypophosphatemia and Its Characterization in Hyp Mice. Calcif. Tissue Int. 2009, 85, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Skrinar, A.; Dvorak-Ewell, M.; Evins, A.; Macica, C.; Linglart, A.; Imel, E.A.; Theodore-Oklota, C.; San Martin, J. The Lifelong Impact of X-Linked Hypophosphatemia: Results From a Burden of Disease Survey. J. Endocr. Soc. 2019, 3, 1321–1334. [Google Scholar] [CrossRef]
- Hughes, M.; Macica, C.; Meriano, C.; Doyle, M. Giving Credence to the Experience of X-Linked Hypophosphatemia in Adulthood: An Interprofessional Mixed-Methods Study. J. Patient-Centered Res. Rev. 2020, 7, 176–188. [Google Scholar] [CrossRef]
- Herrou, J.; Picaud, A.S.; Lassalle, L.; Pacot, L.; Chaussain, C.; Merzoug, V.; Hervé, A.; Gadion, M.; Rothenbuhler, A.; Kamenický, P.; et al. Prevalence of Enthesopathies in Adults With X-linked Hypophosphatemia: Analysis of Risk Factors. J. Clin. Endocrinol. Metab. 2021, 107, e224–e235. [Google Scholar] [CrossRef]
- Gjørup, H.; Kjaer, I.; Beck-Nielsen, S.S.; Poulsen, M.R.; Haubek, D. A radiological study on intra- and extra-cranial calcifications in adults with X-linked hypophosphatemia and associations with other mineralizing enthesopathies and childhood medical treatment. Orthod. Craniofacial Res. 2016, 19, 114–125. [Google Scholar] [CrossRef]
- Connor, J.; Olear, E.A.; Insogna, K.; Katz, L.; Baker, S.; Kaur, R.; Simpson, C.A.; Sterpka, J.; Dubrow, R.; Zhang, J.H.; et al. Conventional Therapy in Adults With X-Linked Hypophosphatemia: Effects on Enthesopathy and Dental Disease. J. Clin. Endocrinol. Metab. 2015, 100, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Karaplis, A.C.; Bai, X.; Falet, J.-P.R.; Macica, C.M. Mineralizing Enthesopathy Is a Common Feature of Renal Phosphate-Wasting Disorders Attributed to FGF23 and Is Exacerbated by Standard Therapy in Hyp Mice. Endocrinology 2012, 153, 5906–5917. [Google Scholar] [CrossRef] [PubMed]
- Cauliez, A.; Zhukouskaya, V.V.; Hilliquin, S.; Sadoine, J.; Slimani, L.; Miceli-Richard, C.; Briot, K.; Linglart, A.; Chaussain, C.; Bardet, C. Impact of Early Conventional Treatment on Adult Bone and Joints in a Murine Model of X-Linked Hypophosphatemia. Front. Cell Dev. Biol. 2020, 8, 591417. [Google Scholar] [CrossRef]
- Lafage-Proust, M.-H. What are the benefits of the anti-FGF23 antibody burosumab on the manifestations of X-linked hypophosphatemia in adults in comparison with conventional therapy? A review. Ther. Adv. Rare Dis. 2022, 3, 263300402210747. [Google Scholar] [CrossRef]
- Lamb, Y.N. Burosumab: First Global Approval. Drugs 2018, 78, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagga, S.; Venkat, S.; Sorsby, M.; Liu, E.S. Insights into the Molecular and Hormonal Regulation of Complications of X-Linked Hypophosphatemia. Endocrines 2023, 4, 151-168. https://doi.org/10.3390/endocrines4010014
Jagga S, Venkat S, Sorsby M, Liu ES. Insights into the Molecular and Hormonal Regulation of Complications of X-Linked Hypophosphatemia. Endocrines. 2023; 4(1):151-168. https://doi.org/10.3390/endocrines4010014
Chicago/Turabian StyleJagga, Supriya, Shreya Venkat, Melissa Sorsby, and Eva S. Liu. 2023. "Insights into the Molecular and Hormonal Regulation of Complications of X-Linked Hypophosphatemia" Endocrines 4, no. 1: 151-168. https://doi.org/10.3390/endocrines4010014
APA StyleJagga, S., Venkat, S., Sorsby, M., & Liu, E. S. (2023). Insights into the Molecular and Hormonal Regulation of Complications of X-Linked Hypophosphatemia. Endocrines, 4(1), 151-168. https://doi.org/10.3390/endocrines4010014