Abstract
Congenital or monogenic hyperinsulinism (HI) is a group of rare genetic disorders characterized by dysregulated insulin secretion and is the most common cause of persistent hypoglycemia in children. Knowledge of normal glucose homeostasis allows for a better understanding of the underlying pathophysiology of hyperinsulinemic hypoglycemia, facilitating timely diagnosis and management. The goal of management is to prevent cerebral insults secondary to hypoglycemia, which can result in poor neurologic outcomes and intellectual disability. Responsiveness to diazoxide, the first-line pharmacologic therapy for persistent hypoglycemia, is also the first step to distinguishing the different genotypic causes of monogenic hyperinsulinism. Early genetic testing becomes necessary when monogenic HI is strongly considered. Knowledge of specific gene mutations allows the determination of a clinical prognosis and definite therapeutic options, such as identifying those with focal forms of hyperinsulinism, who may attain a complete cure through surgical removal of specific affected parts of the pancreas. However, the lack of identifiable cause in a considerable number of patients identified with HI suggests there may be other genetic loci that are yet to be discovered. Furthermore, continued research is needed to explore new forms of therapy, particularly in severe, diazoxide-nonresponsive cases.
1. Introduction
Hyperinsulinism is defined as the elevation of insulin levels in blood and is the most frequent cause of hypoglycemia in infants and children. The state of insulin excess can be transient or permanent, depending on underlying acquired or genetic conditions. Congenital or monogenic hyperinsulinism (HI), which is the focus of this review, is caused by a group of rare monogenic conditions characterized by dysregulated insulin secretion and is the most common etiology of persistent hypoglycemia in children. The incidence of monogenic HI is about 1 in every 40,000 live births, with a higher incidence of up to 1 in every 2500 in highly consanguineous populations [1]. Since its initial description by MacQuarrie [2] in 1954 as ‘idiopathic hypoglycemia of infancy’, advances in medicine and technology have allowed for a better understanding of the disorder. This paved the way for improved diagnostic and treatment modalities, specific to underlying genetic pathology. However, in more than half of patients identified with HI, the exact cause is still unknown, which is suggestive of other genetic causes that are yet to be discovered [3].
2. Glucose Homeostasis
Understanding HI and its mechanisms may be difficult without a good knowledge of glucose homeostasis and its role in cellular metabolism. Glucose is an essential nutrient and the main source of energy for many cells in the human body. Blood glucose levels, therefore, must be maintained in a physiologic range to provide this steady source, a process also known as glucose homeostasis [4]. Main sources of blood glucose include dietary intake and gluconeogenic release of various endogenous stores within a human body. Glucagon, cortisol, and insulin are the main counter-regulatory hormones that regulate glucose homeostasis with the help of major organs such as the liver, skeletal muscle, pancreas, and adipocytes [5]. The pancreatic Islets of Langerhans, contain endocrine beta cells, which create, secrete or store insulin, and endocrine alpha cells that create, secrete or store glucagon, to help maintain blood glucose in physiologic ranges. After meals, insulin triggers the uptake of glucose by the liver, kidney, muscles, and adipose tissues for use and storage, thus modulating the rise of blood glucose. In between meals, glucagon triggers the release of the stored glucose to maintain blood glucose levels [6]. Beta cells release insulin in a biphasic manner corresponding to increasing glucose levels; the initial release of preformed insulin being dependent on the rate of change in serum glucose, and the subsequent sustained release being dependent on insulin synthesis [6,7].
Glucose enters the beta cells via glucose transporter 2 (GLUT2), and the increase in intracellular glucose and glucose metabolism causes an increase in the adenosine triphosphate/adenosine diphosphate (ATP/ADP) ratio. This results in the closing of potassium channels in the beta cells, trapping potassium inside and resulting in cell depolarization and the release of calcium within the cell [8]. The calcium-dependent functions of the cell are activated upon depolarization, one of which facilitates the exocytosis of insulin vesicles, i.e., insulin secretion. In addition to directly triggering insulin secretion, elevated glucose increases serum glucacon-like peptide 1 (GLP-1) and gastric inhibitory peptide (GIP) levels, which mediate insulin secretion via cyclic adenosine monophosphate (cAMP) signaling in the beta cells [9,10].
Once released, insulin acts via the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathways, regulating glucose transport and gluconeogenesis, as well as lipid and glycogen synthesis. As insulin binds to its receptor (InsR) on the target organs, InsR kinase is activated, and recruitment of insulin receptor substrates (IRS) ensues. This then results in recruitment and activation of PI3K, causing rapid phosphorylation of phosphatidylinositol 4,5-biphosphate (PIP2), which then generates the lipid second messenger phosphatidylinositol (3,4,5)-triphosphate (PIP3). PIP3 recruits Akt to the plasma membrane, where it is activated by phosphorylation and regulates glucose utilization through both the forkhead protein box O1 (FoxO1) and CREB-regulated transcription coactivator 2 (CRTC2). Glucogenic gene expression is suppressed by phosphorylation of FoxO1 and CRTC2, which induces their nuclear exclusion and cytoplasmic retention through binding to 14-3-3 protein.
In the fasting state, glucagon, which is the main counterregulatory hormone, initiates glycogenolysis and gluconeogenesis. It then binds to the hepatic glucagon receptor (GCGR), forming a glucagon–receptor complex. As a result, GCGR goes through conformational changes that can activate Gsα (G protein), which in turn activates adenylate cyclase that generates cAMP (second messenger). cAMP-dependent protein kinase (PKA) is subsequently stimulated, allowing for phosphorylation of the cAMP-response element-binding protein (CREB), which then binds to CRE and activates the transcription of CRE-bearing genes (PEPCK and G6PC gene). Glucocorticoid can also promote hepatic gluconeogenesis by binding to its intracellular receptor. Ligand binding causes the complex to enter the nucleus where it activates the expression of genes that control gluconeogenic enzymes, including pyruvate carboxylase (PC), phosphoenolpyruvate carboxykinase (PEPCK), Fructose 1,6-biphosphatase (FBPase), Glucose 6-phosphatase (G6Pase) [11]. Recent studies suggest that pancreatic delta cells have a specific role in controlling blood glucose by regulating alpha and beta cells via other hormones, namely somatostatin. Delta cells assist the beta cells in more precise control of glucose levels through somatostatin, an inhibitor of insulin and glucagon, as well as indirect regulation through ghrelin and long-chain free fatty acids. Delta cells and the molecules they regulate act in a paracrine manner on the neighboring alpha and beta cells to reach a homeostatic set point for glucose in the body [12].
Other endogenous factors can also influence the effectiveness of insulin, particularly glucocorticoids. Glucocorticoids exert an inhibitory effect on insulin’s ability to regulate glucose uptake into cells, resulting in persistent elevations in blood glucose [13,14]. Release of glucocorticoids as well as other counterregulatory hormones (epinephrine, norepinephrine) usually indicates an elevated metabolic state and concurrent increased need for glucose in the cells. The human brain, specifically neuronal cells that have glucose transporters that are less sensitive to insulin, is highly dependent on circulating blood glucose to support cellular metabolism. The absence of or decrease in circulating glucose, or hypoglycemia, and unavailability of ketones from fat metabolism, can result in irreversible brain damage during the prenatal and perinatal periods.
Maintaining normal glucose homeostasis is a vital process in the fetal-to-neonatal transitional phase. Physiologic glucose levels are the result of several neuroendocrine and metabolic regulatory processes, wherein a defect in any of these could result in varying degrees of hypoglycemia and subsequent pathologic consequences. At birth, there is a transition from constant glucose and amino acid supplies from the mother to a state of glucose deficiency from intermittent oral intake. The first few hours after birth are characterized by a fall in insulin levels, which remain low for several days [15]. Suppression of insulin secretion as a result of an infant’s relative fasting state is important in regulating hepatic glycogenolysis, gluconeogenesis, and ketogenesis. Failure of this hormonal response results in increased glucose consumption, a decrease in fat metabolism and ketogenesis, and abnormal reaction to glucagon during hypoglycemia.
3. Hyperinsulinism—General Overview
Hyperinsulinism (HI), which can be acquired or genetic, is a significant etiology of hypoglycemia in infants and children. Although it presents more often during the immediate neonatal period, it may also manifest later in life. Of the acquired forms, maternal diabetes mellitus and perinatal stress are the most common causes that present soon after birth. Infants of mothers with diabetes would have transient HI that may last for a few days [16], while HI from perinatal stress, such as prematurity, intrauterine growth retardation, small for gestational age, or perinatal asphyxia, may take several days to weeks before resolving [17,18]. Persistence of hypoglycemia requiring intervention with intravenous fluids with higher glucose infusion rates as well as pharmacologic therapy to maintain euglycemia is an alert for HI associated with genetic causes, including monogenic disorders (Table 1) and syndromes associated with HI (Table 2). Monogenic HI remains the most common etiology of persistent hypoglycemia in infants and children. The different genes associated with monogenic forms of HI will be discussed further in detail in this review (Figure 1).

Table 1.
Summary of monogenic hyperinsulinism disorders.
Table 2.
Syndromes associated with hyperinsulinemic hyperinsulinemia.
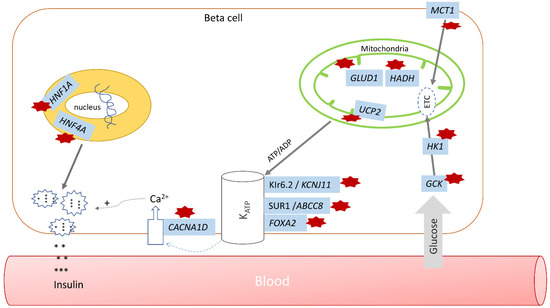
Figure 1.
Pathways regulating insulin secretion in hyperinsulinism. Affected genes are highlighted in blue; hyperinsulinism pathway defects are noted by red stars (GCK—glucokinase HI, HK1—HK1 HI, MCT1—MCT1 HI, GLUD1—hyperinsulinism-hyperammonemia (HI/HA) syndrome, HADH—mitochondrial short-chain L-3-hydroxyacyl-CoA dehydrogenase, or SCHAD, UCP2—UCP2 HI, CACNA1D—CACNA1D HI, KCNJ11 and ABCC8—monogenic HI, FOXA2—FOXA2 HI, HNF1A and HNF4A—HNF1A and HNF4A HI; * denotes insulin.
4. Monogenic Hyperinsulinism
4.1. Pancreatic β-Cell KATP Channel Defects
Inactivating mutations in the ABCC8 and KCNJ11 genes located on chromosome 11p15.1 are the most common and severe forms of monogenic HI. The ABCC8 gene encodes for the outer, sulfonylurea receptor 1 (SUR1) subunits of the KATP channels, which controls Kir6.2 protein activity. The SUR1 subunits function as binding sites for KATP channel openers. The KCNJ11 gene encodes for the inner pore-making, Kir6.2 channel proteins, which allow potassium influx across the β-cell membrane that then triggers depolarization of the cell membrane. This then activates the voltage-gated calcium channels, subsequently causing an insulin release [29,30]. Of note, activating mutations of these genes cause the opposite phenotype of dysregulated insulin secretion, also known as monogenic diabetes. These mutations can be recessively or dominantly inherited. Mutations that result in residual KATP activity lead to diazoxide-responsive forms, while those with significantly diminished channel activity cause diazoxide-unresponsive phenotypes [31]. Depending on the severity of impairment of channel activity and the phenotype, HI from KATP channel defects can be classified as: recessive, diazoxide-unresponsive; dominant, diazoxide-unresponsive; and dominant, diazoxide-responsive. Recessive mutations are more commonly identified in infants and children with monogenic HI, resulting in more severe clinical features, as they interfere with protein expression or channel trafficking, leading to a significantly reduced to an almost absent number of channels on the plasma membrane. Dominant mutations allow normal passage along the channels to the plasma membrane but with partial or completely impaired channel activity, which then results in varying phenotypes of diazoxide responsiveness [32]. This group of HI may also be classified by histological forms: a diffuse form that affects all pancreatic β-cells resulting from monoallelic-dominant or biallelic-recessive mutations, or a focal form with localized islet cell adenomatous hyperplasia that is caused by somatic loss of the maternal chromosome 11p15.5 region by uniparental disomy that unmasks a paternally inherited KATP channel mutation at 11p15.1 [33,34]. Diazoxide-unresponsive, diffuse forms frequently require near-total pancreatectomy for adequate glycemic control. However, for focal disease not responsive to diazoxide, surgical excision of the identified localized lesion may afford a cure in 97% of patients [35]. Because of the ability to determine therapeutic options and a clinical prognosis based on gene mutations, early genetic testing becomes necessary when monogenic HI is strongly considered.
4.2. Glutamate Dehydrogenase (GDH) Hyperinsulinism
Mutation of the glutamate dehydrogenase 1 (GLUD1) causes hyperinsulinism-hyperammonemia (HI/HA) syndrome and is the second most common cause of monogenic/congenital HI. The GLUD1 gene encodes for the enzyme GDH, which catalyzes the deamination of α-ketoglutarate and ammonia [36]. GLUD1 gain-of-function mutations cause increased activity of the GDH enzyme, mediated by the allosteric sensitivity to glucose triphosphate (GTP) inhibition or ADP activation. Together with normal leucine-mediated allosteric activation, this altered activity causes fasting and protein-induced hyperinsulinemic hypoglycemia [26]. Fasting hypoglycemia may be mild, and children can tolerate fasting for up to 8–12 h, with protein-sensitive hypoglycemia being more severe, occurring within 30–90 min of having a meal [37]. Effects of the activating mutation in the kidneys result in persistent hyperammonemia, usually elevated at levels 2–5 times more than normal, with symptoms such as lethargy, headaches, or vomiting [38]. Neurologic abnormalities, such as seizure disorder, behavior issues, and learning disabilities, occur at a higher rate in this group of patients, which is unrelated to hypoglycemia and hyperammonemia but postulated to be related to the direct effect of GDH activity in the brain [34,39]. GDH hyperinsulinism is the most common form of diazoxide-responsive HI [40]. Dietary modification, such as carbohydrate preloading and avoidance of protein without carbohydrate consumption, are also helpful in preventing hypoglycemia.
4.3. Glucokinase Hyperinsulinism
Activating mutations of GCK on chromosome 7p13 result in a less common monogenic HI. Glucokinase is a glucose sensor in the pancreatic β-cell. In glucokinase hyperinsulinism, there is an increased affinity of glucokinase for glucose and decreased threshold of glucose levels to stimulate insulin secretion. Fifteen mutations have been associated with glucokinase HI, occurring as dominantly inherited or sporadic, and there are varying degrees of clinical presentation, depending on the mutation, with the majority of cases being unresponsive to diazoxide [41]. Clinically, it may manifest soon after birth as severe hypoglycemia; however, recognition is more commonly delayed until late infancy or early childhood. Presentation in adulthood is also not uncommon. Affected individuals have a history of being large for gestational age, caused by growth-promoting effects of fetal insulin secretion. While pancreatectomy has been utilized to control hypoglycemia, most patients require additional medical management, such as combined diazoxide and long-acting somatostatin analog [42], while others may require continuous feedings.
4.4. SCHAD Hyperinsulinism
This rare form of autosomal recessive, monogenic hyperinsulinism involves the impaired regulation of GDH activity as a result of inactivating mutations in the HADH gene on chromosome 4q25, which encodes for the mitochondrial short-chain L-3-hydroxyacyl-CoA dehydrogenase, or SCHAD. Clinical presentation shares some similarities with GDH hyperinsulinism, with fasting as well as protein-induced hypoglycemia, however with no demonstrable hyperammonemia or neurodevelopmental manifestations. Other biochemical changes noted are increased plasma 3-hydroxybutyryl-carnitine and urine 3-hydroxyglutarate, reflective of impaired SCHAD enzyme activity. However, unlike in individuals with other fatty acid oxidation defects, those with SCHAD hyperinsulinism do not exhibit hepatic, cardiac, or skeletal dysfunction [43,44]. Most cases respond to diazoxide.
4.5. HNF1A and HNF4A Hyperinsulinism
Hepatocyte nuclear factors 1-α and 4-α are transcription factors expressed in pancreatic β-cells as well as in hepatocytes, intestinal epithelial cells, and renal tubular cells. They are encoded by HNF1A and HNF4A, respectively, and heterozygous mutations cause familial monogenic diabetes. This form of HI causes progressively diminishing insulin secretion and typically manifests before the age of 25 years. The same loss-of-function mutation causes hyperinsulinism during fetal, neonatal, and early childhood stages, resulting in fetal macrosomia and varying degrees of hyperinsulinism, ranging from transient to severe persistent forms [45,46,47].
4.6. UCP2 Hyperinsulinism
UCP2 is an inner mitochondrial carrier protein encoded by the UCP2 gene, expressed in pancreatic islet cells and other tissues. It mediates the proton leak across the inner mitochondrial membrane, which subsequently inhibits ATP generation through mitochondrial oxidative metabolism and decreases glucose-mediated insulin secretion. As such, inactivating heterozygous mutations would augment glucose oxidation, increasing intracellular ATP synthesis and subsequently, hyperinsulinism [48,49]. Clinically, this form of HI can present as transient to prolonged or persistent forms.
4.7. MCT1 Hyperinsulinism
A dominant gain of function mutations in the promoter region of SLC16A1 resulting in increased expression of Monocarboxylate transporter 1 (MCT1) protein produces exercise-induced hyperinsulinism [50,51]. MCT1 is involved in the transport of pyruvate and lactate across the β-cell membrane. Under normal conditions, pancreatic β-cell expression of MCT1 is suppressed to prevent stimulation of insulin secretion. Increased expression of MCT1 leads to glycolysis-generated pyruvate that stimulates insulin secretion inappropriately, even during anaerobic exercise, when low glucose concentrations are expected. Management includes increased carbohydrate intake during periods of exercise. Most affected individuals respond to diazoxide, but may not be complete; thus, dietary modifications are necessary.
4.8. HK-1 Hyperinsulinism
Somatic overexpression of Hexokinase 1 (HK-1) has been reported to cause hyperinsulinemic hypoglycemia [52,53]. Under physiologic conditions, the expression of the HK1 gene, which encodes for the enzyme HK-1, is suppressed in the pancreatic β-cells. Gain-of-function mutation leads to a lowered glucose threshold for insulin secretion.
4.9. FOXA2 Hyperinsulinism
Mutations in the FOXA2 gene, which encodes for forkhead box A2 transcription factor (FOXA2), cause congenital hyperinsulinism, hypopituitarism, cranial dysmorphism, and defects of organs of endodermal origins [54,55].
4.10. CACNA1D Hyperinsulinism
Another recently discovered and rare form of monogenic HI is from mutation of the CACNA1D gene, which encodes Ca2+ channels which regulate insulin release from beta cells. A case report published in 2017 described a child with developmental delays, severe hypotonia, mild aortic insufficiency, and persistent hyperinsulinemic hypoglycemia that was responsive to diazoxide [56].
5. Clinical Presentation, Diagnosis, and General Approach to Management of Hyperinsulinemic Hypoglycemia (HH)
Patients with hyperinsulinemic hypoglycemia can present with varied symptomatology, including nonspecific symptoms, such as poor feeding, increased hunger, sweating, and palpitations, as well as neuroglycopenic manifestations, such as stupor, loss of consciousness, seizures, or coma. Although it often presents during the immediate neonatal period, it may also manifest during late infancy, childhood, and very rarely, in adulthood. Macrosomia, or being large for gestational age, is a common finding in neonates, as an effect of fetal insulin secretion causing cellular hyperplasia from the effects of insulin-like growth factor 1 receptor signaling. Persistent, severe hypoglycemia requiring higher glucose infusion rates to maintain normoglycemia should raise suspicion for HH. Obtaining critical samples during a hypoglycemic episode (<50 mg/dL) and observing the glycemic response to administration of glucagon are crucial in the clinical diagnosis. Biochemically, HH is diagnosed with evidence of inappropriately elevated or normal concentrations of insulin despite the low plasma glucose concentration. An elevated c-peptide level drawn at the same time as insulin and glucose will confirm that the high insulin is not factitious, such as from exogenous administration of insulin medication. Other findings include an inappropriately low plasma beta-hydroxybutyrate and free fatty acids, as well as an increase of >30 mg/dL in glucose levels after administering glucagon. Other biochemical clues to more specific forms of HH are described in the individual gene mutations, as above, such as elevated serum ammonia in patients with GDH hyperinsulinism or raised plasma hydroxybutyrylcarnitine and urinary 3-hydroxybutyrate in HADH hyperinsulinism.
The general therapeutic approach is to maintain the glucose level above 70 mg/dL, which is the threshold for activation of neuroendocrine responses to hypoglycemia [57]. The main goal of therapy is to prevent cerebral insults secondary to hypoglycemia, which can result in intellectual disability in ~30% affected patients [58]. Delivery of continuous glucose infusion at a higher rate than the typical 6–8 mg/kg/min is often needed. Diazoxide is the first-line pharmacologic therapy, which acts by opening KATP channels in the pancreatic β-cells, resulting in decreased insulin secretion. Responsiveness to diazoxide is also the first step in distinguishing the different genotypic causes of monogenic hyperinsulinism. Patients unresponsive to diazoxide then undergo the first tier of genetic testing, which includes ABCC8, KCNJ11, and GCK [24]. Referral to a center that can perform rapid testing or urgent genetic analysis is important in identifying those with a focal form of hyperinsulinism who may benefit from surgical resection of specific affected sites. Patients responsive to diazoxide therapy and deemed to have persistent hyperinsulinism are recommended to undergo further genetic testing to include ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, HNF4A, HNF1A, and MCT1 [24].
Other pharmacologic agents for those who are diazoxide-unresponsive include octreotide and other longer-acting somatostatin analogs [59,60,61,62,63]. Octreotide is a long-acting somatostatin analog that decreases insulin secretion by binding to somatostatin receptors 2 and 5. Octreotide is given as subcutaneous injections every 6–8 h, or via subcutaneous infusion using insulin pumps. Newer pharmacologic therapies include sirolimus and glucagon-like peptide-1 receptor (GLP-1) antagonists. Sirolimus is an mTOR inhibitor that is primarily used as an immunosuppressive agent but has shown efficacy in the treatment of diazoxide-unresponsive HI. The exact mechanism is still unknown, but proposed mechanisms include inhibition of noted overexpression of p-mTOR on acinar cells of patients with diffuse HI [64], as well as decreased insulin release as a result of decreased intracellular Ca2+ and altered mitochondrial activity [65]. Although several case reports [66,67,68,69] have established the safety and efficacy of sirolimus on this subset of patients, potential adverse effects related to its immunosuppressive effects remain a significant concern. Exendin-(9-39) is a specific GLP-1 receptor agonist that has been shown to inhibit insulin secretion in mice [70] and maintain normal glucose levels in infants with KATP mutations [71].
Advances in genetic testing and imaging modalities have improved the management of focal disease, where surgery is still the treatment modality of choice. Further imaging with 18F-DOPA-PET/CT is recommended for all patients requiring surgery, allowing better identification of focal areas to be resected for long-term cure and prevention of risk of diabetes later in life [72]. This imaging technique has been shown to have a sensitivity of 88% and specificity of 94%, which allows differentiation between focal and diffuse lesions [73,74]. DOPA decarboxylase enzyme is expressed in pancreatic β-cells, and increased activity of this enzyme as demonstrated by an increased uptake of L-DOPA, allows for visualization of affected areas on enhanced computed tomography imaging.
6. Conclusions
Timely consideration of directed genetic testing has proven to be essential in the diagnosis and management of monogenic hyperinsulinism. However, the lack of a specific genetic diagnosis in almost half of identified HI patients and challenges in managing those who are diazoxide-nonresponsive prove that further research on this group of genetic disorders is needed.
Author Contributions
(1) Conception and design: S.K., E.G.C.; (2) Administrative support: S.K., E.G.C.; (3) Provision of study materials or patients: E.G.C., S.K.; (4) Manuscript writing: E.G.C., S.K., C.S., D.M. (5) Final approval of manuscript: E.G.C., S.K., C.S., D.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De León, D.D.; Stanley, C.A. Mechanisms of Disease: Advances in diagnosis and treatment of hyperinsulinism in neonates. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 57–68. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, I. Idiopathic spontaneously occurring hypoglycemia in infants; clinical significance of problem and treatment. AMA Am. J. Dis. Child. 1954, 87, 399–428. [Google Scholar] [CrossRef]
- Kapoor, R.R.; Flanagan, S.E.; Arya, V.B.; Shield, P.; Ellard, S.; Hussain, K. Clinical and molecular characterization of 300 patients with congenital hyperinsulinism. Eur. J. Endocrinol. 2013, 168, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Ann. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Han, H.S.; Kim, M.J. Transcriptional regulators of hepatic gluconeogenesis. Arch. Pharm. Res. 2013, 36, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C.; Ishiyama, N.; Nenquin, M.; Ravier, A.M.; Jonas, J. Signals and pools underlying biphasic insulin secretion. Diabetes 2002, 51 (Suppl. 1), S60–S67. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, L.W.; Habener, J.F. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Morgan, L.M.; Tredger, J.A.; Deacon, S.; Wright, J.; Marks, V. Glucagon-like peptide-1 (7-36) amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: Acute post-prandial and 24-h secretion patterns. J. Endocrinol. 1993, 138, 159–166. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2019, 9, 802. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, T.; Donaldson, C.J.; Cáceres, E.; Hunter, E.A.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates soomatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769–776. [Google Scholar] [CrossRef]
- Morgan, S.A.; Sherlock, M.; Gathercole, L.L.; Lavery, G.G.; Lenaghan, C.; Bujalska, I.J.; Laber, D.; Yu, A.; Convey, G.; Mayers, R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 2009, 58, 2506–2515. [Google Scholar] [CrossRef]
- Gathercole, L.L.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W. Glucocorticoid modulation of insulin signaling in human subcutaneous adipose tissue. J. Clin. Endocrinol. Metab. 2007, 92, 4332–4339. [Google Scholar] [CrossRef]
- Sperling, M.A.; DeLamater, P.V.; Phelps, D.; Fiser, H.R.; Oh, W.; Fisher, D.A. Spontaneous and amino acid stimulated glucagon secretion in the immediate postnatal period. Relation to glucose and insulin. J. Clin. Investig. 1974, 53, 1159–1166. [Google Scholar] [CrossRef]
- Farquhar, J.W. The child of the diabetic woman. Arch. Dis. Child. 1959, 34, 76–96. Available online: https://pubmed.ncbi.nlm.nih.gov/13628237/ (accessed on 5 March 2022). [CrossRef]
- Collins, J.E.; Leonard, J.V. Hyperinsulinism in asphyxiated and small-for-dates infants with hypoglycaemia. Lancet 1984, 324, 311–313. [Google Scholar] [CrossRef]
- Hoe, F.M.; Thornton, P.S.; Wanner, L.A.; Steinkrauss, L.; Simmons, R.A.; Stanley, C.A. Clinical features and insulin regulation in infants with a syndrome of prolonged neonatal hyperinsulinism. J. Pediatr. 2006, 148, 207–212. [Google Scholar] [CrossRef]
- Hussain, K.E.; Shepherd, R.M. Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann syndrome due to defects in the function of pancreatic beta-cell adenosine triphosphate-sensitive potassium channels. J. Clin. Endocrinol. Metab. 2005, 90, 4376–4382. [Google Scholar] [CrossRef]
- Faundes, V.; Goh, S.; Akilapa, R. Clinical delineation, sex differences, and genotype-phenotype correlation in pathogenic KDM6A variants causing X-linked Kabuki syndrome type 2. Genet. Med. 2021, 23, 1202–1210. [Google Scholar] [CrossRef]
- Ben Harouch, S.; Klar, A.; Falik Zaccai, T.C. INSR-Related Severe Syndromic Insulin Resistance. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2018; pp. 1993–2022. [Google Scholar] [PubMed]
- Matsuo, T.; Ihara, K.; Ochiai, M. Hyperinsulinemic hypoglycemia of infancy in Sotos syndrome. Am. J. Med. Genet. A 2013, 161, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.E.; Boodhansingh, K.E.; Li, C. Congenital Hyperinsulinism in Infants with Turner Syndrome: Possible Association with Monosomy X and KDM6A Haploinsufficiency. Horm. Res. Paediatr. 2018, 89, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Moravej, H.; Altassan, R.; Jaeken, J. Hypoglycemia in CDG patients due to PMM2 mutations: Follow up on hyperinsulinemic patients. JIMD Rep. 2019, 51, 76–81. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tegtmeyer, L.C.; Rust, S.; van Scherpenzeel, M. Multiple phenotypes in phosphoglucomutase 1 deficiency. N. Engl. J. Med. 2014, 370, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.; Timothy, K.W.; Golden, A. Update on the Molecular Genetics of Timothy Syndrome. Front. Pediatr. 2021, 9, 668546. [Google Scholar] [CrossRef]
- Giurgea, I.; Bellanné-Chantelot, C.; Ribeiro, M. Molecular mechanisms of neonatal hyperinsulinism. Horm. Res. 2006, 66, 289–296. [Google Scholar] [CrossRef]
- Sajorda, B.J.; Gonzalez-Gandolfi, C.X.; Hathaway, E.R. Simpson-Golabi-Behmel Syndrome Type 1. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2006; pp. 1993–2022. [Google Scholar] [PubMed]
- Thomas, P.; Ye, Y.; Lightner, E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum. Mol. Genet. 1996, 5, 1809–1812. Available online: https://pubmed.ncbi.nlm.nih.gov/8923010/ (accessed on 5 March 2022). [CrossRef]
- Thomas, P.; Cote, G.; Wohllk, N. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 1995, 268, 426–429. Available online: https://pubmed.ncbi.nlm.nih.gov/7716548/ (accessed on 5 March 2022). [CrossRef]
- Pinney, S.; MacMullen, C.; Becker, S.L. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J. Clin. Investig. 2008, 118, 2877–2886. Available online: https://pubmed.ncbi.nlm.nih.gov/18596924/ (accessed on 5 March 2022). [CrossRef]
- De León, D.D.; Thornton, P.S.; Stanley, C.A. Hypoglycemia in the newborn and infant. In Pediatric Endocrinology, 4th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2014; pp. 157–185. [Google Scholar]
- De Franco, E.; Saint-Martin, C.; Brusgaard, K. Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum. Mutat. 2020, 41, 884–905. [Google Scholar] [CrossRef]
- Rosenfeld, E.; Ganguly, A.; De Leon, D. Congenital hyperinsulinism disorders: Genetic and clinical characteristics. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 682–692. Available online: https://pubmed.ncbi.nlm.nih.gov/31414570/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Adzick, N.S.; Leon, D.D.D.; States, L.J. Surgical Treatment of Congenital Hyperinsulinism: Results from 500 Pancreatectomies in Neonates and Children. J. Pediatr. Surg. 2019, 54, 27. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6339589/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Kelly, A.; Ng, D.; Ferry, R. Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 3724–3728. Available online: https://pubmed.ncbi.nlm.nih.gov/11502802/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Hsu, B.Y.L.; Kelly, A.; Thornton, P.S. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J. Pediatr. 2001, 138, 383–389. [Google Scholar] [CrossRef]
- Palladino, A.A.; Stanley, C.A. The hyperinsulinism/hyperammonemia syndrome. Rev. Endocr. Metab. Disord. 2010, 11, 171–178. Available online: https://pubmed.ncbi.nlm.nih.gov/20936362/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Bahi-Buisson, N.; Roze, E.; Dionisi, C. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev. Med. Child. Neurol. 2008, 50, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Snider, K.E.; Becker, S.; Boyajian, L. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Sayed, S.; Matchinsky, F.M.; Stanley, C.A. Hyperinsulinism due to activating mutations of glucokinase. In Monogenic Hyperinsulinism Hypoglycemia Disorders; Stanley, C.A., De León, D.D., Eds.; Karger: Basel, Switzerland, 2012; pp. 146–157. [Google Scholar]
- Wabitsch, M.; Lahr, G.; Van de Bunt, M. Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycaemia of infancy. Diabet. Med. 2007, 24, 1393–1399. Available online: https://pubmed.ncbi.nlm.nih.gov/17976205/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Clayton, P.; Eaton, S.; Aynsley-Green, A. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J. Clin. Investig. 2001, 108, 457–465. Available online: https://pubmed.ncbi.nlm.nih.gov/11489939/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Hussain, K.; Clayton, P.T.; Krywawych, S. Hyperinsulinism of infancy associated with a novel splice site mutation in the SCHAD gene. J. Pediatr. 2005, 146, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.; Boj, S.; Steele, A. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007, 4, 760–769. Available online: https://pubmed.ncbi.nlm.nih.gov/17407387/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Fajans, S.S.; Bell, G.I. Macrosomia and neonatal hypoglycaemia in RW pedigree subjects with a mutation (Q268X) in the gene encoding hepatocyte nuclear factor 4α (HNF4A). Diabetologia 2007, 50, 2600–2601. [Google Scholar] [CrossRef] [PubMed]
- Arya, V.B.; Rahman, S.; Senniappan, S. HNF4A mutation: Switch from hyperinsulinaemic hypoglycaemia to maturity-onset diabetes of the young, and incretin response. Diabet. Med. 2014, 31, e11–e15. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.T.; Boodhansingh, K.E.; Paradies, E. Novel hypoglycemia phenotype in congenital hyperinsulinism due to dominant mutations of uncoupling protein 2. J. Clin. Endocrinol. Metab. 2017, 102, 942–949. [Google Scholar] [CrossRef] [PubMed]
- González-Barroso, M.M.; Giurgea, I.; Bouillaud, F. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS ONE. 2008, 3, e3850. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.; Sylow, L.; Sun, G. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic β-cells leads to relative hyperinsulinism during exercise. Diabetes 2012, 61, 1719–1725. Available online: https://pubmed.ncbi.nlm.nih.gov/22522610/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Otonkoski, T.; Jiao, H.; Kaminen-Ahola, N. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic β cells. Am. J. Hum. Genet. 2007, 81, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S.; Ganapathy, K.; Bradfield, J. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm. Res. Paediatr. 2013, 80, 18–27. Available online: https://pubmed.ncbi.nlm.nih.gov/23859901/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Henquin, J.; Sempoux, C.; Marchandise, J.; Godecharles, S.; Guiot, Y.; Nenquin, M.; Rahier, J. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of β-cells. Diabetes 2013, 62, 1689–1696. Available online: https://pubmed.ncbi.nlm.nih.gov/23274908/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Giri, D.; Vignola, M.L.; Gualtieri, A. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum. Mol. Genet. 2017, 26, 4315–4326. [Google Scholar] [CrossRef]
- Vajravelu, M.E.; Chai, J.; Krock, B. Congenital Hyperinsulinism and Hypopituitarism Attributable to a Mutation in FOXA2. J. Clin. Endocrinol. Metab. 2018, 103, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Vairo, F.; Johnson, M.B. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr. Diabetes 2017, 18, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.; Stanley, C.; De Leon, D. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J. Pediatr. 2015, 167, 238–245. Available online: https://pubmed.ncbi.nlm.nih.gov/25957977/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Menni, F.; de Lonlay, P.; Sevin, C. Neurologic outcomes of 90 neonates and infants with persistent hyperinsulinemic hypoglycem ia. Pediatrics 2001, 107, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Erstad, B. Octreotide, a new somatostatin analogue. Clin. Pharm. 1989, 8, 255–273. Available online: https://pubmed.ncbi.nlm.nih.gov/2653711/ (accessed on 5 March 2022). [PubMed]
- Hosokawa, Y.; Kawakita, R.; Yokoya, S. Efficacy and safety of octreotide for the treatment of congenital hyperinsulinism: A prospective, open-label clinical trial and an observational study in Japan using a nationwide registry. Endocr. J. 2017, 64, 867–880. Available online: https://pubmed.ncbi.nlm.nih.gov/28701683/ (accessed on 5 March 2022). [CrossRef] [PubMed]
- Van Der Steen, I.; Van Albada, M.E.; Mohnike, K. A Multicenter Experience with Long-Acting Somatostatin Analogues in Patients with Congenital Hyperinsulinism. Horm Res. Paediatr. 2018, 89, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Corda, H.; Kummer, S.; Welters, A. Treatment with long-acting lanreotide autogel in early infancy in patients with severe neonatal hyperinsulinism. Orphanet. J. Rare Dis. 2017, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Yorifuji, T.; Kawakita, R.; Hosokawa, Y. Efficacy and safety of long-term, continuous subcutaneous octreotide infusion for patients with different subtypes of KATP-channel hyperinsulinism. Clin. Endocrinol. 2013, 78, 891–897. [Google Scholar] [CrossRef]
- Alexandrescu, S.; Tatevian, N.; Olutoye, O. Persistent hyperinsulinemic hypoglycemia of infancy: Constitutive activation of the mTOR pathway with associated exocrine-islet transdifferentiation and therapeutic implications. Int. J. Clin. Exp. Pathol. 2010, 3, 691–705. [Google Scholar]
- Yang, S.B.; Lee, H.Y.; Young, D.M. Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J. Mol. Med. 2012, 90, 575–585. [Google Scholar] [CrossRef]
- Senniappan, S.; Alexandrescu, S.; Tatevian, N. Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N. Engl. J. Med. 2014, 370, 1131–1137. [Google Scholar] [CrossRef]
- Abraham, M.B.; Shetty, V.B.; Price, G. Efficacy and safety of sirolimus in a neonate with persistent hypoglycaemia following near-total pancreatectomy for hyperinsulinaemic hypoglycaemia. J. Pediatr. Endocrinol. Metab. 2015, 28, 1391–1398. [Google Scholar] [CrossRef]
- Minute, M.; Patti, G.; Tornese, G. Sirolimus Therapy in Congenital Hyperinsulinism: A Successful Experience Beyond Infancy. Pediatrics 2015, 136, e1373–e1376. [Google Scholar] [CrossRef] [PubMed]
- Korula, S.; Chapla, A.; Priyambada, L. Sirolimus therapy for congenital hyperinsulinism in an infant with a novel homozygous KCNJ11 mutation. J. Pediatr. Endocrinol. Metab. 2018, 31, 87–89. [Google Scholar] [CrossRef]
- De León, D.D.; Li, C.; Delson, M.I. Exendin-(9–39) corrects fasting hypoglycemia in SUR-1−/− mice by lowering cAMP in pancreatic beta-cells and inhibiting insulin secretion. J. Biol. Chem. 2008, 283, 25786–25793. [Google Scholar] [CrossRef]
- Calabria, A.C.; Li, C.; Gallagher, P.R. GLP-1 receptor antagonist exendin-(9–39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes 2012, 61, 2585–2591. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.S. Recent updates in the management of infants and children with hyperinsulinism. Curr. Opin. Pediatr. 2021, 33, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Avatapalle, B.; Padidela, R. Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin. Endocrinol. 2013, 78, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.J.; Boddaert, N.; Delzescaux, T. Functional imaging of the pancreas: The role of [18F]fluoro-L-DOPA PET in the diagnosis of hyperinsulinism of infancy. Endocr. Dev. 2007, 12, 55–66. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).