Abstract
Thyroid cancer (TC) represents the most common endocrine malignancy, with an increasing incidence all over the world. Papillary TC (PTC), a differentiated TC subtype, is the most common and, even though it has an excellent prognosis following radioiodine (RAI) ablation, it shows an aggressive behavior in 20–30% of cases, becoming RAI-resistant and/or metastatic. On the other side, anaplastic thyroid carcinoma (ATC), the most undifferentiated TC, is a rare but devastating disease, indicating that progression of differentiated to undifferentiated forms of TC could be responsible for RAI-resistance and increased mortality. The epithelial-to-mesenchymal transition (EMT) plays a pivotal role in both tumor progression and resistance to therapy. Moreover, during tumor progression, cancer cells modify their metabolism to meet changed requirements for cellular proliferation. Through these metabolic changes, cancer cells may adopt cancer stem cell-like properties and express an EMT phenotype. EMT, in turn, can induce metabolic changes to which cancer cells become addicted. Here we review metabolic reprogramming in TC highlighting the role of EMT with the aim to explore a potential field to find out new therapeutic strategies for advanced-stage PTC. Accordingly, we discuss the identification of the metabolic enzymes and metabolites, critical to TC progression, which can be employed either as predicting biomarkers of tumor response to RAI therapy or possible targets in precision medicine.
1. Introduction
Thyroid cancer (TC) represents the most common endocrine malignancy all over the world, with a steady increase in both the incidence and the mortality rate for the more aggressive forms [1]. According to the most recent epidemiologic studies in United States, TC incidence increased, on average, 3.6% per year during the period 1974–2013, mainly due to an increase in the incidence of papillary thyroid carcinoma (PTC) [1], and it has been estimated that by 2030 TC will be the fourth leading cancer diagnosis in the United States [2]. Accordingly, a recent deep analysis of the Global Burden of Disease 2019 database has calculated that the global incidence of TC has continued to increase in the past three decades [3]. Some of the highest TC incidence worldwide has been reported in Italy where, under the age of 45, TC was the second most common cancer among women (after breast cancer), and the fifth most common among men [4]. The most frequent TC (84% of all TC) is PTC, a differentiated TC (DTC) deriving from epithelial follicular cells. It is generally characterized by an indolent growth and a good prognosis after adjuvant radioiodine (RAI) treatment; the 5-year relative survival rate for patients who had TC diagnosed during the period 2008–2014 was 98%, and it refers mainly to the most prevalent PTC [5]. However, 20–30% of PTC cases show a more aggressive behavior and patients experience relapse/persistence and/or development of lymph node and visceral metastases with consequent increased mortality, despite the use of targeted therapeutic options, such as tyrosine kinase inhibitors (TKI), including sorafenib and lenvatinib [6,7]. During 1994–2013, incidence-based mortality increased 2.9% per year for advanced-stage PTC [1]. Due to the high global incidence of PTCs, the percentage of those RAI-resistant (RAI-R) has a significant impact and it is therefore imperative to find new therapeutic strategies. The aim of our review is to analyze the possibility that the intercross between epithelial-to-mesenchymal transition (EMT) and metabolism could be exploited to find such strategies. These aggressive forms of PTC exhibit loss of differentiation characteristics, including loss of sodium iodine symporter expression/function, resulting in RAI treatment failure and high mortality. At the molecular level, this loss of differentiation is related to the degree of activation of the mitogen-activated protein kinase (MAPK), which is highest in tumors with BRAF mutations [8].
On the other side, anaplastic thyroid carcinoma (ATC), the most undifferentiated TC, is a rare but devastating disease. It accounts for only 2–5% of all TC cases and is associated with a median overall survival (OS), greatly improved in the last years thanks to the targeted therapy, of 15.7 months, a median 1-year survival of 59%, and a median 2-year survival of 42%, despite aggressive multimodal management [9,10,11]. Current management of ATC consists primarily of surgical resection, combined with adjuvant chemoradiation followed by targeted therapy (dabrafenib and trametinib therapy in patients harboring the BRAF V600E mutation) [12]. The pathogenesis of ATC is still debated. Most studies support a gradual dedifferentiation from DTC to poorly differentiated thyroid carcinoma (PDTC), and eventually to ATC, with the progressive accumulation of somatic pro-cancer mutations. This is supported by the fact that 18–37% of ATC cases result from longstanding goiters or DTC lesions, where ATC occurs concurrently in 30–89% of cases, and ATC sometimes develops following treatment failure of DTC and PDTC. Genomic analyses have further demonstrated shared mutations between co-existing ATC and DTC or PDTC lesions, suggesting a common parent cell [13]. Another theory states that ATC could arise from cancer stem cells (CSCs) that are derived from adult stem cells present within a thyroid niche having accumulated genetic mutations that drive the tumor development [14]. For both theories, EMT plays a pivotal role. In fact, a DTC could lead to ATC as a result of either a dedifferentiation process or the development of CSCs, and both depend on EMT. CSCs are in turn the main responsible of cancer resistance [15], and therefore EMT is a cellular process associated with both tumor progression and TC resistance to therapy. Hence, understanding the biology of EMT and the reverse mesenchymal-to-epithelial transition (MET) process should lead to the design of more effective drugs to target cancer cells, including CSCs.
During tumor progression, cancer cells modify their metabolism to meet the changed requests for cellular proliferation [16]. Several studies have shown similar metabolic alterations occurring in and between cancer cells, including changes in glucose metabolism that result in the Warburg effect, and an increase in biosynthetic activities (such as the synthesis of nucleotides, lipids and amino-acids), which are important for cellular proliferation [15]. Through these metabolic changes, cancer cells may adopt CSC-like properties and express an EMT phenotype [17]. In particular, most tumor cells show highly activated glycolysis through increased expression of glycolytic enzymes or the expression of enzyme isoforms that are not expressed in normal differentiated cells [18]. Indeed, increased glycolysis and glutaminolysis are necessary to maintain rapid cell proliferation, tumor progression, and resistance to cell death [19]. Consequently, tumor cells secrete acidic products, such as lactic acid, thus creating an acidic microenvironment, which has been reported to induce EMT in tumor cells [20,21]. EMT, in turn, strengthen the glycolytic pathway by inducing lactate dehydrogenase gene expression [22].
2. The Warburg and Reverse Warburg Effects
Carcinoma cells show preferential use of lactate-generating glycolysis over the more energy-efficient route of oxidative phosphorylation (OXPHOS), which produces more ATP per glucose molecule than glycolysis [23,24,25,26,27]. This altered metabolism, named “Warburg effect”, implies that cancer cells have increased glucose uptake and lactate secretion, and allows cancer cells to gain an advantage in terms of growth and survival, possibly due to increased carbon utilization, hypoxic adaptation, and increased rate of ATP production [28]. More recently, similar metabolic changes have been described in cancer-associated fibroblasts (CAFs) present in the tumor microenvironment (TME), often as a result of oxidative stress induced by hydrogen peroxide secreted by cancer cells. CAFs in turn increase their own production of reactive oxygen species (ROS), resulting in the induction of aerobic glycolysis and consequent production and secretion of lactate and pyruvate. These metabolites are transferred to cancer cells via inflammation, where they are metabolized into mitochondria to generate new ATP, which assists tumor progression. This metabolic interplay between different tumor cell compartments is called “reverse Warburg effect” and facilitates cancer cell anabolism through catabolic reactions pursued by the TME [29,30,31,32,33]. The reverse Warburg effect can occur not only between CAFs and tumor cells but also between different tumor cells, one of which being hypoxic and hypersecreting intermediate catabolites such as lactate and glutamine. Metabolic coupling with glycolysis occurring in some cancer cells and OXPHOS in other cancer cells promotes cell proliferation and survival. In this multi-compartment metabolism, a key role is played by the lactate monocarboxylate transporters MCT-1 and MCT-4, which mediate the influx into the cell and the efflux from the cell, respectively [34] (Figure 1).
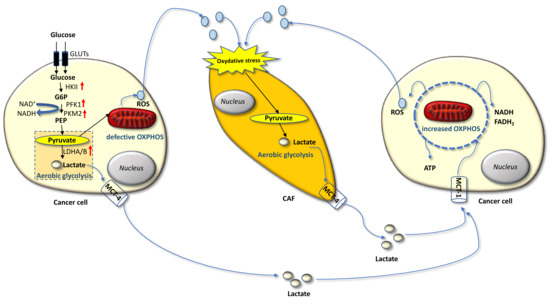
Figure 1.
Warburg and reverse Warburg effect. The cancer cell on the left is undergoing the Warburg effect consisting in the metabolic switch from OXPHOS to aerobic glycolysis, which implies increased glucose uptake and secretion of lactate. Cancers cells also establish a metabolic coupling with cancer-associated fibroblasts (CAFs) and other cancer cells: secretion of reactive species, such as hydrogen peroxide in the tumor microenvironment (TME) induces oxidative stress into a neighboring CAF, which hence engages aerobic glycolysis and generates lactate. This in turn is secreted into the TME and fuels OXPHOS in the cancer cell on the right, thus getting efficient ATP production and promoting survival and proliferation. Lactate monocarboxylate transporters mediate efflux (MCT-4) and the influx (MCT-1) of the lactate from and into the cell.
3. Metabolic Reprogramming in Thyroid Cancer
Thyroid gland is actively involved in thyroid hormone synthesis, an oxidative process that requires a lot of energy. For this reason, thyroid cells are always metabolically active and employ mitochondria to produce the necessary energy through OXPHOS while releasing ROS. Therefore, to counteract the high levels of ROS, they have evolved several antioxidant systems, which are enhanced in DTC but result inactivated in poorly differentiated TC where, consequently, an increase in oxidative damage is observed [35,36]. One of the major antioxidative systems is the Warburg effect that enhances biosynthetic fluxes and antioxidant defense during rapid proliferation of cancer cells. This metabolic reprogramming is regulated by transcription factors such as the hypoxia inducible factor 1 alpha (HIF-1α) that activates either glycolytic enzymes or glucose and lactate transporters while inhibiting OXPHOS [37,38]. Consistently, overexpression of HIF-1α, as well as hexokinase 2 (HK2), phosphoglycerate kinase (PGK), glucose-6-phosphate dehydrogenase (G6PDH), lactate dehydrogenase A (LDHA), glucose transporter 1 (GLUT1), and MCT4 has been observed in TC, associated with distant metastases [39,40,41,42,43,44]. The thyroid TME, consisting of fibroblasts, cells of immune system and endothelial cells, also contributes to the metabolic reprogramming of TC cells. In particular, experimental evidence supports a metabolic symbiosis between cancer cells and CAFs in TC, consisting of the aforementioned reverse Warburg effect [42,45]. Indeed, MCT4 was found to be overexpressed in stromal cells associated with advanced PTC and ATC [42].
3.1. Glucose Metabolism
Metabolic rewiring towards an enhanced glycolytic phenotype primarily involves increased glucose uptake and glycolysis flux, mitochondrial dysfunction, and a more acidic TME, playing a critical role in tumor aggressiveness. In other words, malignant tumor cells alter their glucose metabolism to enhance aerobic glycolysis so that they can maintain their metastatic potential.
In a recent study, glucose metabolic gene expression data in PTCs from The Cancer Genome Atlas (TCGA), including 52 normal tissues and 486 PTCs, were analyzed, showing a significant upregulation of the pyruvate kinase PKM2, the isocitrate dehydrogenase IDH2, the hexokinase HK2 and lactate dehydrogenase LDHA in tumors versus normal tissues. LDHA expression levels, in particular, positively correlate with the tall cell variant, which has a more aggressive clinical behavior compared with the classical PTCs and the more advanced tumor-node-metastasis (TNM) stage. Consistently, in 185 PTCs analyzed by immunohistochemistry, high expression levels of LDHA were associated with aggressive clinicopathological features and poor prognosis, and LDHA expression level resulted an independent prognostic marker for PTCs as it is the TNM stage [46].
3.2. Aminoacidic Metabolism
Amino acids metabolism has a critical role in maintaining cellular metabolic homeostasis. Among all amino acids, glutamine has the greatest consumption during tumor progression and is considered the most important substrate of the cancer cells. It has an essential role in nucleotide and non-essential amino acids synthesis, as well as in providing substrates for the tricarboxylic acid (TCA) cycle, which fuels tumor growth [47]. In particular, TCA cycle is maintained by glutamic acid derived from the conversion of glutamine through the process of glutaminolysis. Consistently, glutamic acid has been found increased in the plasma of patients with thyroid nodules, consisting of 19 PTCs and 16 multinodular goiters, compared to 20 healthy controls [48]. In this pilot study, a panel of significantly altered metabolites, including some associated with amino acids metabolism, such as cysteine and cystine as well as glutamic acid, was identified by untargeted gas chromatography-mass spectrometry in the plasma of patients with PTC nodules compared to healthy subjects. Differently from glutamic acid, cysteine and cystine were decreased in PTC patients and their levels correlated with the tumor stage [48].
Conversely, in a previous study, cysteine and most amino acids were found significantly up-regulated in PTC tissue (collected from 57 patients) compared to adjacent non-tumor tissue [49]. Cysteine is a precursor for glutathione (GSH) biosynthesis, which plays an essential role in supporting intracellular redox homeostasis by extinguishing ROS from mitochondrial respiration. Cancer cells require exogenous cysteine for GSH synthesis to protect themselves from ROS in order to maintain cell proliferation and resistance to apoptosis [50]. Therefore, decreased plasma levels of cysteine and cystine in patients with thyroid nodules may be explained by the higher consumption of cysteine in the cancer cells. Consistently, in the study by Abooshahab and coworkers, significantly altered metabolites between PTC nodules and healthy persons were also associated with GSH biosynthesis. Overall, they found that the metabolism of about 11 amino acids, including metabolites related to GSH biosynthesis, but also methionine, glycine, serine, threonine, and phenylalanine, had been changed in plasma of patients with PTC nodules compared to healthy subjects. Moreover, the TCA cycle, fatty acids (FA), and purine and pyrimidine metabolism were significantly changed as well [48].
3.3. Lipid Metabolism
Lipids can affect cellular functions including cell cycle, proliferation, growth, and differentiation, by serving as second messenger, thus leading to carcinogenesis. Additionally, they can promote the interaction between cancer cells and adjacent immune cells, supporting tumor growth and progression [51].
In the aforementioned study by Abooshahab and coworkers [48], major alternations of long- and medium-chain FA metabolism, suggestive of an increased FA β-oxidation, were detected in patients with PTC nodules. Reprogramming of lipid metabolism is now recognized as a hallmark of carcinogenesis as are other metabolic changes, such as those related to glucose and glutamine [52], being tightly related to the proliferation, invasion, migration, radiosensitivity, and chemosensitivity of several tumors, including TC. Consistently, in a recent study performed with a total of 497 PTC patients from the Cancer Genome atlas (TCGA) database, lipid metabolism-related genes allowed the identification of molecular subtypes in PTC related to different clinical features, such as the time to relapse, immune score, and patients’ outcome [53]. Furthermore, by studying 50 PTCs and their matched normal thyroid tissues, the same group previously demonstrated that the histone lysine methyltransferase KMT5A acts as an oncogene in PTC, where it correlates with extrathyroidal extension, lymph node metastasis and advanced pathological stage, by upregulating key molecules involved in lipid metabolism, including sterol regulatory element binding protein 1 (SREBP1), Stearoyl-CoA Desaturase (SCD), FAS and Acetyl-CoA carboxylase (ACC) [54].
Interestingly, Wojakowska and coworkers identified a number of FAs and FA esters, including lauric, pentadecanoic, hexadecanoic, heptadecanoic, nonadecanoic, eicosanoic, decanoic, ricinoleic, and monostearin, able to differentiate malignant versus benign thyroid lesions. In more details, using a combination of gas chromatography and mass spectrometry, they analyzed tissue specimens from seven follicular thyroid carcinomas (FTC), four classical variants of PTC (CV-PTC), four follicular variants of PTC (FV-PTC), six medullary thyroid carcinomas (MTC), six ATC, three follicular thyroid adenomas and five normal controls, identifying 28 metabolites, including lipids, carboxylic acids, and saccharides, whose levels were significantly different among different types of thyroid tumors. Some of them, such as increased lactic acid and reduced FA, in particular, were able to discriminate TC from normal tissue, and others, such as myo-inositol phosphate, succinic acid and the above-mentioned FAs, could differentiate malignant versus benign lesions; furthermore, downregulation of gluconic acid and upregulation of citric acid could discriminate CV-PTC from FV-PTC, while decanoic acid ester could differentiate FTC from FV-PTC [55].
4. Thyroid Cancer Progression and Reciprocal Role of Epithelial-Mesenchymal Transition and Metabolic Rewiring
Activation of EMT, a process by which epithelial cancer cells acquire mesenchymal features, is a key determinant of cancer progression toward an invasive and metastatic phenotype. By acquiring mesenchymal features, cancer cells, in fact, lose cell-to-cell junctions and gain the capacity to migrate and invade the basal lamina thanks to a complex reprogramming of transcription through epigenetic changes. In TC progression, the tumor cells undergo EMT, becoming spindle shaped and invading tumor stroma. Molecular changes include reduced E-cadherin expression levels and increased expression of Snail, Slug, Twist, Paired Related Homeobox 1 (PRRX1), and other EMT-related genes. Hence, first intravasation into the blood and/or lymphatic vessels and then extravasation in distant metastatic sites, such as the lymph nodes and lungs, occur. After a variable time in the quiescence state, the tumor cells are subjected to MET to colonize distant organs forming secondary tumors (Figure 2). During this last phase there is a decrease in the expression of Twist and PRRX1 and an increase in the expression of epidermal growth factor (EGFR) and c-Met [56]. Indeed, well-differentiated TC and normal thyroid express high levels of E-cadherin, but do not commonly express Snail and Twist [57]. However, the leading front of PTCs, as well as ATCs, frequently express EMT markers, such as vimentin and Snail, Slug and Twist, but not E-cadherin [56,58,59].
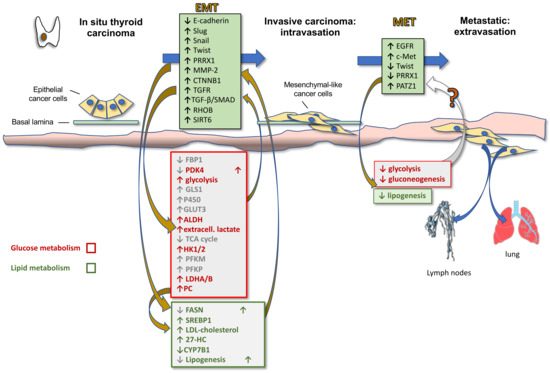
Figure 2.
Thyroid cancer progression: reciprocal role of EMT and metabolic reprogramming. The cartoon illustrates the phases of thyroid cancer progression, from in situ to invasive carcinoma and metastatic tumor, highlighting the molecular actors of EMT as well as their reciprocal relationship with metabolic players. Upregulation (arrow up)/downregulation (arrow down) of proteins demonstrated in other cancers but not yet validated in TC is shown in gray.
During EMT cancer cells also acquire stem cell features that allow them to resist to different treatment options. Based on the CSC hypothesis of TC development, normal follicular cells that accumulate errors can give rise to differentiated cancers, which in turn can develop into undifferentiated cancers following the enrichment of CSCs through the EMT process [13]. This is likely the reason why patients with ATCs, which consist of CSCs and non-CSCs, usually have a relapse after surgery and conventional chemotherapy and radio-iodine [56].
More recently, it has become clear that EMT is also involved in metabolic rewiring needed for the increased energetic demand of the mesenchymal cells compared to their epithelial counterparts due to the increased motility and invasion ability. In fact, EMT induction in epithelial mammary cells by Twist expression upregulates the expression of 44 metabolic genes, including dihydropyrimidine dehydrogenase (DPYD), an enzyme involved in pyrimidine catabolism, that in turn upregulates EMT [60]. Therefore, it is likely that metabolic rewiring is required for completeness of EMT. Other metabolic pathways modulated by EMT include glycolysis, lipid metabolism, mitochondrial metabolism and glutaminolysis. Specifically, it has been shown that EMT induction suppresses the expression of multiple metabolic proteins, including fructose-1.6-bisphosphatase 1 (FBP1), thus resulting in increased glycolysis [61], fatty acid synthase (FASN) and ACC, thus resulting in decreased lipogenesis [62], nucleoside transporter [63], and pyruvate dehydrogenase kinase 4 (PDK4) [64], whilst enhancing the expression of glutaminase 1(GLS1) [65], enzymes of glutathione metabolism, cytochrome P450, aldehyde dehydrogenase, thus accounting for the increased chemoresistance [66], and GLUT3 [67]. On the other side, these metabolic alterations sustain the Warburg effect and induce EMT by enhancing glycolysis and blocking the TCA cycle. In particular, upregulation of (i) GLUT1 and GLUT3 glucose transporters activates matrix metallopeptidase MMP-2, which in turn induces EMT and invasiveness; (ii) HK1 and HK2 hexokinase, involved in the first step of glycolysis, activates Snail and Slug, which in turn induces EMT; (iii) PFKM and PFKP, rate-limiting enzymes of glycolysis, directly induce EMT; (iv) LDHA and LDHB, associated with enhanced glycolysis and lactate production, as well as extracellular lactate, activate Snail and therefore EMT [17].
More specifically in TC, recent studies have shown certain metabolic changes involved in EMT induction and tumor progression (Table 1). Liu et al., examining public datasets and a local cohort of patients, also assisted by in vitro studies, demonstrated that PTC metastases can be mediated by pyruvate carboxylase (PC), the enzyme that catalyzes the carboxylation of pyruvate to form oxaloacetate, through induction of a transforming growth factor β TGF-β-mediated EMT [68]. They also showed that PC increases FA synthesis, which then promotes TC progression and metastases. In particular, PC induces upregulation of multiple genes of the FA synthesis signaling pathway, including SREBP1c and FASN, which in turn are responsible for the increased lipogenesis and are required for the aggressiveness of TC cells [69]. Furthermore, LDHA, the key enzyme that accumulates for the Warburg effect, was identified by Hou et al. as a candidate target gene for PTC. In fact, knockdown experiments in PTC cell lines revealed that LDHA promotes the transcription of EMT-related genes, including catenin beta 1 (CTNNB1), ras homologue family member B (RHOB) and TGF receptor 1 (TGFR1), to promote migration and invasion. The mechanism involves the release of intracellular acetyl-CoA, which enhances the histone acetylation of these EMT-related genes [46]. Consistent with these results, another study showed that LDHA and glycolysis were critical for PTC progression [70]. Still on the role of glycolysis in TC progression, Ren et al. demonstrated that the circular RNA circCCDC66 promotes TC progression by sponging miR-211-5p and thereby increasing the expression of PDK4, which in turn promotes glycolysis [71].

Table 1.
Studies linking metabolic reprogramming to EMT in thyroid cancer progression.
Similarly, previous studies have shown that Sirtuin 6 (SIRT6), which is upregulated in PTC, induces both the Warburg effect, through the upregulation of ROS, and the EMT [72,73,74]. Subsequently, the same authors demonstrated that SIRT6 can also repress the Warburg effect in PTC cell lines. In fact, they showed that SIRT6 acts through accumulation of ROS and consequent increase of endoplasmic reticulum stress and autophagy. Autophagy, in turn, mediates the degradation of GLUT1, thus suppressing the Warburg effect [75]. Indeed, they have shown, through in vitro and in vivo experiments, that a weaker inhibition of ROS activates the Warburg effect by suppressing autophagy, while a stronger inhibition of ROS activates autophagy and repress the Warburg effect [75].
The interplay between lipid metabolism and EMT in TC clearly emerged in the study by Revilla et al., showing that cholesterol and the intra-tumor accumulation of its metabolite 27-hydroxycholesterol (27-HC) promote progression of PTC. In brief, they reported that patients with more aggressive tumors (high-risk PTC, PDTC and ATC) have decreased levels of serum low-density lipoprotein (LDL) cholesterol and apolipoprotein B associated with an increase of intracellular 27-HC and a decrease in the mitochondrial enzyme 25-hydroxycholesterol 7-alpha-hydroxylase (CYP7B1), which is responsible for 27-HC catabolism. Furthermore, they demonstrated that intracellular LDL cholesterol promotes proliferation and migration, while overexpression of CYP7B1 arrested growth and decreased migration of an ATC cell line [76].
Finally, we recently found that ATC cells, in which a partial reversion of EMT was induced by POZ/BTB And AT Hook Containing Zinc Finger 1 (PATZ1) overexpression [77], have reduced levels of several proteins involved in glycolysis/gluconeogenesis, FA metabolism and amino acid biosynthesis, suggesting that the tumor suppressor role of PATZ1 in thyroid carcinogenesis could act through metabolic changes involved in EMT [78]. Overall, the data provided demonstrate that TC progression involves both EMT and metabolic rewiring and suggest that the two processes are related to each other.
5. Clinical Perspectives and Conclusions
The most challenging issue in RAI-R TC is establishing when a patient should be considered RAI-R and when to initiate treatment with other therapeutic options, such as TKI, including sorafenib and lenvatinib [7]. Prediction of an aggressive and RAI-R tumor will aid to avoid ineffective and therefore useless radio-therapy, thus preserving the patient’s well-being and leading to a significant economic saving for the National Health System. Moreover, it is possible that, in an advanced stage of carcinogenesis such as that one of an aggressive PTC, X-rays enhance tumor progression by increasing the risk of accumulating further adverse events and leading to bystander and off-target effects [7,79]. On the other side, early prediction of tumor aggressiveness will ensure that the patient is rapidly enrolled in alternative more specific target therapies.
Exploiting the metabolic rewiring in tumors to treat cancer is an emerging but still poorly explored field in oncology. Here we reviewed the metabolic enzymes and metabolites so far identified as critical for TC progression, which can be employed either as predicting biomarkers of tumor response to RAI therapy or possible targets in precision medicine. Subtle metabolic rewiring of TC cells can be, indeed, exploited in drug development. In particular, manipulation of these metabolite levels through enzymatic regulators may be a new therapeutic option. Moreover, it could suggest dietary support measures during TC treatment.
Author Contributions
Conceptualization, M.F.; methodology, M.F., S.B., L.C.; writing—original draft preparation, M.F.; writing—review and editing, M.F., S.B., L.C.; supervision, M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank Antonella Napolitano for editing support. Fondazione AIRC per la Ricerca sul Cancro is acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in thyroid cancer incidence and mortality in the United States, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Bao, W.Q.; Zi, H.; Yuan, Q.Q.; Li, L.Y.; Deng, T. Global burden of thyroid cancer and its attributable risk factors in 204 countries and territories from 1990 to 2019. Thorac. Cancer 2021, 12, 2494–2503. [Google Scholar] [CrossRef] [PubMed]
- Dal Maso, L.; Lise, M.; Zambon, P.; Falcini, F.; Crocetti, E.; Serraino, D.; Cirilli, C.; Zanetti, R.; Vercelli, M.; Ferretti, S.; et al. Incidence of Thyroid Cancer in Italy, 1991–2005: Time trends and age-period-cohort effects. Ann. Oncol. 2011, 22, 957–963. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef]
- Pacini, F. Which patient with thyroid cancer deserves systemic therapy and when? Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 291–294. [Google Scholar] [CrossRef]
- Aashiq, M.; Silverman, D.A.; Na’ara, S.; Takahashi, H.; Amit, M. Radioiodine-refractory thyroid cancer: Molecular basis of redifferentiation therapies, management, and novel therapies. Cancers 2019, 11, 1382. [Google Scholar] [CrossRef]
- Prasongsook, N.; Kumar, A.; Chintakuntlawar, A.V.; Foote, R.L.; Kasperbauer, J.; Molina, J.; Garces, Y.; Ma, D.; Wittich Neben, M.A.; Rubin, J.; et al. Survival in response to multimodal therapy in anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 2017, 102, 4506–4514. [Google Scholar] [CrossRef] [PubMed]
- Ljubas, J.; Ovesen, T.; Rusan, M. A systematic review of phase II targeted therapy clinical trials in anaplastic thyroid cancer. Cancers 2019, 11, 943. [Google Scholar] [CrossRef]
- Maniakas, A.; Dadu, R.; Busaidy, N.L.; Wang, J.R.; Ferrarotto, R.; Lu, C.; Williams, M.D.; Gunn, G.B.; Hofmann, M.-C.; Cote, G.; et al. Evaluation of overall survival in patients with anaplastic thyroid carcinoma, 2000–2019. JAMA Oncol. 2020, 6, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Kebebew, E.; Brierley, J.; Brito, J.P.; Cabanillas, M.E.; Clark, T.J., Jr.; Di Cristofano, A.; Foote, R.; Giordano, T.; Kasperbauer, J.; et al. 2021 American Thyroid Association Guidelines for management of patients with anaplastic thyroid cancer. Thyroid 2021, 31, 337–386. [Google Scholar] [CrossRef]
- Davies, T.F.; Latif, R.; Minsky, N.C.; Ma, R. The emerging cell biology of thyroid stem cells. J. Clin. Endocrinol. Metab. 2011, 96, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Borah, A.; Raveendran, S.; Rochani, A.; Maekawa, T.; Kumar, D.S. Targeting self-renewal pathways in cancer stem cells: Clinical implications for cancer therapy. Oncogenesis 2015, 4, e177. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Morcavallo, A.; Giuliano, S.; Belfiore, A. Thyroid cancer development and progression: Emerging role of cancer stem cells. Minerva Endocrinol. 2012, 37, 103–115. [Google Scholar]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of metabolic reprogramming in Epithelial-Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef]
- Coelho, R.G.; Fortunato, R.S.; Carvalho, D.P. Metabolic reprogramming in thyroid carcinoma. Front. Oncol. 2018, 8, 82. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Zhang, Y.; Cao, Y.; Wei, H.; Wu, Z. Upregulation of lactate-inducible snail protein suppresses oncogene-mediated senescence through p16 INK4a inactivation. J. Exp. Clin. Cancer Res. 2018, 37, 39. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D. TLR4-mediated galectin-1 production triggers epithelial-mesenchymal transition in colon cancer cells through ADAM10- And ADAM17-associated lactate production. Mol. Cell Biochem. 2017, 425, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hou, Y.; Yuan, J.; Tang, S.; Zhang, H.; Zhu, Q.; Du, Y.; Zhou, M.; Wen, S.; Xu, L.; et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget 2015, 6, 25755–25769. [Google Scholar] [CrossRef]
- Curry, J.M.; Tuluc, M.; Whitaker-Menezes, D.; Ames, J.A.; Anantharaman, A.; Butera, A.; Leiby, B.; Cognetti, D.M.; Sotgia, F.; Lisanti, M.P.; et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle 2013, 12, 1371–1384. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar]
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Power surge: Supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012, 15, 4–5. [Google Scholar] [CrossRef]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Whitaker-Menezes, D.; Dasgupta, A.; Philp, N.J.; Lin, Z.; Gandara, R.; Sneddon, S.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: Stromal MCT4 predicts poor clinical outcome in triple negative breast cancers. Cell Cycle 2012, 11, 1108–1117. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vãgran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Karbownik-Lewiålnska, M.; Kokoszko-Bilska, A. Oxidative damage to macromolecules in the thyroid—Experimental evidence. Thyroid Res. 2012, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Rovcanin, B.R.; Gopcevic, K.R.; Kekic, D.L.; Zivaljevic, V.R.; Diklic, A.D.; Paunovic, I.R. Papillary thyroid carcinoma: A malignant tumor with increased antioxidant defense capacity. Tohoku J. Exp. Med. 2016, 240, 101–111. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenzaet, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Klaus, A.; Fathi, O.; Tatjana, T.; Bruno, N.; Oskar, K. Expression of hypoxia-associated protein HIF-1α in follicular thyroid cancer is associated with distant metastasis. Pathol. Oncol. Res. 2018, 24, 289–296. [Google Scholar] [CrossRef]
- Burrows, N.; Babur, M.; Resch, J.; Williams, K.J.; Brabant, G. Hypoxia-inducible factor in thyroid carcinoma. J. Thyroid Res. 2011, 2011, 17. [Google Scholar] [CrossRef]
- Chen, M.; Shen, M.; Li, Y.; Liu, C.; Zhou, K.; Hu, W.; Xu, B.; Xia, Y.; Tang, W. GC-MS-based metabolomic analysis of human papillary thyroid carcinoma tissue. Int. J. Mol. Med. 2015, 36, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Nahm, J.H.; Kim, H.M.; Koo, J.S. Glycolysis-related protein expression in thyroid cancer. Tumor Biol. 2017, 39, 3. [Google Scholar] [CrossRef]
- Grabellus, F.; Nagarajah, J.; Bockisch, A.; Schmid, K.W.; Sheu, S.-Y. Glucose transporter 1 expression, tumor proliferation, and iodine/glucose uptake in thyroid cancer with emphasis on poorly differentiated thyroid carcinoma. Clin. Nucl. Med. 2012, 37, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Haber, R.S.; Weiser, K.R.; Pritsker, A.; Reder, I.; Burstein, D.E. Glut1 glucose transporter expression in benign and malignant thyroid nodules. Thyroid 1997, 7, 363–667. [Google Scholar] [CrossRef]
- Gill, K.S.; Tassone, P.; Hamilton, J.; Hjelm, N.; Luginbuhl, A.; Cognetti, D.; Tuluc, M.; Martinez-Outschoorn, U.; Johnson, J.M.; Curry, J.M. Thyroid cancer metabolism: A review. J. Thyroid Disord. Ther. 2016, 5, 200. [Google Scholar] [CrossRef]
- Hou, X.; Shi, X.; Zhang, W.; Li, D.; Hu, L.; Yang, J.; Zhao, J.; Wei, S.; Wei, X.; Ruan, X.; et al. LDHA induces EMT gene transcription and regulates autophagy to promote the metastasis and tumorigenesis of papillary thyroid carcinoma. Cell Death Dis. 2021, 12, 347. [Google Scholar] [CrossRef]
- Luo, M.; Brooks, M.; Wicha, M.S. Asparagine and glutamine: Co-conspirators fueling metastasis. Cell Metab. 2018, 27, 947–949. [Google Scholar] [CrossRef] [PubMed]
- Abooshahab, R.; Hooshmand, K.; Razavi, S.A.; Gholami, M.; Sanoie, M.; Hedayati, M. Plasma metabolic profiling of human thyroid nodules by Gas Chromatography-Mass Spectrometry (GC-MS)-based untargeted metabolomics. Front. Cell Dev. Biol. 2020, 16, 8–385. [Google Scholar] [CrossRef]
- Xu, Y.; Zheng, X.; Qiu, Y.; Jia, W.; Wang, J.; Yin, S. Distinct metabolomic profiles of papillary thyroid carcinoma and benign thyroid adenoma. J. Proteome Res. 2015, 14, 3315–3321. [Google Scholar] [CrossRef]
- Combs, J.A.; DeNicola, G.M. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers 2019, 11, 678. [Google Scholar] [CrossRef]
- Matsushita, Y.; Nakagawa, H.; Koike, K. Lipid metabolism in oncology: Why It matters, how to research, and how to treat. Cancers 2021, 13, 474. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Xu, M.; Sun, T.; Wen, S.; Zhang, T.; Wang, X.; Cao, Y.; Wang, Y.; Sun, X.; Ji, Q.; Shi, R.; et al. Characteristics of lipid metabolism-related gene expression-based molecular subtype in papillary thyroid cancer. Acta Biochim. Biophys. Sin. 2020, 52, 1166–1170. [Google Scholar] [CrossRef]
- Liao, T.; Wang, Y.J.; Hu, J.Q.; Wang, Y.; Han, L.T.; Ma, B.; Shi, R.L.; Qu, N.; Wei, W.J.; Guan, Q.; et al. Histone methyltransferase KMT5A gene modulates oncogenesis and lipid metabolism of papillary thyroid cancer in vitro. Oncol. Rep. 2018, 39, 2185–2192. [Google Scholar] [CrossRef]
- Wojakowska, A.; Chekan, M.; Marczak, Ł.; Polanski, K.; Lange, D.; Pietrowska, M.; Widlak, P. Detection of metabolites discriminating subtypes of thyroid cancer: Molecular profiling of FFPE samples using the GC/MS approach. Mol. Cell. Endocrinol. 2015, 417, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hardin, H.; Lloyd, R.V. Cancer stem-like cells and thyroid cancer. Endocr. Relat. Cancer 2014, 21, T285–T300. [Google Scholar] [CrossRef] [PubMed]
- Buehler, D.; Hardin, H.; Shan, W.; Montemayor-Garcia, C.; Rush, P.S.; Asioli, S.; Chen, H.; Lloyd, R.V. Expression of epithelial–mesenchymal transition regulators SNAI2 and TWIST1 in thyroid carcinomas. Mod. Pathol. 2013, 26, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.G.; Vicente-Dueñas, C.; González-Herrero, I.; Anderson, C.; Flores, T.; Hughes, S.; Tselepis, C.; Ross, J.A.; Sánchez-García, I. Snail family transcription factors are implicated in thyroid carcinogenesis. Am. J. Pathol. 2007, 171, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Vasko, V.; Espinosa, A.V.; Scouten, W.; He, H.; Auer, H.; Liyanarachchi, S.; Larin, A.; Savchendo, V.; Francis, G.L.; de la Chapelle, A.; et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc. Natl. Acad. Sci. USA 2007, 104, 2803–2808. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Freinkman, E.; Comb, W.C.; Cantor, J.R.; Tam, W.L.; Thiru, P.; Kim, D.; Kanarek, N.; Pacold, M.E.; Chen, W.W.; et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014, 158, 1094–1109. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Daemen, A.; Hatzivassiliou, G.; Arnott, D.; Wilson, C.; Zhuang, G.; Gao, M.; Liu, P.; Boudreau, A.; Johnson, L.; et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab. 2014, 2, 20. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Couto, K.; Jha, A.; Choe, S.; Wang, A.; Woo, H.-K.; Steadman, M.; DeLaBarre, B.; Gross, S.; Driggers, E.; et al. Mesenchymal phenotype predisposes lung cancer cells to impaired proliferation and redox stress in response to glutaminase inhibition. PLoS ONE 2014, 9, e115144. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, L.; Liu, Y.; Zhao, Q.; Pan, Y.; Zhang, Y. Value of pyruvate carboxylase in thyroid fine-needle aspiration wash-out fluid for predicting papillary thyroid cancer lymph node metastasis. Front. Oncol. 2021, 11, 643416. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, X.; Pan, Y.; Liu, Y.; Zhang, Y. Pyruvate carboxylase promotes thyroid cancer aggressiveness through fatty acid synthesis. BMC Cancer 2021, 21, 722. [Google Scholar] [CrossRef]
- Huo, N.; Cong, R.; Sun, Z.J.; Li, W.C.; Zhu, X.; Xue, C.Y.; Chen, Z.; Ma, L.Y.; Chu, Z.; Han, Y.C.; et al. STAT3/LINC00671 axis regulates papillary thyroid tumor growth and metastasis via LDHA-mediated glycolysis. Cell Death Dis. 2021, 12, 799. [Google Scholar] [CrossRef]
- Ren, H.; Song, Z.; Chao, C.; Mao, W. circCCDC66 promotes thyroid cancer cell proliferation, migratory an invasive abilities and glycolysis through the miR-211-5p/PDK4 axis. Oncol. Lett. 2021, 21, 416. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Hu, J.Q.; Liu, L.; Zhang, T.-T.; Sun, G.-H.; Shi, R.-L.; Ji, Q.-H. SIRT6 is upregulated and associated with cancer aggressiveness in papillary thyroid cancer via BRAF/ERK/Mcl1 pathway. Int. J. Oncol. 2017, 50, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Yang, Z.; Huang, R.; Min, Z.; Ye, M. SIRT6 promotes the Warburg effect of papillary thyroid cancer cell BCPAP through reactive oxygen species. OncoTargets Ther. 2019, 12, 2861–2868. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yu, W.; Huang, R.; Ye, M.; Min, Z. SIRT6/HIF-1a axis promotes papillary thyroid cancer progression by inducing epithelial-mesenchymal transition. Cancer Cell Int. 2019, 19, 17. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, R.; Wei, X.; Yu, W.; Min, Z.; Ye, M. SIRT6-autophagy-Warburg effect axis in papillary thyroid cancer. Front. Oncol. 2020, 10, 1265. [Google Scholar] [CrossRef]
- Revilla, G.; de Pablo Pons, M.; Baila-Rueda, L.; García-León, A.; Santos, D.; Cenarro, A.; Magalhaes, M.; Blanco, R.M.; Moral, A.; Pérez, J.I.; et al. Cholesterol and 27-hydrocholesterol promote thyroid carcinoma aggressiveness. Sci. Rep. 2019, 9, 10260. [Google Scholar] [CrossRef]
- Chiappetta, G.; Valentino, T.; Vitiello, M.; Pasquinelli, R.; Monaco, M.; Palma, G.; Sepe, R.; Luciano, A.; Pallante, P.; Palmieri, D.; et al. PATZ1 acts as a tumor suppressor in thyroid cancer via targeting p53-dependent genes involved in EMT and cell migration. Oncotarget 2015, 6, 5310–5323. [Google Scholar] [CrossRef]
- Chiappetta, G.; Vinh, J.; Paris, D.; Palomba, L.; Motta, A.; Battista, S.; Cerchia, L.; Fedele, M. Uncovering the downstream signaling landscape responsible for PATZ1-mediated reversion of malignant phenotype in anaplastic thyroid cancer cells: A metabolic perspective. In Proceedings of the SIBBM 2021—Frontiers in Metabolic Research, Online, 7–10 June 2021. [Google Scholar]
- Pouget, J.P.; Georgakilas, A.G.; Ravanat, J.L. Targeted and off-target (bystander and abscopal) effects of radiation therapy: Redox mechanisms and risk/benefit analysis. Antioxid. Redox Signal. 2018, 29, 1447–1487. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).