Abstract
Endocrine axes (prolactin, thyroid and adrenal axes) directly and indirectly modulate and drive human female central functions, mainly behavior and reproduction. Though having distinct abilities, they greatly act both at peripheral as well as at neuroendocrine levels, so as to participate in the control of reproduction. Any event that changes these balanced activities produces specific peripheral signals that induce abnormal functions centrally, thus triggering menstrual disorders such as oligomenorrhea or amenorrhea. It is clear that the knowledge of the relationships that exist between the different endocrine axes becomes essential for the choice of therapeutical approach. This review aims to focus on the main aspects of the physiopathology of the endocrine diseases that might be at the basis of that interference with female reproductive capacity.
Keywords:
hypothyroidism; PRL; hyperthyroidism; hyperandrogenism; adrenal axis; anovulation; reproductive axis 1. Introduction: Endocrine System and Reproductive Ability
Female biology is a complex system ruled by various and different endocrine axes: despite each axis having its own different role, they still manage to work together effectively [1]. As for the biological functions, the good performance of the reproductive system depends on the balanced activity of various elements of the axis, (i.e., the hypothalamus, the pituitary and the ovary) and the other hormonal axes.
The pituitary gland has distinct and diversified cell nuclei (gonadotropic, thyrotropic, lactotrophs, somatotropic and corticotropic cells), which control/regulate all the endocrine axes in concert and modulate the reproductive axis.
During the menstrual cycle, the LH secretion profile shows pulsating changes with a variable amplitude and frequency, thus enabling the recruitment and the choice of follicles. Many alterations in reproductive capacity are primarily linked to a modification of those fine mechanisms that regulate ovulation induction and follicular recruitment. It is well known that gonadotropins secretion is under the modulation of a number of steroids (i.e., estrogen, androgens, cortisol, etc.) as well as of neuropeptides (i.e., opioids, catecholamines, neuropeptides) that act directly on GnRH or kisspeptin neurons as well as on gonadotrope cells.
The whole reproductive system depends on GnRH neurons’ secretory activity and around the receptors expression, which are modulated by positive and negative factors so as to activate or impair their functions. The biggest driving force of GnRH discharge is kisspeptin, whose receptors are expressed on GnRH neurons [2,3]. As for many neuroendocrine systems, kisspeptin is extremely sensitive to the many signal coming from the periphery—mainly metabolic signals such as Ghrelin, cholecystokinin, leptin and insulin—and it greatly affected by changes coming from the gastro-intestinal tract [4]. Therefore, also all these modulators of kisspeptin and GnRH release indirectly affect the release of gonadotropins. In fact, a number of neuropeptides and neurotransmitters released inside the central nervous system (CNS) act on the modulation of the release of GnRH, thus affecting the reproductive functions. Stressful situations such as psychological, physical or metabolic stressors, together with weight fluctuations, such as loss of weight or obesity, can induce an impaired neuroendocrine function at the hypothalamic level through the impaired production of amines, beta-endorphin, and serotonin [5]. All of them can interfere in the regulation of GnRH, thus influencing the reproductive capacity overall [6]. Obviously, gonadal steroids also play a crucial role in the modulation of the GnRH-induced gonadotropin secretion, since estrogen and androgens are able to greatly interfere in the amount of LH released from the pituitary through the feedback control system [7]. The major role exerted by estrogen has been recently demonstrated in some physiopathological conditions since estriol administration was able to induce a greater response of gonadotrope cells to both exogenous and endogenous GnRH stimulation [8,9].
In addition, impairments of the main endocrine axes that regulate our biology (thyroid, adrenal and prolactin axis) have been demonstrated to cause changes in the reproductive ability and in the menstrual cyclicity. Our biological system is dynamic and interactive, based on genetic and epigenetic relationships and conditioned by environmental factors in most cases.
2. Impaired Gonadotropin Release, Altered Menstrual Cyclicity and Body Weight
Whenever the menstrual cyclicity is altered, a menstrual and ovulatory impairment occurs. Menstrual irregularity is defined when cycles occur within 21 days or longer than 40 days; the elements that can interfere with menstrual cyclicity are various and greatly non-specific: it is important to know what they are and how they act.
The endocrine factors that trigger the chronic anovulation and disrupt the control system of the reproductive axis are: hyperandrogenic states, of adrenal or ovarian origin, hypo- or hyperthyroidism and hyperprolactinemia; peripheral signals, in particular the balance of metabolism, are extremely important and can also interfere greatly with the control of reproduction [1,10].
In fact, obesity and malnutrition, the two opposite aspects of dysmetabolism, are particularly critical because they block the reproductive system, inducing impaired hypothalamic activity aspects as a defensive mechanism to avoid pregnancy and adverse negative pregnancy-induced side effects. The quality and quantity of the nutrients of our diet strongly affect the reproductive capacity since they modulate, directly and indirectly through specific hormonal signals from the various organs of the gastro-intestinal tract, the hypothalamic functions [11,12]. The incidence of overweight–obesity in the Italian population is around 25%, being 5% in 1952: reproductive biology has certainly been affected by this trend. In fact, a number of studies have been carried out on how reduced or increased BMI are responsible for impaired reproductive function through the reduced or excessive release of the specific neuroendocrine modulators that act on the kisspeptin–GnRH neurons and impair the LH secretory pattern and reproduction [2,13]. Moreover, a number of studies clearly report that a lack or excess of adequate elements from nutrition create specific impaired neuroendocrine responses that are intended to reduce or block follicle recruitment and then reproduction [5,14,15,16].
It is clear that a good weight control can improve in any case the functional performance of our reproductive axis. In particular, though adipose tissue is not an endocrine axis, it manages the synthesis of hormones like an endocrine organ. It is clear that a situation of overweight up to obesity can change not only the adiponoktines and leptin produced, but also the steroids milieu. Any excess of these substances is able to interfere in the hypothalamic functions, together with insulin acting on kisspeptin neurons, thus affecting GnRH neurons’ activity [17,18]. Moreover, adipocytes produce androstenedione, estrone and estriol, and can absorb therapeutically administered steroids [15]. Such estrogen and progestogen uptake permits their release back into circulation in variable moments and amounts later, causing severe fluctuations of plasma concentrations [7]. This fact impacts the optimal function of contraceptive steroids, especially in overweight/obese women, since the side effect of occur intermenstrual spotting frequently occurs (15% in overweight compared to 3% in normal-weight) [7].
It is therefore very important to teach patients to follow healthy eating attitude, in order to limit the deposition of adipose tissue and to have an optimal body weight. In addition, excess weight also affects the muscle, the neuroendocrine systems, the brain cortex, the thalamus, the hippocampus (mood, sleep, behavior), and the autonomic system in general [15].
3. Hyperandrogenism
Androgens originate from both the ovary and the adrenal gland. In primates, androgens are essential for the function of the cerebral cortex, because they increase critical abilities such as logic, memory, adaptation, libido and sexuality [19].
In fact, the production of androgens from the ovary is reduced with menopause: this causes the gradual decrease in these abilities, in particular libido and sexuality. On the contrary, in men, androgens tend to be more stable throughout their lifespan. However, the adrenal gland decreases its activity in a chronologically similar way in both men and women.
Gonadal steroids are essential regulators of reproductive function. Studies performed on knockout mice lacking an androgen receptor at the ovarian level demonstrated that folliculogenesis does not operate properly in the absence of the androgen signal [20,21]. On the other hand, follicle development is impaired when there is an excess of androgens; this happens with polycystic ovary syndrome (PCOS), a condition showing both hyperandrogenism and anovulation and often, infertility [20]. The relevance of the androgen signal not only at the ovarian level but also centrally has been disclosed properly on specific animal models in which the lack of androgen receptors or their blockade using flutamide (an androgen receptor blocker) restore a normal neuroendocrine control of gonadotropin release and follicle recruitment, avoiding a PCOS-like situation [22].
Hyperandrogenism can also be triggered by a chronic situation of stress, and, if coupled with ovarian hyperandrogenism, might generate a series of consequences.
The adrenal gland releases cortisol, the stress hormone, with precise circadian rhythms. Cortisol can modulate the reproductive axis with central actions, affecting the synthesis of gonadotropins, and on the other hand, can act directly on the ovary, interfering with the synthesis and release of estrogen and, during the luteal phase, of progesterone. Stressful situations cause cortisol elevation, and excess cortisol slows down the reproductive axis due to the direct effect of cortisol on hypothalamic nuclei: when stress becomes chronic, menstrual disorders occur more frequently, even up to amenorrhea [23]. In fact, high levels of glucocorticoids inhibit GnRH neurons’ activity, thus affecting gonadotrope cells and gonadal function [24]. CRH (corticotrophin-releasing hormone) infusion during the mid-luteal phase reduces plasma LH and FSH levels, while their concentrations return to normal levels as soon as CRH infusion is stopped. Interestingly, CRH infusion did not alter the gonadotropin response to GnRH bolus, thus confirming that CRH acts at the hypothalamic level [25].
The key point is a restraint action on aromatase, the enzyme of granulosa cells that converts androgens (androstenedione and testosterone) into estradiol. Aromatase is sensitive to alterations such as an excess of androgens, prolactin and hyperinsulinemia. By modulating the activity of aromatase, the androgen levels change accordingly [15].
Additionally, stress through adrenal gland hyperactivation increases cortisol and through this gluconeogenesis, thus favoring obesity and hyperandrogenism; this leads to a growing endocrine impairment and might favor greater cardiovascular risk, the primary cause of death in women over 50, in the following 30 years.
Moreover, the excess of cortisol interferes with insulin and growth factors: too much gluconeogenesis contributes to the higher availability of glucose and insulin, promoting mechanisms that predispose one to a greater oncological risk [26,27].
In PCOS, hyperandrogenism is the key element that induces anovulation. It is well known that PCOS occurs when there is the presence of at least two out of three revised Rotterdam Consensus Conference criteria (i.e., hyperandrogenemia, oligo/amenorrhea, polycystic ovaries upon ultrasound) [28]. Too many androgens limit the ovarian function, making ovaries assume the classic PCO morphology. If we assume that a PCO ovary is the only cause of the hyperandrogenic state, we might miss the real cause of anovulation.
Another element to consider is hyperinsulinemia, classically present in obese PCOS: insulin stimulates GnRH release and consequently gonadotropins secretion that stimulate the production of ovarian steroids (especially androstenedione) with inhibitory action on aromatase, resulting in an excess of androgens not converted to estrogen. Insulin also inhibits sex hormone-binding globulin (SHBG) with an increase in free androgens (Figure 1). It is clear that hyperinsulinemia represents a fundamental issue in the case of PCOS, since it favors the condition of hyperandrogenism with greater amounts of free plasma androgens [29] and specific negative effects on liver function [17,30].
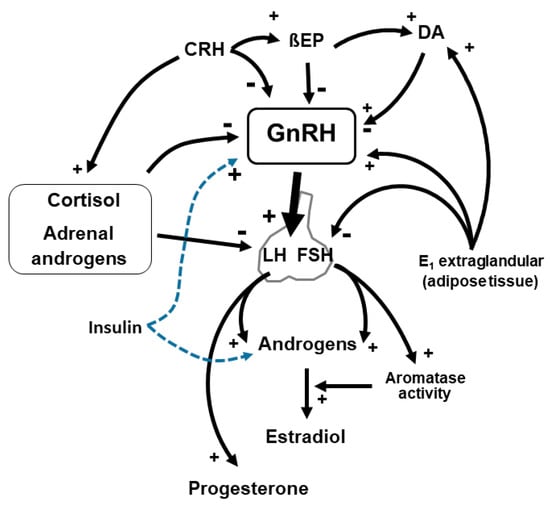
Figure 1.
GnRH-secreting neurons undergo to a number of modulations, positive and negative. Stress, through CRH activation, determines a slowing down of GnRH secretion together with opioid peptides (βendorphin, BEP). Similarly, adrenal gland hormones play a negative role on hypothalamic and hypophyseal function. Insulin, especially in hyperinsulinemic PCOS patients, stimulates ovarian androgens synthesis and LH release, thus improving the ovarian impairment. DA: dopamine, CRH: corticotrophin-releasing hormone, E1: estrone, βEP: beta endorphin.
When evaluating a hyperandrogenic and obese woman with PCOS, we have to keep in mind that there might be also other factors triggering the increase in androgens [31]. In fact, PCOS women might be hyperandrogenic, with the contribution of the adrenal gland even though it does not have an impaired adrenal function: the PCOS clinical condition is often characterized by an increased psychological stress that triggers the adrenal activity [32].
As reported by Farah-Eways et al., PCOS patients, other than having higher levels of androgens produced at the ovarian level, also may show an increase in cortisol and 17-OHP plasma levels [33]. Performing the ACTH test, these authors verified that no pathological alterations were present, and these women were simply more stressed and had a more activated adrenal gland in baseline conditions, with no abnormal response to ACTH stimulation. This is an important condition to keep in mind when looking into therapeutic choices [33].
To better understand how the adrenal gland acts, a study on the co-secretion of gonadotropins with androgen (androstenedione) and cortisol with androgens (androstenedione) was conducted on two groups of PCOS patients: one hyperandrogenic from ovarian overproduction, and the other hyperandrogenic from stress-induced hyperproduction.
In the first group, the LH and androstenedione plasma profile were found to be co-secreted, showing a parallel trend. Since LH drives the ovary, androstenedione peaks follow those of LH, thus indicating that this androgen is predominantly of ovarian origin. The other group of patients presented an opposite situation: while the LH pulsatile profile did not show variability, the androstenedione profile decreased its plasma levels within 2 h and, in addition, the profiles of cortisol and androstenedione showed a parallel trend, both starting high at the beginning of the test, and then lowering in a parallel manner, with cortisol and androstenedione pulses being co-secreted. This indicates that, although a part of androstenedione had an ovarian origin, in these patients, a greater part comes from the hyperactivated adrenal gland [34].
It is also important to remember that true adrenal hyperandrogenism should be suspected when elevated DHEAS and 17-OHP basal hormone levels are found together with androstenedione. This occurs when an enzymatic defect of the adrenal gland, hyperandrogenism, arises from the deficiency of one of these enzymes: 21-hydroxylase, 11β-hydroxylase or 3β/-hydroxysteroid-dehydrogenase.
With these enzyme defects, the direct pathway of cortisol biosynthesis is impaired: the adrenal gland, however, tries to arrive at the synthesis of cortisol through other pathways, but this leads to the excess of the intermediate products upstream of cortisol, leading to a greater release of DHEAS, 17-OHP and androstenedione.
Despite the fact that PCOS is 40 to 50 times more frequent than NC-CAH in fertile women than in women with hyperandrogenism, it is essential to check for NC-CAH in all patients with apparent PCOS. Ethnicity has little impact on the prevalence of PCOS [35,36]. Levels of 17-OHP, DHEAS, androstenedione and cortisol plasma have to be checked. In the case that cortisol is normal, but 17-OHP and androstenedione are too high, an issue at the adrenal gland is suspected [15].
Plasma levels of 17-OHP above 200 ng/dL (6 nmol/L) strongly indicates the presence of NC-CAH, whereas plasma concentrations below 200 ng/dL (6 nmol/L) do not sustain the presence of NC-CAH. To prove the suspicion, an ACTH stimulation test has to be performed. A 17-OHP response equal or higher than 1500 ng/dL (43 nmol/L) under ACTH stimulation confirms the suspicion of NC-CAH [37,38].
With 17-OHP plasma levels being increased during the preovulatory or luteal phase of the menstrual cycle, plasma evaluation has to be performed no later than 8–10 days after the beginning of the menstrual bleeding, or any time if the patient is amenorrheic.
4. Prolactin Disorders
Prolactin (PRL) is a protein hormone whose role is to stimulate milk production in female mammals [39]. PRL is under the control of the tuberoinfundibular dopaminergic system (TIDA) that directly controls PRL release from the gonadotropes inhibiting PRL secretion [40]. PRL secretion also depends in part on the direct stimulation of TRH that classically controls the TSH section and thyroid function [41] (Figure 2). The stimuli of eating, estrogen treatment, mating, ovulation and nursing induce PRL release from lactotrope cells in the pituitary gland. PRL release is in a pulsatile manner and modulates metabolism, the immune system and pancreatic development.
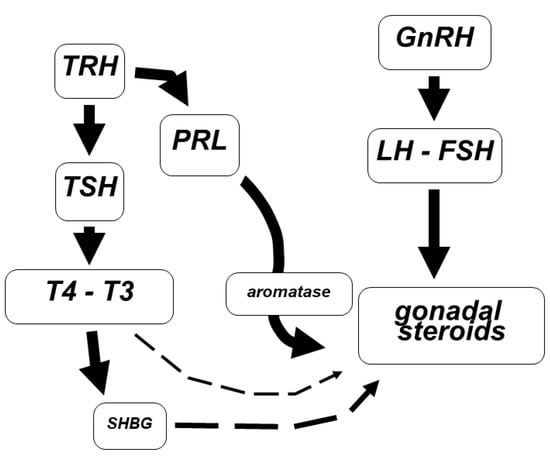
Figure 2.
Schematic representation of the interconnection between PRL and thyroid axes. TRH is able to induce both PRL and T4 secretion. Whatever event reduces or increases TRH release leads to a change in the concentrations of SHBG as well as in the expression of aromatase, so as to impair the gonadal steroids synthesis and binding.
Though mostly released by the anterior pituitary, PRL has also extra-pituitary origins such as from the brain, prostate, immune cells, skin, adipose tissue and human decidua [39]. PRL reaches the target cells through the circulatory system and acts on specific receptors, located on the plasma membrane [42].
PRL also acts as a neuropeptide when it crosses the blood–brain barrier: it gets though a PRLRs-independent mechanism and modulates the hypothalamus, central functions, behavior, arousal and sexuality [43,44].
With no endocrine target organ to provide feedback control, the biological evolution of PRL made it regulate its own secretion within the hypothalamus, modulating the hypothalamic releasing and inhibiting factors that control their secretions. PRL suppress its further secretion acting on tuberoinfundibular (TIDA) neurons located in the arcuate nucleus that stimulate the synthesis and release of dopamine into the portal vasculature at the median eminence [45]. Dopamine acts on D2 receptors to inhibit PRL synthesis and release. Other PRL-releasing factors are: thyrotropin-releasing hormone (TRH), serotonin (5-HT), vasoactive intestinal peptide (VIP), and arginine vasopressin (AVP) [46].
Together with DA, PRL exerts negative feedback on its secretion and GnRH production, thus explaining how hyperprolactinemia interferes with human reproduction [39].
PRL is a putative disruptor of the normal neuroendocrine control of the ovarian cycle. Therefore, it is one of the many hormones involved in the control of follicle maturation, in the evolution of the luteal phase, in the interference of FSH-induced aromatization and in the modulation of GnRH secretion.
This observation confirms the role of PRL in the management of the ovarian cycle, both centrally and at the ovarian level. An excess of PRL up to the extent of hyperprolactinemia can interfere with female and male reproduction. Hyperprolactinemia decreases GnRH and, subsequently, LH secretion, decreasing pulse frequency and amplitude, thus affecting gonadal steroids production and inducing menstrual irregularities.
PRL affects the production of gonadal steroids from the granulosa cells, mainly slowing aromatase activity; hyperandrogenism is a frequent occurrence, paralleling the picture of PCOS. Such effects may impair ovulation, creating a predisposition to infertility, decreasing libido, and frequently inducing galactorrhea [47,48].
The effects of PRL on fertility have been studied in animal models that confirmed that reproduction needs PRL signaling. Both short and long isoforms of PRLRs are expressed by the granulosa, interstitial and luteal cells at the ovarian level, as well as in the endometrium, myometrium and decidua in the uterus. Kisspeptin secreting neurons are essential for pubertal maturation in humans and are the main inductors of GnRH secretion. Kisspeptin is probably involved as a potential mediator of prolactin’s actions on fertility [49].
Kisspeptin and prolactin are able to modulate each other, but in different ways according to the species. In fact, kisspeptin induces PRL prolactin release in rats when estradiol levels are high, while in women, the concomitant pulsatile release of PRL with kisspeptin-induced LH occurs when they are hypogonadal [41,50].
In response to stress, PRL acts negatively on the hypothalamic–pituitary axis, modulating GnRH release as well as the adrenal activity through the modulation of corticotrophin-releasing hormone (CRH)–ACTH secretions [43].
PRL is tightly linked to emotional responsiveness, acting on the modulation of anxiety and depressive disorders. These events take place based on the actions of PRL on neurogenesis in the hippocampal area. Acute stressor events, in female mice, suppress the activation of kisspeptin neurons and LH pulsatile release is reduced. When the same stress is chronically administered during the day, mice showed higher corticosterone levels and the lengthening of the estrous cycles. Such chronic stressor situations can induce hyperprolactinemia, thus participating to impaired fertility in women [51,52].
PRL promotes leptin resistance and increased appetite, hyperphagia and insulin resistance: under pathological hyperprolactinemia, obesity, abnormal glucose tolerance and hyperinsulinemia due to insulin resistance have been observed. Indeed, low PRL levels may induce metabolic changes, while high PRL levels just below or above the conventional hyperprolactinemic limit (25 ng/mL) promote metabolic abnormalities, leading to diabetes and obesity-induced metabolic risks.
Weight gain (up to obesity) and insulin resistance occur frequently when PRL plasma levels are 70–100 ng/mL or higher: this tends to occur during hyperprolactinemia due to pituitary adenomas, hypothyroidism or under specific medications (i.e., antidepressants, prokinetics, antipsychotics).
Circulating levels of PRL in women are usually around 10–15 ng/mL: when these levels are above 25 ng/mL, they are conventionally defined as hyperprolactinemia [39].
Recently, it has been reported that patients suffering from polycystic ovary syndrome (PCOS) have higher PRL levels (18–20 ng/mL), despite not having hyperprolactinemia.
In the case of stress (fear, psychological stress, etc.), PRL plasma levels increase rapidly (up to 90–100 ng/mL). Pregnancy and lactation promote the increase in PRL levels up to 200 ng/mL. PRL levels around or above 25 ng/mL should be reevaluated by inserting a heparin indwelling in a forearm vein and sampling three times at 15–20 min intervals (i.e., time 0, 15–20 and 30–40 min) to account for pulsatility [47].
When hyperprolactinemia is found, a precise anamnestic investigation has to assess whether any other disease is present (liver or kidney impairments, or hypothyroidism). It is also important to clarify what kind of medications have been taken in the preceding days or weeks before the PRL testing. Lifestyle, body mass index (BMI), excessive physical activity and stressful situations have to be monitored. If PRL plasma levels are above 50 ng/mL, a pituitary magnetic resonance imaging (MRI), preferably enhanced with gadolinium, should be performed in order to disclose the presence of a pituitary adenoma or any other sellar/parasellar mass, which might be causing stalk compression [47,48].
In general, hyperprolactinemia around or above 100 ng/mL is a possible indicator of a secreting adenoma; untreated hypothyroidism can rarely also mimic an adenoma: this requires TSH to be checked [39].
Despite being secreted by different cells, prolactin and TSH are related, because they are both stimulated by TRH. In the presence of any functional defect that induces hypothyroidism, TSH is higher due to an increase in the release of TRH at the hypothalamic level, and, consequently, PRL becomes elevated.
In case of dysfunctional hyperprolactinemia, the treatment is quite simple: cabergoline decreases prolactin levels slowly, decreases androgens in circulation, and increases gonadotropins.
As a dopamine agonist, cabergoline is better than bromocriptine in controlling PRL levels in hyperprolactinemic men and women with sexual dysfunctions [39].
5. Thyroid Dysfunctions
The thyroidal axis is one of the pivotal endocrine axes. The release of thyrotropin-releasing hormone (TRH) from the hypothalamus directly induces TSH secretion from pituitary cells. TSH acts directly on follicular cells of the thyroid, thus permitting the production of thyroidal hormone T4 and T3. Thyroidal hormones are mainly involved in metabolic pathways, and through these actions, they modulate cell functions at any level [53].
Thyroid diseases are quite frequent; more common in women than men suffer from them, and they may interfere with the reproductive system [54]. In fact, thyroid hormones can impair reproduction both directly and indirectly through several actions: the increase in liver production of sex hormone-binding globulin (SHBG), of ovarian testosterone and androstenedione secretion and the reduction in the clearance of all gonadal steroids, accelerating the conversion of androgens to estrone.
Receptors for thyroid hormones are at the level of oocytes, where these hormones act synergically with FSH to induce the production of sexual hormones [55]. Thyroid dysfunctions result in the high frequency of abnormal menstrual bleeding due to ovulatory impairments. In fact, triiodothyronine (T3) acts on folliculogenesis, modulating the P13K/Akt pathway [56]. Due to this, hypothyroidism is associated with a reduced fertility rate despite the recognition of the endometrial expression of deiodinase type 3 and placental increases in T3 [57,58].
Other than the fact that estrogens induce SHBG secretion, they also increase the concentrations of thyroxine binding globulin (TBG) [59]. In fact, pregnancy and oral contraceptive pills significantly increase TBG concentrations [60]. TBG concentrations increase greatly with the menopausal transition during menopause and later with the aging process, as well as for the hypoestrogenic condition [61].
Hyperthyroidism can impair menstrual bleeding, inducing hypomenorrhea and polimenorrea, while hypothyroidism induces oligomenorrhea. Frequently, these menstrual irregularities occur prior to the identification of thyroid disease [62].
6. Hypothyroidism
The most common cause of hypothyroidism is iodine deficiency [63], but in most cases, hypothyroidism is due to autoimmune thyroiditis [64]. Hypothyroidism can also occur because of the destruction of thyroid tissue due to radioactive iodine therapy for thyroid cancer, thyroidectomy, and rare diseases (such as scleroderma and amyloidosis). Hypothyroidism may also occur due to lithium treatment, or iodine-containing drugs. Central hypothyroidism is a rare disorder caused by hypothalamic–pituitary diseases.
The symptoms of hypothyroidism are lethargy, pretibial myxedema, hypothermia, and the tendency to gain weight due to the accumulation of sugar derivatives that are deposited in the extracellular matrix adsorbing water.
After 10 years of disease, in 15% of cases, hypothyroidism with high antibodies correlates with a greater risk of early ovarian failure, due to a higher incidence of anti-thyroid and anti-ovarian antibodies [1]. Oligomenorrhea is the classic menstrual abnormality induced by hypothyroidism that may be diagnosed for the highest TSH plasma levels. This is very frequent in case of recurrent abortion.
At the basis of the infertility state induced by hypothyroidism is the impaired peripheral estrogen metabolism, the occurrence of hyperprolactinemia and abnormal GnRH secretion that affects gonadotropin episodic secretions [65].
Due to the slowing of the metabolic clearance of gonadal steroids, hypothyroidism increases androstenedione and estrone plasma levels and permits the increase in peripheral aromatization [45,46]. Moreover, with SHBG being decreased, both total testosterone and E2, and their unbound fractions are increased. Impairments of steroid metabolism disappear when a euthyroid state is restored with an appropriate L-thyroxine substitutive treatment [66] (Figure 1).
In the case of hypothyroidism, GnRH levels are usually normal, though impaired LH pulsatile release has been reported with a delayed LH response to a GnRH stimulation test [67,68]. In this latter case, serum PRL concentration was reported to be increased due a higher discharge of the hypothalamic TRH that stimulates both TSH and PRL secretions [69].
7. Autoimmune Thyroid Disease
Thyroid autoimmunity is the most frequent autoimmune disease (5–20%) in women of fertile age and is the cause of hypothyroid function. It is characterized by the presence of anti-thyroid antibodies, which include anti-thyroperoxidase and anti-thyroglobulin antibodies. It may remain latent, asymptomatic, or even undiagnosed for years.
One kind of antibodies (anti-TSH) have an allosteric conformation similar to TSH, and for this reason can stimulate the receptor by inducing high fT3 and fT4, and consequently increase both all the thyroid hormone concentrations and their actions, mainly acting on metabolism in the presence of an almost zero TSH. For the diagnosis, it is important to assay the antibodies in the plasma [15].
Thyroid autoimmunity is more common in females than in males. This seems to be related to the action of both estrogens and androgens on the immune system. In fact, the female/male ratio of patients with Graves’ disease decreases after 60 years. Early menarche or a late menopause are considered risk factors for the development of an autoimmune thyroiditis, probably because of a greater exposure to estrogen during the reproductive life [70,71].
Serum thyroid autoantibodies are detectable in up to 25% of women over the age of 60, and autoimmune hypothyroidism is eight to nine times more frequent in women than in men, increasing with age [53].
Symptoms of autoimmune thyroid disease can simulate the onset of early menopause since antibodies against the ovary might produce hot flashes, amenorrhea, moodiness and insomnia.
8. Hyperthyroidism
Hyperthyroidism classically shows undetectable levels of serum TSH, and elevated FT3 and FT4 concentrations. In subclinical hyperthyroidism, the serum TSH level is low or very low, but FT3 and FT4 concentrations are normal.
The most frequent cause of hyperthyroidism, in iodine-sufficient areas, is Graves’ disease. On the contrary, toxic adenoma (Plummer adenoma) and toxic multinodular goiter are highly frequent where iodine intake is low [72]. Graves’ disease is triggered by the high synthesis of an immunoglobulin that binds the TSH receptor. Usually such a disease occurs during the second up to the fourth decade [73]. The toxic adenomas can induce thyrotoxicosis due to the growth of benign monoclonal thyroid cells able to hyper-produce thyroid hormones. Usually, only a single functioning nodule develops, but in some cases more than one nodule can grow (toxic multinodular goiter) and this is more frequent after the age of 60 [74].
Sometimes, thyrotoxicosis starts due to a postpartum thyroiditis or subacute thyroiditis, or due to unintentional excessive replacement therapy in hypothyroid patients or to intentional TSH suppressive therapy for benign or malignant thyroid disease. The hypersecretion of TSH for a trophoblastic tumor or metastatic thyroid carcinoma is very rare.
Amenorrhea is a classic and precocious symptom associated with hyperthyroidism [75]. Together with thyrotoxicosis there is the increase in serum levels of SHBG and also frequently of estrogen during all phases of the menstrual cycle (compared with normal women) [76]. This is because the metabolic clearance rate of estradiol is reduced in hyperthyroidism, and this is related to the increased binding of E2 to SHBG [77]. Similarly, androgen metabolism also changes, since plasma levels of testosterone and androstenedione are increased together with higher production rates of both testosterone and androstenedione. The increased estrogenic tone is in part also sustained by the higher conversion rate of androstenedione to estrone and of testosterone to E2, thus participating in the maintenance of the impaired gonadotropin secretion all along the menstrual cycle [78,79,80]. LH plasma levels are increased, while the pulsatile secretion does not differ greatly from healthy subjects. Once treatment is started with antithyroid drugs, LH goes back to normal levels [81].
It is relevant to point out that thyroid dysregulation often induces a PCO-like picture. Though it cannot be excluded a possible link due to common genetic and autoimmune factors, the feature of the impaired androgenic milieu that hyperthyroidism may show is at the basis of such morphology at the ultrasound scan.
Metabolic changes in PCOS are frequent [82]. Hashimoto’s thyroiditis (HT) triggers metabolic impairments due to the thyroid dysfunction; similarly, insulin resistance may occur, and if combined with a PCOS phenotype, may trigger cardiovascular risks. The coexistence of HT and PCOS may exaggerate metabolic dysfunctions that usually might be disclosed by higher values of body weight, glucose level, insulin, HOMA-IR index and lipid profile [83].
If a thyroid dysfunction is suspected, fT3, fT4, TSH should be controlled together with the antibodies’ plasma levels; in the case that TSH is very low, anti-TSH antibodies must be essayed, and an ultrasound scan of the thyroid gland has to be performed.
If a defect of fT3 is found, we have to exclude a hypothyroid condition. This is the case of stress-induced amenorrhea when the “low T3 syndrome” is triggered and the activity of the thyroid gland is decreased by reducing the T4 toT3 conversion, with normal TSH plasma levels. This happens because a distinct deiodinase, activated in these cases, leads to the synthesis of a biologically inactive T3, called reverse T3 (rT3). In these patients with stress-induced amenorrhea or functional hypothalamic amenorrhea (FHA), levothyroxine should not be administered, since the low fT3 level is a defensive event to prevent from excess energy consumption, and it has to be checked for eventual abnormal eating attitude.
Additionally, the metabolic profile should be analyzed, evaluating the insulin and amylase levels and the hormonal basal levels of the reproductive axis in order to find the primary cause of the hormonal disease [15].
In conclusion, the endocrine environment and axes are responsible both for reproductive function as well as for its failure. Normal reproductive function is affected by such a situation, since it is dependent on the equilibrium among the many endocrine and neuroendocrine factors so as to modulate the hypothalamic centers where the reproductive function is triggered.
Author Contributions
Conceptualization: A.D.G.; writing—original draft preparation, V.T.; writing—review and editing, C.B.; supervision: A.D.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors report no conflict of interest.
References
- Gargiulo, A.R. Yen & Jaffe’s Reproductive Endocrinology, 8th ed.; Elsevier Publ.: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Podfigurna, A.; Maciejewska-Jeske, M.; Meczekalski, B.; Genazzani, A.D. Kisspeptin and LH pulsatility in patients with functional hypothalamic amenorrhea. Endocrine 2020, 70, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Meczekalski, B.; Katulski, K.; Czyzyk, A.; Podfigurna-Stopa, A.; Maciejewska-Jeske, M. Functional hypothalamic amenorrhea and its influence on women’s health. J. Endocrinol. Investig. 2014, 37, 1049–1056. [Google Scholar] [CrossRef]
- Gordon, C.M.; Ackerman, K.E.; Berga, S.L.; Kaplan, J.R.; Mastorakos, G.; Misra, M.; Murad, M.H.; Santoro, N.F.; Warren, M.P. Functional Hypothalamic Amenorrhea: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2017, 102, 1413–1439. [Google Scholar] [CrossRef]
- Genazzani, A.D.; Petraglia, F.; Bonati, M.; Genazzani, A.R. Developmental of endogenous opioids system and control of gonadotropin secretion. In Frontiers in Endocrinology 191–197; Bergadà, C., Moguilevsky, J.A., Eds.; Ares-Serono Symposia Publication: Rome, Italy, 1995. [Google Scholar]
- Genazzani, A.D. Neuroendocrine aspects of amenorrhea related to stress. Pediatr. Endocrinol. Rev. 2005, 2, 661–668. [Google Scholar] [PubMed]
- Hall, J.E. The Female Reproductive System, Infertility, and Contraception. In Harrison’s Endocrinology; McGraw-Hill Education: New York, NY, USA, 2011; pp. 178–193. [Google Scholar]
- Genazzani, A.D.; Santagni, S.; Chierchia, E.; Rattighieri, E.; Campedelli, A.; Prati, A.; Federica, R.; Tommaso, S. Estimation of instantaneous secretory rates and intrinsic characteristics of luteinizing hormone secretion in women with Kallmann syndrome before and after estriol administration. Reprod. Biol. 2011, 11, 284–293. [Google Scholar] [CrossRef]
- Genazzani, A.D.; Meczekalski, B.; Podfigurna-Stopa, A.; Santagni, S.; Rattighieri, E.; Ricchieri, F.; Chierchia, E.; Simoncini, T. Estriol administration modulates luteinizing hormone secretion in women with functional hypothalamic amenorrhea. Fertil. Steril. 2012, 97, 483–488. [Google Scholar] [CrossRef]
- Genazzani, A.; Chierchia, E.; Santagni, S.; Rattighieri, E.; Farinetti, A.; Lanzoni, C. Hypothalamic amenorrhea: From diagnosis to therapeutical approach. Ann. d’Endocrinologie 2010, 71, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.J.; Geisler, C.; Heymsfield, S.B.; Bosy-Westphal, A. Recent advances in understanding body weight homeostasis in humans. F1000Research 2018, 7, 1025. [Google Scholar] [CrossRef]
- Fontana, R.; Della Torre, S. The Deep Correlation between Energy Metabolism and Reproduction: A View on the Effects of Nutrition for Women Fertility. Nutrients 2016, 8, 87. Available online: https://pubmed.ncbi.nlm.nih.gov/26875986/ (accessed on 26 July 2021). [CrossRef]
- Katulski, K.; Podfigurna, A.; Czyzyk, A.; Meczekalski, B.; Genazzani, A. Kisspeptin and LH pulsatile temporal coupling in PCOS patients. Endocrine 2018, 61, 149–157. [Google Scholar] [CrossRef]
- Genazzani, A.R.; Petraglia, F.; Gamba, O.; Sgarbi, L.; Greco, M.M.; Genazzani, A.D. Neuroendocrinology of the menstrual cycle. In Gynecologic Endoscopic Surgery 48–54 (1994); The New York Academy of Sciences: New York, NY, USA, 1997. [Google Scholar]
- Genazzani, A.D.; Santagni, S.; Rattighieri, E.; Chierchia, E.; Despini, G.; Prati, A.; Ricchieri, F. PCOS and Insulin Resistance (IR): From Lifestyle to Insulin Sensitizers; Springer: Berlin/Heidelberg, Germany, 2015; pp. 11–23. [Google Scholar]
- Genazzani, A.D.; Despini, G.; Bonacini, R.; Prati, A. Functional Hypothalamic Amenorrhea as Stress Induced Defensive System; Springer: Berlin/Heidelberg, Germany, 2017; pp. 111–118. [Google Scholar]
- Genazzani, A.D.; Prati, A.; Simoncini, T.; Napolitano, A. Modulatory role of D-chiro-inositol and alpha lipoic acid combination on hormonal and metabolic parameters of overweight/obese PCOS patients. Eur. Gynecol. Obs. 2019, 1, 29–33. [Google Scholar]
- Skorupskaite, K.; George, J.T.; Anderson, R.A. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum. Reprod. Update 2014, 20, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Joseph, D.N.; Whirledge, S. Stress and the HPA Axis: Balancing Homeostasis and Fertility. Int. J. Mol. Sci. 2017, 18, 2224. [Google Scholar] [CrossRef] [PubMed]
- Astapova, O.; Minor, B.M.N.; Hammes, S.R. Physiological and Pathological Androgen Actions in the Ovary. Endocrinology 2019, 160, 1166–1174. [Google Scholar] [CrossRef]
- Sen, A.; Hammes, S.R. Granulosa Cell-Specific Androgen Receptors Are Critical Regulators of Ovarian Development and Function. Mol. Endocrinol. 2010, 24, 1393–1403. [Google Scholar] [CrossRef]
- Walters, K.A.; Gilchrist, R.; Ledger, W.; Teede, H.J.; Handelsman, D.J.; Campbell, R.E. New Perspectives on the Pathogenesis of PCOS: Neuroendocrine Origins. Trends Endocrinol. Metab. 2018, 29, 841–852. [Google Scholar] [CrossRef]
- Whirledge, S.; Cidlowski, J.A. Glucocorticoids and Reproduction: Traffic Control on the Road to Reproduction. Trends Endocrinol. Metab. 2017, 28, 399–415. [Google Scholar] [CrossRef]
- Magiakou, M.A.; Mastorakos, G.; Webster, E.; Chrousos, G.P. The hypothalamic-pituitary-adrenal axis and the female reproductive system. Ann. N. Y. Acad. Sci. 1997, 816, 42–56. [Google Scholar] [CrossRef]
- Barbarino, A.; De Marinis, L.; Tofani, A.; della Casa, S.; D’Amico, C.; Mancini, A.; Corsello, S.M.; Sciuto, R.; Barini, A. Corticotropin-Releasing Hormone Inhibition of Gonadotropin Release and the Effect of Opioid Blockade. J. Clin. Endocrinol. Metab. 1989, 68, 523–528. [Google Scholar] [CrossRef]
- Ortega, M.T.; McGrath, J.A.; Carlson, L.; Poccia, V.F.; Larson, G.; Douglas, C.; Sun, B.Z.; Zhao, S.; Beery, B.; Vesper, H.W.; et al. Longitudinal Investigation of Pubertal Milestones and Hormones as a Function of Body Fat in Girls. J. Clin. Endocrinol. Metab. 2021, 106, 1668–1683. [Google Scholar] [CrossRef]
- Genazzani, A.D. Inositol as putative integrative treatment for PCOS. Reprod. Biomed. Online 2016, 33, 770–780. [Google Scholar] [CrossRef]
- Fauser, B.C.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S.; et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2012, 97, 28–38.e25. [Google Scholar] [CrossRef]
- Genazzani, A.D.; Prati, A.; Chierchia, E.; Santagni, S. Gli stati iperandrogenici. Boll. Ginecol. Endocrinol. 2014, 8, 35–44. [Google Scholar]
- Genazzani, A.D. Expert’s opinion: Integrative treatment with inositols and lipoic acid for insulin resistance of PCOS. Gynecol. Reprod. Endocrinol. Metab. 2020, 1, 146–157. Available online: https://gremjournal.com/journal/03-2020/experts-opinion-integrative-treatment-with-inositols-and-lipoic-acid-for-insulin-resistance-of-pcos/ (accessed on 26 July 2021).
- Genazzani, A. Metformin administration modulates and restores luteinizing hormone spontaneous episodic secretion and ovarian function in nonobese patients with polycystic ovary syndrome. Fertil. Steril. 2004, 81, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Lizneva, D.V.; Gavrilova-Jordan, L.; Walker, W.; Azziz, R. Androgen excess: Investigations and management. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 37, 98–118. [Google Scholar] [CrossRef]
- Farah-Eways, L.; Reyna, R.; Knochenhauer, E.S.; Bartolucci, A.A.; Azziz, R. Glucose action and adrenocortical biosynthesis in women with polycystic ovary syndrome. Fertil. Steril. 2004, 81, 120–125. [Google Scholar] [CrossRef]
- Genazzani, A.D.; Petraglia, F.; Pianazzi, F.; Volpogni, C.; Genazzani, A.R. The concomitant release of androstenedione with Cortisol and luteinizing hormone pulsatile releases distinguishes adrenal from ovarian hyperandrogenism. Gynecol. Endocrinol. 1993, 7, 33–41. [Google Scholar] [CrossRef]
- Moran, C.; Azziz, R. 21-Hydroxylase-Deficient Nonclassic Adrenal Hyperplasia: The Great Pretender. Semin. Reprod. Med. 2003, 21, 295–300. [Google Scholar] [CrossRef]
- Ding, T.; Hardiman, P.J.; Petersen, I.; Wang, F.-F.; Qu, F.; Baio, G. The prevalence of polycystic ovary syndrome in reproductive-aged women of different ethnicity: A systematic review and meta-analysis. Oncotarget 2017, 8, 96351–96358. [Google Scholar] [CrossRef] [PubMed]
- Speiser, P.W.; Azziz, R.; Baskin, L.S.; Ghizzoni, L.; Hensle, T.W.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Montori, V.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2010, 95, 4133–4160. [Google Scholar] [CrossRef]
- Azziz, R.; Dewailly, D.; Owerbach, D. Clinical review 56: Nonclassic adrenal hyperplasia: Current concepts. J. Clin. Endocrinol. Metab. 1994, 78, 810–815. [Google Scholar] [CrossRef]
- Nappi, R.E.; Di Ciaccio, S.; Genazzani, A.D. Prolactin as a neuroendocrine clue in sexual function of women across the reproductive life cycle: An expert point of view. Gynecol. Endocrinol. 2021, 37, 490–496. [Google Scholar] [CrossRef]
- Timmerman, W.; Deinum, M.E.; Poelman, R.T.; Westernik, B.H.; Schuiling, G.A. Characterisation of the DA-ergic system in the mediobasal hypothalamus: A new approach to simultaneously monitor the release of DA from the TIDA neurons and the PRL secretion from the adenohypophysis in awake rats. Brain Res. 1994, 657, 275–280. [Google Scholar] [CrossRef]
- Cetel, N.; Yen, S.S.C. Concomitant Pulsatile Release of Prolactin and Luteinizing Hormone in Hypogonadal Women*. J. Clin. Endocrinol. Metab. 1983, 56, 1313–1315. [Google Scholar] [CrossRef]
- Bole-Feysot, C.; Goffin, V.; Edery, M.; Binart, N.; Kelly, P.A. Prolactin (PRL) and its receptor: Actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr. Rev. 1998, 19, 225–268. [Google Scholar] [CrossRef] [PubMed]
- Torner, L. Actions of Prolactin in the Brain: From Physiological Adaptations to Stress and Neurogenesis to Psychopathology. Front. Endocrinol. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S.E.; Wyatt, A.; Herbison, R.E.; Knowles, P.J.; Ladyman, S.R.; Binart, N.; Banks, W.A.; Grattan, D.R. Prolactin transport into mouse brain is independent of prolactin receptor. FASEB J. 2016, 30, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.A.; Frawley, L.S.; Neill, J.D. Neuroendocrine Control of Prolactin Secretion. Annu. Rev. Physiol. 1983, 45, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S.; Polonsky, K.S.; Larsen, P.R.; Kronenberg, H.M. Williams Textbook of Endocrinology, 13th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Melmed, S.; Casanueva, F.F.; Hoffman, A.R.; Kleinberg, D.L.; Montori, V.; Schlechte, J.; Wass, J.A.H. Diagnosis and Treatment of Hyperprolactinemia: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2011, 96, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Kalsi, A.K.; Halder, A.; Jain, M.; Chaturvedi, P.K.; Sharma, J.B. Prevalence and reproductive manifestations of macroprolactinemia. Endocrine 2019, 63, 332–340. [Google Scholar] [CrossRef]
- De Roux, N.; Genin, E.; Carel, J.C.; Matsuda, F.; Chaussain, J.L.; Milgrom, E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. USA 2003, 100, 10972–10976. [Google Scholar] [CrossRef]
- Ribeiro, A.; Leite, C.M.; Kalil, B.; Franci, C.R.; Anselmo-Franci, J.A.; Szawka, R.E. Kisspeptin Regulates Tuberoinfundibular Dopaminergic Neurones and Prolactin Secretion in an Oestradiol-Dependent Manner in Male and Female Rats. J. Neuroendocr. 2015, 27, 88–99. [Google Scholar] [CrossRef]
- Yang, J.A.; Song, C.I.; Hughes, J.K.; Kreisman, M.J.; Parra, R.A.; Haisenleder, D.J.; Kauffman, A.S.; Breen, K.M. Acute Psychosocial Stress Inhibits LH Pulsatility and Kiss1 Neuronal Activation in Female Mice.-Abstract-Europe PMC. Endocrinology 2017, 158, 3716–3723. [Google Scholar] [CrossRef]
- Levine, S.; Muneyyirci-Delale, O. Stress-Induced Hyperprolactinemia: Pathophysiology and Clinical Approach. Obstet. Gynecol. Int. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Salvatore, D.; Davies, T.; Schlumberger, M.; Hay, I.; Reed Larsen, P. Thyroid Physiology and Diagnostic Evaluation of Patients with Thyroid Disorders. In Williams Textbook of Endocrinology, 12th ed.; Melmed, S., Polonsky, K.S., Kronenberg, H.M., Eds.; Saunders, Elsevier Science: Amsterdam, The Netherlands, 2011; pp. 327–361. [Google Scholar]
- Poppe, K.; Velkeniers, B.; Glinoer, D. Thyroid disease and female reproduction. Clin. Endocrinol. 2007, 66, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, S.; Rucci, N.; Scaldaferri, M.L.; Masciulli, M.P.; Rossi, G.; Moretti, C.; D’Armiento, M.; Ulisse, S. Thyroid hormone effects on mouse oocyte maturation and granulosa cell aromatase activity. Endocrinology 1999, 140, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Guo, L.; Zhu, B.; Feng, Y.; Yu, S.; An, N.; Wang, X. Effects of 3, 5, 3’-Triiodothyronine (T3) and Follicle Stimulating Hormone on Apoptosis and Proliferation of Rat Ovarian Granulosa Cells. Chin. J. Physiol. 2013, 56, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Korevaar, T.I.M.; Medici, M.; Visser, T.J.; Peeters, R.P. Thyroid disease in pregnancy: New insights in diagnosis and clinical management. Nat. Rev. Endocrinol. 2017, 13, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.A.; Dorfman, D.M.; Genest, D.R.; Salvatore, D.; Larsen, P.R. Type 3 Iodothyronine Deiodinase Is Highly Expressed in the Human Uteroplacental Unit and in Fetal Epithelium. J. Clin. Endocrinol. Metab. 2003, 88, 1384–1388. [Google Scholar] [CrossRef] [PubMed]
- Ain, K.B.; Mori, Y.; Refetoff, S. Reduced Clearance Rate of Thyroxine-Binding Globulin (TBG) with Increased Sialylation: A Mechanism for Estrogen-Induced Elevation of Serum TBG Concentration*. J. Clin. Endocrinol. Metab. 1987, 65, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Steingold, K.A.; Matt, D.W.; DeZiegler, D.; Sealey, J.E.; Fratkin, M.; Reznikov, S. Comparison of Transdermal to Oral Estradiol Administration on Hormonal and Hepatic Parameters in Women with Premature Ovarian Failure. J. Clin. Endocrinol. Metab. 1991, 73, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, D.R.; DeFazio, J.D.; Erlik, Y.; Lu, J.K.; Wolfsen, A.F.; Carlson, H.E.; Hershman, J.M.; Judd, H.L. Pituitary hormones during the menopausal hot flash. Obstet. Gynecol. 1984, 64, 752–756. [Google Scholar] [PubMed]
- Krassas, G.G.; Poppe, K.; Glinoer, D. Thyroid Function and Human Reproductive Health. Endocr. Rev. 2010, 31, 702–755. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Andersson, M. Assessment of iodine nutrition in populations: Past, present, and future. Nutr. Rev. 2012, 70, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Vanderpump, M.P.J.; Tunbrldge, W.M.G.; French, J.M.; Appleton, D.; Bates, D.; Clark, F.; Evans, J.G.; Hasan, D.M.; Rodgers, H.; Tunbridge, W.M.; et al. The incidence of thyroid disorders in the community: A twenty-year follow-up of the Whickham Survey. Clin. Endocrinol. 1995, 43, 55–68. [Google Scholar] [CrossRef]
- Krassas, G.E. Thyroid disease and female reproduction. Fertil. Steril. 2000, 74, 1063–1070. [Google Scholar] [CrossRef]
- Gordon, G.G.; Southren, A.L. Thyroid-hormone effects on steroid-hormone metabolism. Bull. N. Y. Acad. Med. 1977, 53, 241–259. [Google Scholar]
- Longcope, C.; Abend, S.; Braverman, L.E.; Emerson, C.H. Androstenedione and Estrone Dynamics in Hypothyroid Women*. J. Clin. Endocrinol. Metab. 1990, 70, 903–907. [Google Scholar] [CrossRef]
- Valenti, G.; Ceda, G.P.; Denti, L.; Tarditi, E.; Speroni, G. Gonadotropin secretion in hyperthyroidism and hypothyroidism. La Ric. Clin. e Lab. 1984, 14, 53–63. [Google Scholar]
- Honbo, K.S.; van Herle, A.J.; Kellett, K.A. Serum prolactin levels in untreated primary hypothyroidism. Am. J. Med. 1978, 64, 782–787. [Google Scholar] [CrossRef]
- Phillips, D.I.W.; Lazarus, J.H.; Butland, B.K. The influence of Pregnancy and Reproductive span on the occurrence of autoimmune thyroiditis. Clin. Endocrinol. 1990, 32, 301–306. [Google Scholar] [CrossRef]
- Sawin, C.T.; Castelli, W.P.; Hershman, J.M.; McNamara, P.; Bacharach, P. The aging thyroid. Thyroid deficiency in the Framingham Study. Arch. Intern. Med. 1985, 145, 1386–1388. [Google Scholar] [CrossRef]
- Carlé, A.; Pedersen, I.B.; Knudsen, N.; Perrild, H.; Ovesen, L.; Rasmussen, L.B.; Laurberg, P. Epidemiology of subtypes of hyperthyroidism in Denmark: A population-based study. Eur. J. Endocrinol. 2011, 164, 801–809. [Google Scholar] [CrossRef]
- Vitti, P.; Rago, T.; Chiovato, L.; Pallini, S.; Santini, F.; Fiore, E.; Rocchi, R.; Martino, E.; Pinchera, A. Clinical Features of Patients with Graves’ Disease Undergoing Remission After Antithyroid Drug Treatment. Thyroid 1997, 7, 369–375. [Google Scholar] [CrossRef]
- Tonacchera, M.; Vitti, P.; Agretti, P.; Giulianetti, B.; Mazzi, B.; Cavaliere, R.; Ceccarini, G.; Fiore, E.; Viacava, P.; Naccarato, A.; et al. Activating Thyrotropin Receptor Mutations in Histologically Heterogeneous Hyperfunctioning Nodules of Multinodular Goiter. Thyroid 1998, 8, 559–564. [Google Scholar] [CrossRef]
- Kahaly, G.K. Management of Graves Thyroidal and Extrathyroidal Disease: An Update. J. Clin. Endocrinol. Metab. 2020, 105, 3704–3720. [Google Scholar] [CrossRef]
- Akande, E.O.; Hockaday, T.D. Plasma oestrogen and luteinizing hormone concentrations in thyrotoxic menstrual disturbance. Proc. R. Soc. Med. 1972, 65, 789–790. [Google Scholar] [CrossRef][Green Version]
- Ridgway, E.C.; Longcope, C.; Maloof, F. Metabolic Clearance and Blood Production Rates of Estradiol in Hyperthyroidism. J. Clin. Endocrinol. Metab. 1975, 41, 491–497. [Google Scholar] [CrossRef] [PubMed]
- The Male and Female Reproductive System in Thyrotoxicosis-Werner & Ingbar’s The Thyroid: A Fundamental & Clinical Text, 9th Edition. Available online: https://doctorlib.info/medical/thyroid/50.html (accessed on 26 July 2021).
- Southren, A.L.; Olivo, J.; Gordon, G.G.; Vittek, J.; Brener, J.; Rafii, F. The Conversion of Androgens to Estrogens in Hyperthyroidism. J. Clin. Endocrinol. Metab. 1974, 38, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Akande, E.O.; Hockaday, T.D.R.; Pereira, B.; Rosa, L.F.B.C.; Safi, D.A.; Bechara, E.J.H.; Curi, R. Plasma luteinizing hormone Levels in women with thyrotoxicosis. J. Endocrinol. 1972, 53, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Akande, E.O. The effect of oestrogen on plasma levels of luteinizing hormone in euthyroid and thyrotoxic postmenopausal women. BJOG Int. J. Obstet. Gynaecol. 1974, 81, 795–803. [Google Scholar] [CrossRef]
- Dunaif, A. Insulin Resistance and the Polycystic Ovary Syndrome: Mechanism and Implications for Pathogenesis. Endocr. Rev. 1997, 18, 774–800. [Google Scholar]
- Ganie, M.A.; Marwaha, R.K.; Aggarwal, R.; Singh, S. High prevalence of polycystic ovary syndrome characteristics in girls with euthyroid chronic lymphocytic thyroiditis: A case-control study. Eur. J. Endocrinol. 2010, 162, 1117–1122. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).