Appendix A
A detailed description of derivation for the constitutive hyperelastic model without strain amplification is given here. The statistical development of the kinetic theory of a Gaussian polymer chain, and later of an ensemble of chains corresponding to a polymer network, was originally pioneered by Kuhn [
12,
13]. Since a single polymer chain exhibits intricate statistics, one needs to simplify the problem by idealizing the chains’ structure. Kuhn tackled this issue by replacing the actual chain structure with an ideal chain of
equal segments (links), each with the statistical length
with an entirely random orientation in space. The average molecular dimension of this ideal chain is then characterized by the root mean square end-to-end distance
.
One of the main assumptions in the treatment of a Gaussian chain is that that the end-to-end distance of the chain must be much shorter than the fully extended length
of the chain, i.e.,
. In the non-Gaussian treatment of the problem, Kuhn and Grün [
3] abandoned this assumption by proposing the so-called inverse Langevin approximation which accounts for the finite extensibility of a polymer chain. Upon macroscopic deformation of the network, the end-to-end distance of a chain is stretched or compressed by a factor
(usually referred to as the chains stretch). According to the Langevin statistics, the corresponding change in the free energy of a chain due to the displacement of the chain ends (or cross-links) is given in [
35]
where the Boltzmann’s constant is denoted by
, and
is the absolute temperature. The inverse Langevin function is denoted with
. The constant
does not depend on the deformation and is merely a result of the assumption of an energy-free undeformed state.
The interaction of a single chain with its surroundings in a polymer network brings about restrictions on its movement and hence reduces the number of configurations it can adopt. Such interactions, also known as entanglements, can be considered by a so-called tube model first developed by Edwards [
50]. The physical idea behind this well-established model is the assumption of an average constraining potential, which acts equally on each segment of the chain, instead of focusing on discrete constraints. As the result of the uncrossability of the chains in a real polymer network, a single chain is confined to the neighborhood of an initial or mean configuration (also known as the primitive path). The movements of such a chain perpendicular to its contour are faced with great resistance, whereas the movements along the contour are much easier. Accordingly, one can assume that each chain is effectively confined in a tube-like region, where the tube radius characterizes the strength and the deformation dependence of the confinement. The tube radius
takes the value
in the undeformed state. Upon a macroscopic deformation of the polymer network, the tube deforms as well, and as a result of which the initial tube radius
changes into
. The change in the confinement free energy of the chain associated with the change in the tube cross-section area can be written as [
51]
in which
and
is a positive numerical factor depending on the geometry of the tube cross-section.
Consequently, the total strain energy per chain can be calculated as the sum of
associated with the unconstrained motion of the chain between two cross-links and a contribution
due to the constraining action of the tube. We then have [
52]
with
and
as the two micro-variables associated with the motion of the single chain in the polymer network.
In order to obtain the macroscopic strain energy of the network, one needs to find a relationship between the macroscopic deformation and the micro-variables. In continuum mechanics, the right Cauchy–Green tensor
provides direct information about the deformation of a body. The deformation gradient
describes how a line element
originating from position
in a reference configuration changes to
in the current configuration. Hyperelastic material models often depend on the invariants of the right Cauchy–Green tensor, i.e.,
,
and
, where principal stretches
and
represent the eigenvalues of
and square roots of the eigenvalues of
. Hyperelastic materials usually show a decoupled response to volumetric (shape preserving) and deviatoric (volume-preserving) deformations. The multiplicative split of the deformation gradient in a volumetric and isochoric contribution
was first proposed by Flory in [
53]. The isochoric part
satisfies the relation
One can define the corresponding isochoric right Cauchy-Green tensor
, wherein
. Upon the condition of incompressibility
, the invariants of
and
coincide [
48]. In this case, the strain energy function depends solely on
and
.
The total strain energy
of the network is dependent on the orientation and concentration of the single chains constituting the network. Different chain orientations in the network are often accounted for by the introduction of specific polyhedric unit cells, e.g., a cube in the eight-chain model of Arruda and Boyce [
11], a tetrahedron in the four-chain model [
54], and an octahedron in the three-chain model [
14]. Obviously, these cell structures only incorporate a discrete number of chain orientations.
Alternatively, one can consider a network cell of polymer chains with the orientation of their end-to-end vector distributed in a random fashion in the unstrained state. It can be assumed that all chains are joined together from one end at a central junction point and have approximately the same initial end-to-end distance . This suggests that the other end of the chains lies on the surface of a sphere with a total surface area of .
This is also known as a full-network model developed by Wu and van der Giessen [
55,
56]. The initial isotropy of a polymer network in the undeformed state implies an equal distribution of chain orientations in three-dimensional space, i.e., one is faced with a continuous uniform distribution of chain orientations. Under these conditions, the total strain energy
of the network can be obtained by accounting for the contribution
of all chains, with
as their number per unit volume, and their possible orientations. This can be expressed as
where
denotes a deferential area element of a unit sphere
(a sphere with unit radius) with total area
. Note that
and
represent the spherical coordinates in the undeformed state. Hence, the full-network model results in an integration over a unit sphere representing the continuous averaging for an equal distribution of chain distribution. The continuous averaging in Equation (A4) is denoted by the operator
, and is also expressed as the homogenization of the state variable
[
52].
Based on the full-network model, Beatty [
57] proposed an alternative approach which does not require a specific polyhedric unit cell. Accordingly, one can consider a full network of molecular chains with end-to-end vectors distributed in a random manner initially. All the chains are joined together from one end at an arbitrarily chosen junction point, the other end being on the surface of a sphere
with radius
. The end-to-end vector
of a representative chain along a unit vector
in the unstrained state is characterized in terms of the spherical coordinates
by the initial direction cosines
If the network undergoes an affine deformation with principal stretches
and
, the initial end-to-end chain vector changes from
to
, with
as the isochoric deformation gradient. Subsequently, one can express the squared, deformed end-to-end vector length as
The average value of the squared chain stretch over all possible chain orientations, namely
can then be obtained with the aid of the averaging operator
. As shown for Equation (A4), for an initially uniform distribution of chain orientations, this is equivalent to the integration over a unit sphere. Thus, by incorporating Equations (A5) and (A6), we have
with
as the first invariant of the right Cauchy–Green tensor
. The average chains stretch for a chain with an arbitrary initial orientations can be expressed as
which is equivalent to the well-known chain stretch in the eight-chain model of Arruda and Boyce [
11]. It is worth mentioning that this average chain stretch had already been obtained in 1971 by Dickie and Smith [
58] and later in 1989 by Kearsely [
59]. If we only consider the single chain energy
given using Equation (A1), the macroscopic strain energy per unit volume for a homogeneous full-network can be approximated as [
53,
58]
where, as previously found,
. For the sake of comparison, the integral
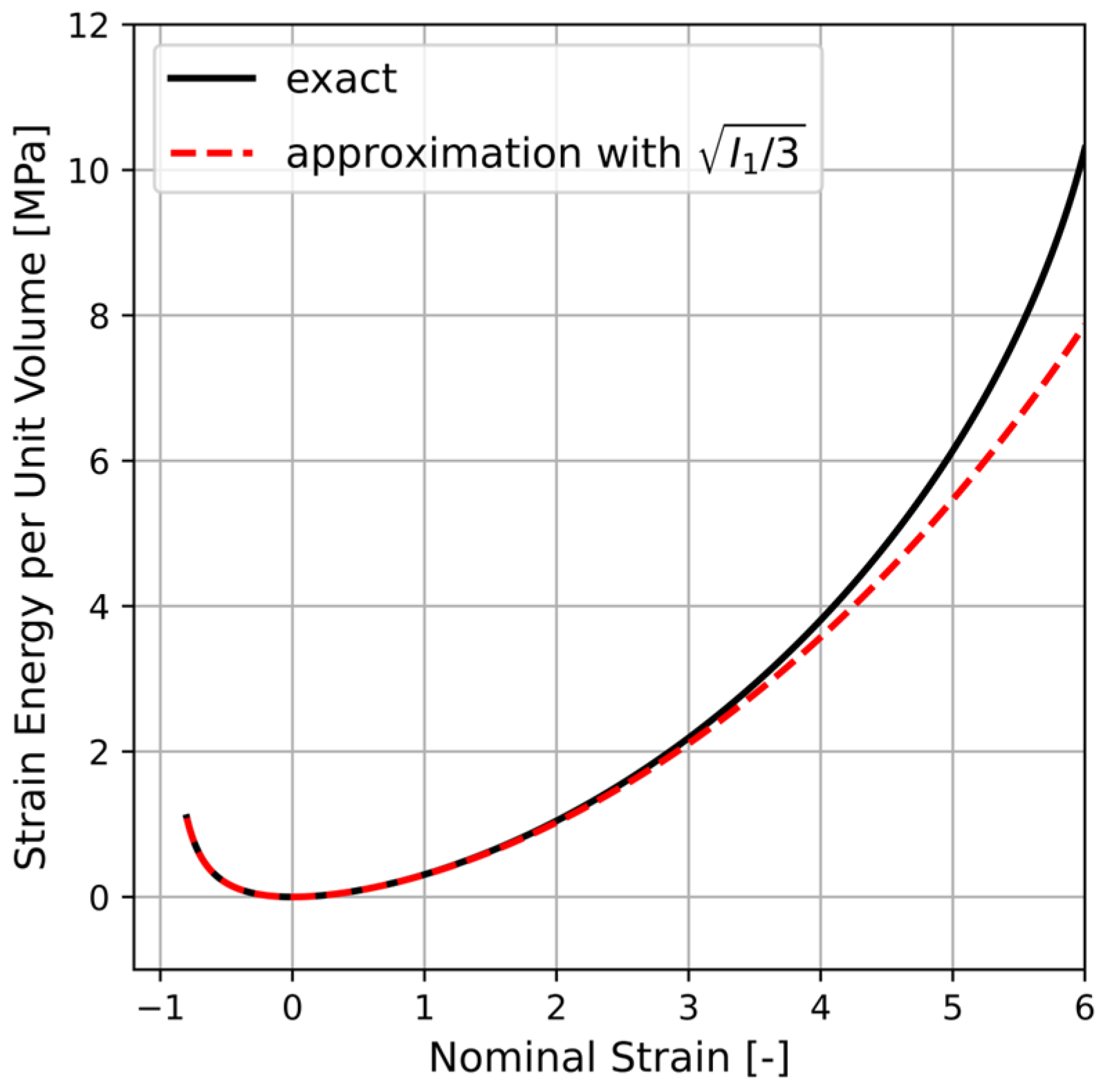
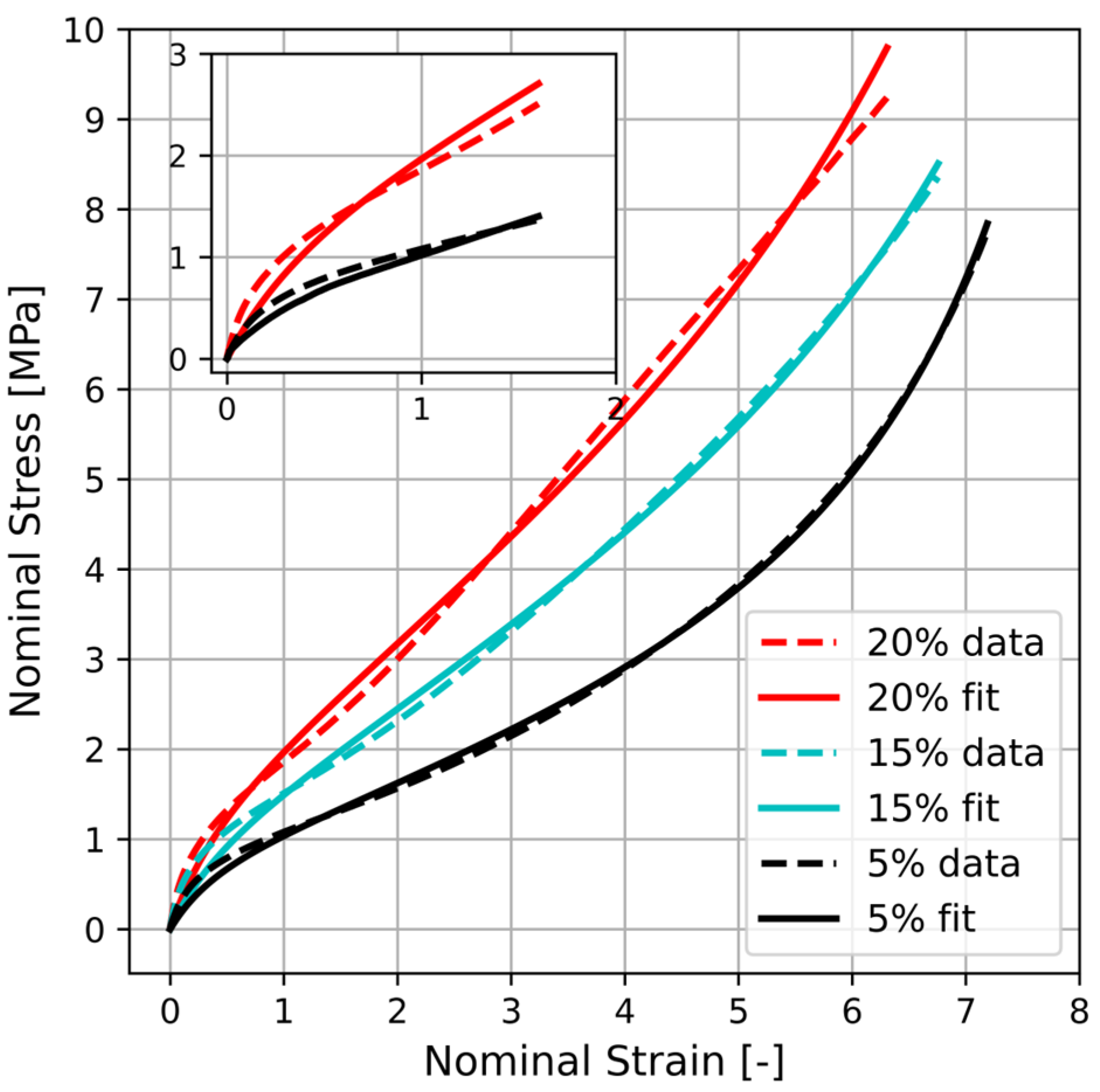
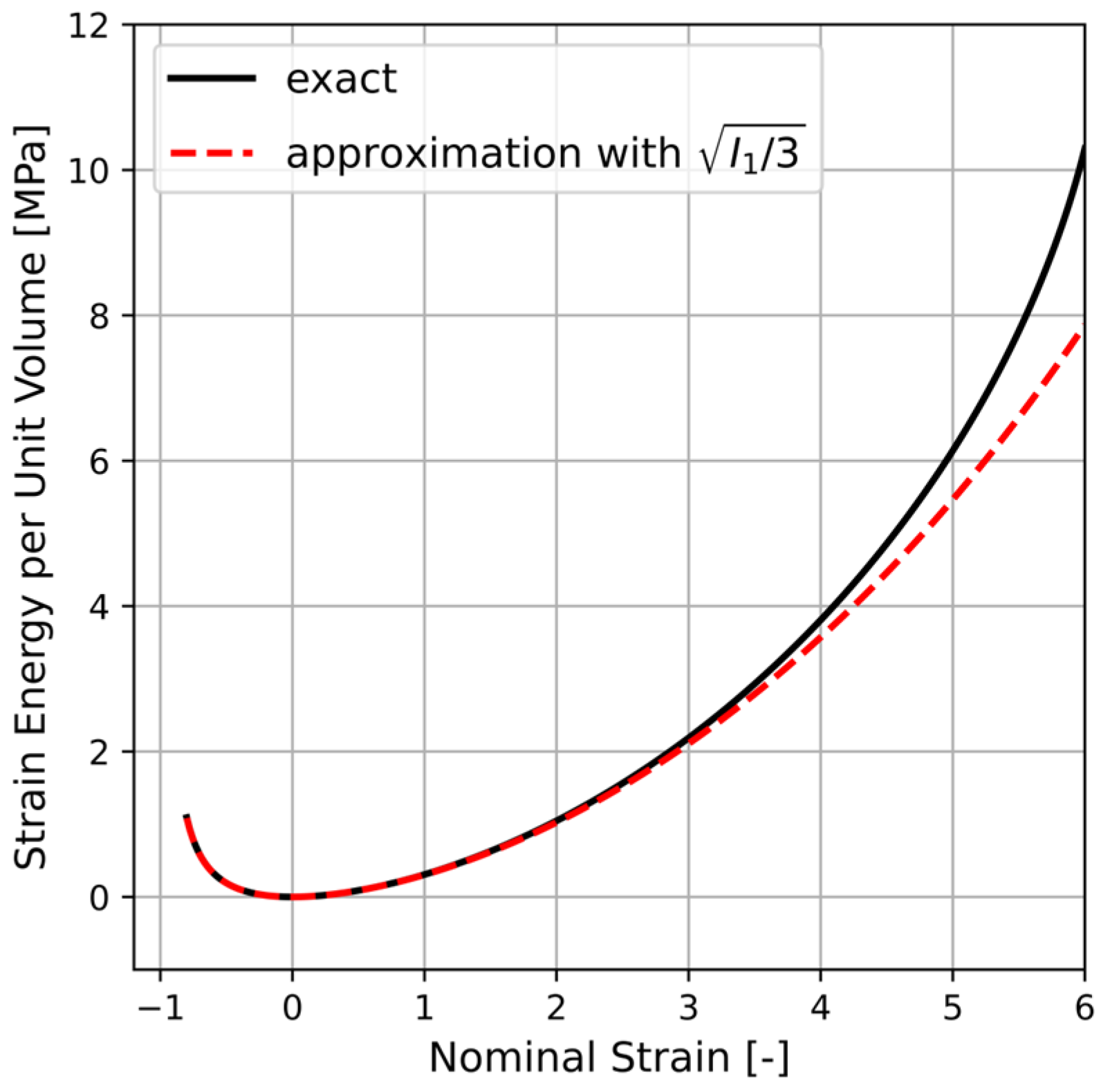
in Equation (A8) is solved numerically in a Python script by incorporating Equation (A1) and the direction cosines given using Equation (A5). The result is indicated by a solid line in
Figure A1 where also
with
(dashed line) is plotted. It is apparent that the approximation given by Equation (A8) is fairly accurate. Hence, by using the concept of averaging of a single chain micro-variable over all possible chain orientations, the corresponding macroscopic strain energy can be obtained.
Figure A1.
Comparison between (solid line) and with (dashed line) where the integral in the former is solved numerically using Python. The chosen constants are and (cf. Equation (A4)).
Figure A1.
Comparison between (solid line) and with (dashed line) where the integral in the former is solved numerically using Python. The chosen constants are and (cf. Equation (A4)).
In analogous matter, one can find the contribution of topological constraints (Equation (A2)) to the strain energy of the network. Hence, a relation between the corresponding micro-variable, i.e., the change in the tube cross-section
, and the macroscopic principal stretches
and
. In continuums mechanic, an area element
characterized by a unit normal vector
in an initial configuration is related to the deformed (current) configuration according to the Nanson’s Formula
where
is the area element in the current configuration with the unit normal
. Multiplying each side (dot product) of Equation (A9) by itself yields the squared areal change
with
and
as the principal values of the inverse of the right Cauchy–Green tensor
. One can assume that the micro-tube deformation, i.e., the reduction in the tube cross-section
coincides with the macroscopic areal change of the material surface element such that
where
in Equation (A9) is substituted by
. In order to account for all possible orientations of the normal vector
, we employ the averaging operator
. Hence, with the help of the relation found above for
and the direction cosine given using Equation (A5), we obtain
in which
is the second invariant of the right Cauchy–Green tensor
. It should be mentioned that Kearsley [
59] was apparently the first to notice the relation between the second strain invariant and the average areal change of a surface element in which the average is taken over all possible orientations. According to Equation (A10), the average reduction in the tube cross-section for a chain with an arbitrary initial orientation can be expressed as
. Subsequently, one can extend Equation (A8) by an extra term that accounts for the effect of topological constraints in the network, i.e.,
. Hence, substitution of the average micro-variables
and
in the single chain strain energies Equations (A1) and (A2) yields the total strain energy per unit volume of the network
The nominal or engineering stress for an incompressible material is given by
with
and
[
48,
60]. In the case where no volume change occurs,
turns out to be a Lagrange multiplier that can be evaluated from the reference state of stress. Note that the strain energy function given using Equation (A11) depends on the first and second invariant of the deformation and hence the chain rule must be applied for the derivation of the nominal stress. In case of a uniaxial deformation,
and the incompressibility condition
imply that
The hydrostatic pressure
can then be found from the boundary condition
. Consequently, the nominal stress in uniaxial deformation corresponding to the strain energy function Equation (A11) may be written as
in which the model parameters
and
are associated with the unconstrained motion of the chains between two cross-links, whereas the constant
is related to the tube constraint that models the entanglements.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}