Abstract
In the realm of advanced material science, multi-principal element alloys (MPEAs) have emerged as a focal point due to their exceptional mechanical properties and adaptability for high-performance applications. This study embarks on an extensive investigation of four MPEAs—CoCrNi, MoNbTa, HfNbTaTiZr, and HfMoNbTaTi—alongside key pure metals (Mo, Nb, Ta, Ni) to unveil their structural and mechanical characteristics. Utilizing a blend of molecular statics and hybrid molecular dynamics/Monte Carlo simulations, the research delves into the impact of chemical short-range order (CSRO) and thermal effects on the fundamental structural parameters and stacking fault energies in these alloys. The study systematically analyzes quantities such as lattice parameters, elastic constants (, , and ), and generalized stacking fault energies (GSFEs) across two distinct structures: random and CSRO. These properties are then evaluated at diverse temperatures (0, 300, 600, 900, 1200 K), offering a comprehensive understanding of temperature’s influence on material behavior. For CSRO, CoCrNi was annealed at 350 K and MoNbTa at 300 K, while both HfMoNbTaTi and HfNbTaTiZr were annealed at 300 K, 600 K, and 900 K, respectively. The results indicate that the lattice parameter increases with temperature, reflecting typical thermal expansion behavior. In contrast, both elastic constants and GSFE decrease with rising temperature, suggesting a reduction in resistance to stability and dislocation motion as thermal agitation intensifies. Notably, MPEAs with CSRO structures exhibit higher stiffness and GSFEs compared to their randomly structured counterparts, demonstrating the significant role of atomic ordering in enhancing material strength.
1. Introduction
Multi-principal element alloys (MPEAs) are an advanced class of metallic materials that have gained significant attention in materials science and engineering. The quest for materials that can withstand harsh environments, such as extreme heat, has led to the innovation of MPEAs, which differ from traditional alloys by using a balanced mix of multiple elements in equiatomic or near-equiatomic proportions at crystalline lattice sites, typically with either a face-centered cubic (FCC) or body-centered cubic (BCC) arrangement. The concept of equiatomic alloys forming stable solid solutions with high configurational entropies, resulting in the absence of complex phases and microstructures, was first introduced by Cantor et al. [1] and Yeh et al. [2], spurring further advancements in MPEAs. MPEAs have gained popularity owing to their fundamental properties, such as their high entropy effect, sluggish diffusion, cocktail effect, and lattice-distortion effect. The high configurational entropy notably decreases the Gibbs free energy within the system, enabling these alloys to avert the creation of brittle phases. This process, in turn, promotes the development of single-phase solid solutions, including FCC, BCC, and hexagonal-close-packed (HCP) structures, within MPEAs [3]. Some MPEAs demonstrate substantially enhanced characteristics compared to conventional alloys. These mechanical characteristics are believed to be a direct result of the unique lattice structures and atomic interactions inherent in MPEAs, resulting in a rich tapestry of deformation mechanisms, including, but not limited to, intricate dislocation activities, stacking fault generation, and the formation of nanotwinning networks [4,5,6].
CoCrNi stands out as a key example of the diverse and enhanced structural properties that MPEAs can provide. MPEAs with an FCC structure, such as CoCrNi, demonstrate remarkable mechanical characteristics including high fracture toughness, work hardening, and resistance to shear-band formation [7]. As a subset of the Cantor alloy [1], CoCrNi is characterized by twinning-dominated deformation and exhibits superior mechanical properties, including an increased tensile strength of 1.3 GPa and a failure strain of 90% [8] at 77 K, thus outperforming the Cantor alloy in these respects which has a 1 GPa tensile strength and a 60% failure strain [9] at the same temperature.
In the meantime, a significant limitation of refractory metallic alloys lies in their weak resistance to oxidation at high temperatures. MPEAs offer a solution by incorporating high levels of elements that enhance oxidation resistance. Much of the research in this area focuses on merging elements with high melting points, which are known for their strength and thermal stability at elevated temperatures (such as Hf, Mo, Nb, Ta, W, and Zr), with elements that improve oxidation resistance, including Al, Cr, Ti, and Si [10]. Senkov et al. [11] successfully created two single-phase MPEAs, MoNbTaW and MoNbTaVW, with near-atomic compositions and a BCC structure. The exceptional strength, durability, and distinctive microstructural features of these alloys indicate their suitability for use in sectors such as aerospace, automotive, and defense, where materials must withstand extreme stress and temperatures. Wadsworth et al. [12] highlight that refractory alloys, including Mo, Nb, Ta, and W, offer extended creep resistance, oxidation tolerance, and improved welding methods, addressing brittleness and oxidation challenges, thus proving valuable for aerospace uses. The two equimolar refractory MPEAs, HfMoNbTaTi and HfNbTaTiZr, demonstrate substantial compressive yield strengths at room temperature, with HfMoNbTaTi exhibiting strengths between 1369–1713 MPa [13,14] and HfNbTaTiZr showing strengths in the range of 900–1073 MPa [15,16,17,18,19]. HfMoNbTaTi shows an impressive strength at high temperatures, reaching 699 MPa at 1473 K [13], while HfNbTaTiZr maintains a considerable ductility at room temperature, ranging from 11% to 25% [19,20,21,22].
Results emerging from these investigations underscore the considerable potential of these alloys, warranting increased exploration and focus. However, the processing and fabrication of these alloys present challenges, being both resource-intensive and costly. Nonetheless, atomistic calculations yield promising perspectives on the mechanical characteristics of various alloys, facilitating the prediction of plastic deformation behaviors that are congruent with experimental results [23,24,25]. An in-depth understanding of dislocation dynamics within metals is essential for a comprehensive grasp of plastic deformation phenomena. In metals, dislocation glide necessitates the disruption and reformation of bonds across crystalline glide planes, analogous to the shearing of a crystal along a plane [26]. The energy required for this shearing is encapsulated in the concept of generalized stacking fault energy (GSFE) [27,28]. GSFE has emerged as a pivotal parameter in the design and development of MPEAs with varied chemical compositions, proving to be invaluable for investigating the effects of different elemental concentrations and compositions on the behavior of plastic deformation [29]. Mayahi et al. [30] show that increasing the Co content in AlCoCrFeNi enhances dislocation-mediated slip and martensitic transformation, influencing plastic deformation. Based on the evaluation of the GSFE curve, this research provides key insights for designing these alloys with an optimal balance of strength and ductility, notably in the AlCoCrFeNi alloy which exhibits superior properties. In CoCrFeNi, an increase in Cr and Fe reduces the GSFE while higher Co and Ni raise it, which impacts the yield stress [31].
MPEAs are characterized by the absence of chemical long-range ordering and often exhibit chemical short-range ordering (CSRO), a characteristic that has been experimentally validated [32,33]. CSRO in MPEAs significantly impacts their dislocation dynamics [34,35,36]. In MoNbTaW, CSRO significantly impacts the mobility of edge and screw dislocations, enhancing the mobility of edge dislocations while reducing the rate of double-kink nucleation in screw dislocations, an effect that diminishes at higher temperatures [36]. In HfNbTaZr, Chen et al. [22] investigated how annealing affects its nanostructure and mechanical properties, such as yield strength and ductility. They found that chemical short-range clustering (SRC) developed during annealing which significantly enhanced the alloy’s strength. These SRCs impact the critically resolved shear stress, influencing the transition from ductile to brittle behavior. Zheng et al. [37] worked on TaNbTi and MoNbTi RMPEAs, and it was found that CSRO greatly impacts dislocation glide, with higher CSRO in MoNbTi leading to increased unstable stacking fault energies (USFE). This elevation in USFE necessitates greater stress for dislocation movement and results in hardening due to the compositional fluctuation.
In our work, we focus on four specific equi-molar, single-phase, single-crystalline MPEAs: CoCrNi, MoNbTa, HfMoNbTaTi, and HfNbTaTiZr. We aim to calculate their lattice parameters, elastic constants , , and , and GSFE. These calculations directly correlate with key mechanical properties, including strength, ductility, and hardness. In particular, calculations of GSFE are crucial for improving the performance of materials, as they help identify deformation mechanisms like slip and twinning [38]. In this research, we analyzed specific parameters for two distinct structures: random and CSRO. Examining the properties of the CSRO structures offers valuable insights into the deformation mechanisms and behavior of materials [39,40,41]. To explore MPEAs exhibiting varying degrees of CSRO, we employed hybrid molecular dynamics (MD) and Monte Carlo (MC) simulations at different annealing temperatures. CoCrNi was annealed at 350 K and MoNbTa at 300 K, whereas both HfMoNbTaTi and HfNbTaTiZr were annealed at three different temperatures: 300 K, 600 K, and 900 K, respectively. Our focus was on deciphering the mechanical properties of these MPEAs under diverse temperature conditions. For some alloys, we gradually increased the environmental temperature from 300 K to 1200 K to assess the properties. Additionally, to contrast the behavior of MPEAs with that of pure metals, we also studied Mo, Nb, Ta, and Ni under the same escalating temperature range.
2. Materials and Methods
In this investigation, molecular statics simulations were conducted using LAMMPS [42]. The embedded-atom method (EAM) potentials are used to describe the interatomic potential. The atomic interactions within the CoCrNi system are represented by the EAM interatomic potential developed by Li et al. [43], which has been used in recent MD studies involving dislocation nucleation and motion [43,44]. The atomic interactions within HfNbTaTiZr and HfMoNbTaTi are represented by the EAM potential developed by Xu et al. [45]. In this study, the atomic interactions employed within MoNbTa were also developed by Xu et al. [46].
The EAM formulation that defines potential energy is given by [47]:
where N represents the number of atoms, signifies the number of neighboring atoms of atom i, V refers to the pair potential, F is the embedding potential, denotes the host electron density at atom i. Additionally, is the magnitude of vector which points from atom i to atom j, i.e.,
where is defined as the local electron density contributed by atom j to atom i. Here, formulations of V and only involve the i type of atom and the distance between atom i and its neighboring atom j, , regardless of the type of atom j is. For example, let atom i be Mo, then V and depend only on for any atom j which may be Nb or Ta. Thus, by using the EAM potentials for pure metals like Mo, Nb, or Ta, a unique “alloy potential” may be developed for every MPEA. For the four MPEAs considered here, this kind of potential will be utilized, as it has been to calculate GSFE in MoNbTi and NbTiZr [48], lattice distortion and chemical disorder in NbTiZr [49] and NbTaV [50,51], lattice parameters and stacking fault in Ti-Al-Nb systems [52], and high-temperature fatigue resistance of the polysynthetic twinned-TiAl single crystal [53].
To check the accuracy of the potential, density functional theory (DFT) simulations were conducted for lattice parameter and elastic constants , , and with random structures to compare the results with atomistic calculations. Details can be found in Appendix A. Table 1 presents the results of atomistic calculations for the MPEAs with random structures at 0 K, which are in good agreement with those obtained from DFT calculations, with the exception of , where a discrepancy is observed. Such discrepancies are known in many metals [54].
For this study, we utilized previously established CSRO structures for CrCoNi [55], as well as the quinary alloys HfMoNbTaTi and HfNbTaTiZr [45]. For this study, we constructed the CSRO structures for MoNbTa. Pure Mo samples were used to create MoNbTa MPEAs with completely random atomic distributions using Atomsk [56]. The Mo atoms were randomly substituted by Nb or Ta atoms until the desired equi-molar composition (Mo:Nb:Ta = 1:1:1) was reached. For the CSRO structures, first, using the hybrid MD/MC technique under the semi-grand-canonical ensemble at 1500 K, the chemical potential differences between Mo and Nb or Ta were determined to be , , respectively. Next, we employed the hybrid MD/MC method under the variance-constrained semi-grand-canonical ensemble for annealing temperatures of 300 K for MoNbTa MPEA. For both random and CSRO structures, the Warren–Cowley (WC) parameters were calculated via
where n means the nth-nearest-neighbor shell of the central i-type atom, denotes the probability of a j-type atom being around an atom of type i within the nth shell, is the concentration of j-type atom, and is the Kronecker delta function [57,58].
In a completely random structure, the value of will be close to zero, whereas a higher magnitude value of would suggest a higher degree of CSRO. The positive value of indicates the segregation of the same species () whereas the negative value of corresponds to the local ordering of the different species ( ).
In all alloys, we use “number+K+MDMC” to denote a structure obtained by hybrid MD/MC at a specific annealing temperature. For CoCrNi, one CSRO structure, annealed at 350 K [55], was considered. For this ternary, , . Figure 1 shows the average , for the 1st nearest neighbor, for the random and CSRO structure of MoNbTa generated at an annealing temperature of 300 K. The value for the random structure is close to zero, and the CSRO structure has a comparatively higher value, as expected. The positive value of and the high negative values of and indicate that these pairs tend to segregate or form local ordering in the structure. In Figure 2, the atomistic structures of random and CSRO annealed at 300 K are presented. For HfNbTaTiZr and HfMoNbTaTi, three different CSRO structures were used, which were annealed at temperatures of 300, 600, and 900 K respectively [45].

Figure 1.
Pairwise chemical WC parameters in MoNbTa.

Figure 2.
Atomistic structures of MoNbTa with random and CSRO structure. Visualization is carried out in OVITO [59].

Table 1.
Lattice parameters (in Å) and effective elastic constants , , and (in GPa) of random structures for four MPEAs using three different methods: experiment (Exp), DFT, and EAM.
Table 1.
Lattice parameters (in Å) and effective elastic constants , , and (in GPa) of random structures for four MPEAs using three different methods: experiment (Exp), DFT, and EAM.
| MPEA | Methods | ||||
|---|---|---|---|---|---|
| CoCrNi | Exp | 3.565 [60] | |||
| DFT | 3.305 [61] | 278.8 [62] | 188.8 [62] | 183.0 [62] | |
| EAM | 3.55644 | 251.644 | 178.246 | 95.134 | |
| DFT | 3.26 | 261.51 | 181.48 | 61.72 | |
| MoNbTa | EAM | 3.243 | 307.17 | 159.46 | 77.51 |
| HfMoNbTaTi | Exp | 3.305 [13,14] | |||
| DFT [45] | 3.305 | 209.95 | 133.92 | 39.68 | |
| EAM [45] | 3.31 | 201.42 | 146.64 | 82 | |
| HfNbTaTiZr | Exp | 3.304 [15] | 172 [63] | 116 [63] | 28 [63] |
| DFT [64] | 3.457 | 160.2 | 124.4 | 62.4 | |
| EAM [45] | 3.405 | 149.63 | 111.83 | 64.67 |
The bulk energy for every cell size is calculated to obtain the lattice parameter of each atomic structure using a sequence of periodic simulated cells of different sizes. In the end, the cohesive energy, is obtained by dividing the total number of atoms in the cell by the least bulk energy among these cells. It has been shown that this strategy, known as the “energy-volume method” yields results that are almost the same as those obtained using the “relaxation method” [65]. On the other hand, the stress–strain approach is used to determine the stiffness tensor C[66]. The effective elastic constants, , , and , at finite temperatures were determined using the Born-matrix method [67]. GSFEs were obtained following our previous work [48].
3. Results and Discussions
3.1. Lattice Parameters
As previously mentioned, we determined the lattice parameter, , for both the random and CSRO structures in four MPEAs, CoCrNi, MoNbTa, HfNbTaTiZr, and HfMoNbTaTi, across a temperature range of 300 to 1200 K. Figure 3a illustrates the correlation between and temperature for CoCrNi with random and CSRO structures and Figure 3b displays MoNbTa in its random and CSRO states and the simple rules of mixtures (SROM) of MoNbTa. It was observed that exhibits an increase with the temperature rise. The expansion of in MPEAs with rising temperature is accentuated by the pronounced lattice distortions caused by the varying atomic sizes and potentials of their constituent elements [68]. The of the random and CSRO structures in CoCrNi and MoNbTa exhibit minimal differences in compared to their random counterparts, suggesting that chemical order may have a negligible effect on thermal expansion in these specific alloys. Mayahi [30] notes that the GSFE can be influenced by alloy composition and ordering, but does not significantly affect . Furthermore, by averaging the values of Mo, Nb, and Ta at each temperature, the lattice parameters for a random MoNbTa with SROM were determined. The graph unequivocally demonstrates that a random MoNbTa with SROM closely resembles the observed values and temperature patterns in the ternary MoNbTa’s random and CSRO structures. The lattice parameter for the random MoNbTa with SROM exhibits smaller values compared to the random MoNbTa structure.
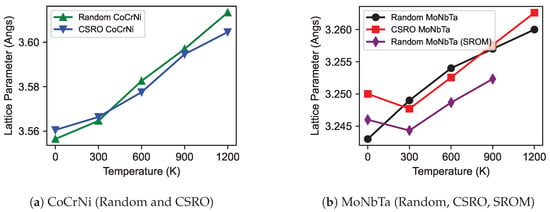

Figure 3.
Relationship between lattice parameter and temperature of MPEAs and pure metals. Data for Mo, Nb, Ta, HfMoNbTaTi, and HfNbTaTiZr at 0 K are taken from previous work.
As displayed in Figure 3c, the progression of for HfMoNbTaTi MPEA at various temperatures reflects a similar increasing pattern to that seen in the previous two MPEAs. For HfMoNbTaTi, the random structure demonstrates a larger compared to the three CSRO structures—300KMDMC, 600KMDMC, and 900KMDMC—which show lower values. As the temperature climbs, these CSRO structures exhibit a consistent and collective increase in . In Figure 3d, the lattice parameters of HfNbTaTiZr in a random structure configuration are presented, demonstrating a consistent increase as the temperature rises.
For pure metals, as observed in Figure 3e, Mo, Ta, Ni, and Nb all show an increase in with temperature, with Mo showing the least change. This behavior reflects the inherent material properties of pure metals, where the atomic bonding and size dictate their thermal expansion characteristics [69]. The lesser change in Mo’s could be due to its higher melting point and stronger bonds, which make it more resistant to thermal expansion. Using our interatomic potential, at 1200 K, Nb exhibited an unstable structure; thus, we did not calculate any properties for Nb at this high temperature.
3.2. Elastic Constants
The elastic constant behavior with varying temperature has been exhibited in Figure 4, Figure 5 and Figure 6. Three elastic constants (, , and ) have been explored in this study. Figure 4a–c illustrate all the elastic behavior trends for CoCrNi (random and CSRO), MoNbTa (random and CSRO), and random MoNbTa based on the simple rule of mixtures (SROM), Figure 5 illustrates all the elastic behavior trends for four structures of HfMoNbTaTi and HfNbTaTiZr; Figure 6 illustrates all the elastic behavior trends for four pure metals (Mo, Nb, Ni, and Ta). In all figures, it is evident that, with temperature, the elastic constants tend to decrease. This behavior is consistent with the fundamental understanding of the fact that, as temperature increases, the atomic vibrations become more pronounced, leading to a reduction in the material’s resistance to deformation, which is reflected in the decrease in elastic constants [37,70]. The elastic constants of the CSRO variants of CoCrNi and MoNbTa showed higher elastic constants than their random counterparts [71]. In the case of the elastic constant , the CSRO and random structures of both MoNbTa and CoCrNi display a similar trend, but both structures of CoCrNi display a pronounced decrease compared to the MoNbTa structures. A comparative study of SROM behavior in MoNbTa with random and CSRO structures is shown in Figure 4. The MoNbTa SROM values are similar to the ternary MoNbTa structures in both the random and CSRO variations. Nevertheless, we are unable to draw firm conclusions from this comparison because these SROM values do not closely match any particular MoNbTa MPEA structure. As a result, it would appear that SROM inadequately represents all of the ternary configurations of MoNbTa. The and for MoNbTa SROM display higher values than random MoNbTa structure, whereas the for MoNbTa SROM shows a lower value.

Figure 4.
Relationship between elastic constants and temperature in CoCrNi and MoNbTa.

Figure 5.
Relationship between elastic constants and temperature in HfMoNbTaTi and HfNbTaTiZr. Data at 0 K are taken from previous work [45].

Figure 6.
Relationship between elastic constants and temperature in four pure metals. Data for Mo [48], Nb [48], and Ta [46] at 0 K are taken from previous work.
Similarly, the four configurations of HfMoNbTaTi and HfNbTaTiZr demonstrate a consistent downward trend in elastic constants as temperature increases. The elastic behavior across the four distinct atomic arrangements reveals a negligible variation. This uniformity in the reduction of elastic constants with temperature, observed in both random and CSRO structures, implies that the alloy’s inherent thermal response predominates over the influence of particular atomic configurations [72].
Figure 6 indicates that the behavior of elastic constants in pure metals, specifically Mo, Nb, Ta, and Ni, parallels the patterns observed in MPEAs. Notably, the shear-related elastic constants, and , demonstrate minor variations across these metals when contrasted with the longitudinal elasticity constant, . The graphs further reveal that Mo exhibits superior stiffness, which can be attributed to its robust atomic bonding and elevated melting temperature.
3.3. Generalized Stacking Fault Energy (GSFE)
GSFE provides an energy map which is critical for understanding the mechanical stability and slip behavior of MPEAs, influenced by their complex elemental interplay. In this study, GSFE curves were generated by shifting the top half of the samples along the z-axis within the {111} or {110} plane. For robust statistical accuracy, GSFE curves on 20 distinct planes are calculated in each alloy. Additionally, we calculated the GSFE curves (not shown here) by shifting two halves of the cell with respect to each other continuously by 20 lattice periodicities within the same plane, but the results are different from those based on distinct planes, especially for the CSRO structures. Within the GSFE assessments, the calculations of USFE and intrinsic stacking fault energy (ISFE) are vital. USFE represents the peak energy barrier against dislocation nucleation, directly correlating to the alloy’s yield strength, while ISFE provides insight into the energy landscape post-slip.
For CoCrNi the GSFEs were computed along the crystallographic direction where the orientations of x, y, and z were , , and , respectively, taking into account both random and CSRO structures, whereas the GSFEs of MoNbTa and the two quinaries (HfMoNbTaTi and HfNbTaTiZr) were calculated along the direction on {110} planes, using simulation cells with orientations of , , and . In Figure 7, the mean GSFE curves of random- and CSRO-structured CoCrNi are presented with their standard deviations, with the unstable SFE () and intrinsic SFE () labeled in the graph. In general, a positive suggests a stable FCC structure, while a negative indicates that the material is more stable in an HCP structure. Our results show that the random structure exhibits a negative , while the of the CSRO structure displays a positive value indicating a more stable FCC structure. Experimentally, the presence of a short-range order in FCC CoCrNi has been confirmed [73], aligning with our findings. In Table 2, we observe the USFE and ISFE values for the CoCrNi alloy in both random and CSRO structures using EAM and DFT calculation. The CSRO structures of CoCrNi show a higher USFE and ISFE due to the possibility that the presence of CSRO will raise the energy barriers for dislocation nucleation and motion. On the other hand, the CSRO structure of MoNbTa shows a lower USFE than the random structure (see Table 3). In Table 3, a DFT calculation for the USFE of a random structure is also depicted. Moreover, the USFE value of the SROM random MoNbTa, which is 938.01 mJ/m, is in good agreement with the values found in the ternary MoNbTa that is both random and CSRO structured. This observation aligns with the findings reported by Romero et al. [74].

Figure 7.
GSFE curves of Random and CSRO structured CoCrNi. The shaded region indicates the stand deviation based on 20 GSFE curves.
Table 2.
USFE and ISFE (in mJ/m) of random and CSRO structures of CoCrNi. For the CSRO structures, EAM-based data were from annealing at 350 K, while DFT-based data were from annealing at 500 K.
Table 3.
USFE (in mJ/m) of random and CSRO structures of MoNbTa.
In Figure 8a, the HfMoNbTaTi alloy shows a clear decrease in USFE with increasing temperature across all structural states, indicating a reduction in resistance to dislocation motion as thermal agitation increases. A similar trend is observed in Figure 8b for the HfNbTaTiZr (random structure), where the USFE decreases as the temperature rises. This is consistent with the typical thermal softening behavior of metals and alloys [41,69,74,77]. Materials usually expand with rising temperatures, increasing the interatomic distances. As a result of this expansion, atomic bonds become weaker and the energy barrier for dislocation movements (as shown by USFE) is lowered. Additionally, rising temperatures intensify atomic vibrations, which further destabilise the lattice, facilitating the development of dislocations and reducing the USFE. In Figure 8c, the USFEs of four structures of HfNbTaTiZr have been displayed at 0 K where it is evident that the CSRO structures have higher values than the random ones. A similar trend was also followed by the four structures of HfMoNbTaTi (see Figure 8a). For the four MPEAs considered in this study, the GSFEs can be significantly affected by the CSRO, contributing to the observed differences in GSFE between CSRO and random structures [45,78].

Figure 8.
Relationship between USFE and temperature of HfMoNbTaTi, HfNbTaTiZr, and pure metals. Data for Mo [48], Nb [48], Ta [65], HfMoNbTaTi [45], and HfNbTaTiZr [45] at 0 K are taken from previous work.
Additionally, Figure 8d showcases the temperature dependence of USFE for pure metals Mo, Ta, Ni, and Nb. The temperature dependence of USFE is significant such that it influences the propensity for cross-slip and twinning behaviors in metals, which are crucial mechanisms for plastic deformation. An increased temperature can lead to more pronounced cross-slip activities [79]. Among the three BCC metals, Mo, with the highest USFE, shows the most substantial resistance to shear deformation at elevated temperatures, while Nb, with the lowest USFE, indicates increased ductility.
4. Conclusions
In this study, we explored the impact of temperature and CSRO on the lattice parameters, elastic constants, and GSFEs of four MPEAs which includes CoCrNi, MoNbTa, HfMoNbTaTi, and HfNbTaTiZr and four pure metals (Mo, Nb, Ta, and Ni). A detailed examination of CSRO structures, annealed at various temperatures, provided deeper insights into the effects of CSRO.
- The investigation revealed that an increase in temperature leads to an expansion in the lattice parameter across all studied MPEAs and pure metals. For CoCrNi and MoNbTa, the lattice parameters showed minimal variation between CSRO and random structures.
- Consistent with expectations, a temperature rise resulted in a decrease in the stiffness of both MPEAs and pure metals, thus reducing their elastic constants. Notably, CSRO structures demonstrated greater stiffness relative to random structured MPEAs. The trend in elastic constants for both random and CSRO structures was similar, with a decrease observed as the temperature increased. Additionally, CSRO structures manifested higher GSFEs than random structures.
- Understanding the role of CSRO and temperature in determining material properties allows for the fine-tuning of alloys to achieve desired mechanical properties, including ductility, strength, and resistance to deformation mechanisms like slip and twinning. It supports the development of more accurate models for predicting material behavior, facilitating the exploration of new alloys and composite materials. This work lays a foundational basis for further investigations into the mechanical characteristics of various alloys, encouraging a more in-depth exploration of dislocation dynamics and the effects of elemental concentrations on plastic deformation behavior.
Author Contributions
Conceptualization, S.X. and W.-R.J.; Methodology, S.X. and W.-R.J.; Software, S.M., W.-R.J. and S.X.; Validation, S.M. and S.X.; Formal Analysis, S.M.; Investigation, S.M. and S.X.; Resources, W.-R.J. and S.X.; Data Curation, S.M., S.X. and W.-R.J.; Writing—Original Draft Preparation, S.M.; Writing—Review Editing, S.X. and W.-R.J.; Visualization, S.M.; Supervision, S.X.; Project Administration, S.X.; Funding Acquisition, S.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by a grant from the Research Council of the University of Oklahoma (OU) Norman Campus. S.M. expresses gratitude for receiving the Ph.D. Recruitment Excellence Fellowship awarded by the Gallogly College of Engineering at OU. Additionally, S.M. and S.X. are grateful for the startup funds provided by OU.
Data Availability Statement
The data presented in this study are openly available at https://github.com/shuozhixu/Modelling_2024 (accessed on 27 February 2024).
Acknowledgments
This work used Bridges-2 at the Pittsburgh Supercomputing Center through allocation # MAT230058 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. Some of the computing for this project was performed at the OU Supercomputing Center for Education & Research (OSCER) at the University of Oklahoma (OU).
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A. Structural Parameters and GSFE of MoNbTa Calculated via DFT
To confirm the accuracy of the alloy potential for MoNbTa, this study also conducted DFT calculations to determine the lattice parameters, elastic constants, and GSFE in MoNbTa, as detailed in Table 1 and Table 3. These DFT calculations were performed using VASP [80] in its random configuration. Our approach aligns with the methodologies referenced in Refs. [81,82]. For the exchange-correlation energy functional, we employed the Perdew–Burke–Ernzerhof version of the generalized gradient approximation [83]. The Brillouin zone integrations were executed using the Monkhorst–Pack grid [84], applying a smearing width of 0.2 eV as per the Methfessel–Paxton technique [85]. k-point meshes of and were utilized for basic structural parameters and GSFE calculations, respectively.
References
- Cantor, B.; Chang, I.T.H.; Knight, P.; Vincent, A.J.B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375-377, 213–218. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured High-Entropy Alloys with Multiple Principal Elements: Novel Alloy Design Concepts and Outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Miracle, D.B.; Senkov, O.N. A critical review of high entropy alloys and related concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Ma, E. Unusual dislocation behavior in high-entropy alloys. Scr. Mater. 2020, 181, 127–133. [Google Scholar] [CrossRef]
- Nöhring, W.G.; Curtin, W.A. Design using randomness: A new dimension for metallurgy. Scr. Mater. 2020, 187, 210–215. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, S.; Ritchie, R.O.; Meyers, M.A. Mechanical properties of high-entropy alloys with emphasis on face-centered cubic alloys. Prog. Mater. Sci. 2019, 102, 296–345. [Google Scholar] [CrossRef]
- Gludovatz, B.; Hohenwarter, A.; Thurston, K.V.S.; Bei, H.; Wu, Z.; George, E.P.; Ritchie, R.O. Exceptional damage-tolerance of a medium-entropy alloy CrCoNi at cryogenic temperatures. Nat. Commun. 2016, 7, 10602. [Google Scholar] [CrossRef]
- Zeng, Z.; Xiang, M.; Zhang, D.; Shi, J.; Wang, W.; Tang, X.; Tang, W.; Wang, Y.; Ma, X.; Chen, Z.; et al. Mechanical properties of Cantor alloys driven by additional elements: A review. J. Mater. Res. Technol. 2021, 15, 1920–1934. [Google Scholar] [CrossRef]
- Senkov, O.N.; Miracle, D.B.; Chaput, K.J.; Couzinie, J.P. Development and exploration of refractory high entropy alloys—A review. J. Mater. Res. 2018, 33, 3092–3128. [Google Scholar] [CrossRef]
- Senkov, O.N.; Wilks, G.B.; Miracle, D.B.; Chuang, C.P.; Liaw, P.K. Refractory high-entropy alloys. Intermetallics 2010, 18, 1758–1765. [Google Scholar] [CrossRef]
- Wadsworth, J.; Nieh, T.G.; Stephens, J.J. Recent advances in aerospace refractory metal alloys. Int. Mater. Rev. 1988, 33, 131–150. [Google Scholar] [CrossRef]
- Tseng, K.K.; Juan, C.C.; Tso, S.; Chen, H.C.; Tsai, C.W.; Yeh, J.W. Effects of Mo, Nb, Ta, Ti, and Zr on Mechanical Properties of Equiatomic Hf-Mo-Nb-Ta-Ti-Zr Alloys. Entropy 2019, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Bian, H.; Aoyagi, K.; Hayasaka, Y.; Yamanaka, K.; Chiba, A. Synergetic strengthening in HfMoNbTaTi refractory high-entropy alloy via disordered nanoscale phase and semicoherent refractory particle. Mater. Des. 2021, 212, 110248. [Google Scholar] [CrossRef]
- Senkov, O.N.; Scott, J.M.; Senkova, S.V.; Miracle, D.B.; Woodward, C.F. Microstructure and room temperature properties of a high-entropy TaNbHfZrTi alloy. J. Alloys Compd. 2011, 509, 6043–6048. [Google Scholar] [CrossRef]
- Senkov, O.N.; Scott, J.M.; Senkova, S.V.; Meisenkothen, F.; Miracle, D.B.; Woodward, C.F. Microstructure and elevated temperature properties of a refractory TaNbHfZrTi alloy. Mater. Sci. Eng. A 2012, 47, 4062–4074. [Google Scholar] [CrossRef]
- Couzinié, J.P.; Lilensten, L.; Champion, Y.; Dirras, G.; Perrière, L.; Guillot, I. On the room temperature deformation mechanisms of a TiZrHfNbTa refractory high-entropy alloy. Mater. Sci. Eng. A 2015, 645, 255–263. [Google Scholar] [CrossRef]
- An, Z.; Mao, S.; Liu, Y.; Wang, L.; Zhou, H.; Gan, B.; Zhang, Z.; Han, X. A novel HfNbTaTiV high-entropy alloy of superior mechanical properties designed on the principle of maximum lattice distortion. J. Mater. Sci. Technol. 2021, 79, 109–117. [Google Scholar] [CrossRef]
- Yasuda, H.Y.; Yamada, Y.; Cho, K.; Nagase, T. Deformation behavior of HfNbTaTiZr high entropy alloy singe crystals and polycrystals. Mater. Sci. Eng. A 2021, 809, 140983. [Google Scholar] [CrossRef]
- Juan, C.C.; Tsai, M.H.; Tsai, C.W.; Hsu, W.L.; Lin, C.M.; Chen, S.K.; Lin, S.J.; Yeh, J.W. Simultaneously increasing the strength and ductility of a refractory high-entropy alloy via grain refining. Mater. Lett. 2016, 184, 200–203. [Google Scholar] [CrossRef]
- Senkov, O.N.; Pilchak, A.L.; Semiatin, S.L. Effect of Cold Deformation and Annealing on the Microstructure and Tensile Properties of a HfNbTaTiZr Refractory High Entropy Alloy. Metall. Mater. Trans. A 2018, 49, 2876–2892. [Google Scholar] [CrossRef]
- Chen, S.Y.; Wang, L.; Li, W.D.; Tong, Y.; Tseng, K.K.; Tsai, C.W.; Yeh, J.W.; Ren, Y.; Guo, W.; Poplawsky, J.D.; et al. Peierls barrier characteristic and anomalous strain hardening provoked by dynamic-strain-aging strengthening in a body-centered-cubic high-entropy alloy. Mater. Res. Lett. 2019, 7, 475–481. [Google Scholar] [CrossRef]
- Fang, Q.; Chen, Y.; Li, J.; Jiang, C.; Liu, B.; Liu, Y.; Liaw, P.K. Probing the phase transformation and dislocation evolution in dual-phase high-entropy alloys. Int. J. Plast. 2019, 114, 161–173. [Google Scholar] [CrossRef]
- Smith, L.T.W.; Su, Y.; Xu, S.; Hunter, A.; Beyerlein, I.J. The effect of local chemical ordering on Frank-Read source activation in a refractory multi-principal element alloy. Int. J. Plast. 2020, 134, 102850. [Google Scholar] [CrossRef]
- Hua, D.; Xia, Q.; Wang, W.; Zhou, Q.; Li, S.; Qian, D.; Shi, J.; Wang, H. Atomistic insights into the deformation mechanism of a CoCrNi medium entropy alloy under nanoindentation. Int. J. Plast. 2021, 142, 102997. [Google Scholar] [CrossRef]
- Hull, D.; Bacon, D.J. Introduction to Dislocations; Butterworth-Heinemann: Oxford, UK, 2001. [Google Scholar]
- Vitek, V. Atomic level computer modelling of crystal defects with emphasis on dislocations: Past, present and future. Prog. Mater. Sci. 2011, 56, 577–585. [Google Scholar] [CrossRef]
- Vitek, V. Structure of dislocation cores in metallic materials and its impact on their plastic behaviour. Prog. Mater. Sci. 1992, 36, 1–27. [Google Scholar] [CrossRef]
- Su, Y.; Xu, S.; Beyerlein, I.J. Density functional theory calculations of generalized stacking fault energy surfaces for eight face-centered cubic transition metals. J. Appl. Phys. 2019, 126, 105112. [Google Scholar] [CrossRef]
- Mayahi, R. An investigation concerning generalized stacking fault behavior of AlCoxCrFeNi high entropy alloys: Insights from first-principles study. J. Alloys Compd. 2020, 818, 152928. [Google Scholar] [CrossRef]
- Jarlöv, A.; Ji, W.; Zhu, Z.; Tian, Y.; Babicheva, R.; An, R.; Seet, H.L.; Nai, M.L.S.; Zhou, K. Molecular dynamics study on the strengthening mechanisms of Cr–Fe–Co–Ni high-entropy alloys based on the generalized stacking fault energy. J. Alloys Compd. 2022, 905, 164137. [Google Scholar] [CrossRef]
- Huang, S.; Luo, S.; Qin, L.; Shu, D.; Sun, B.; Lunt, A.J.G.; Korsunsky, A.M.; Mi, J. 3D local atomic structure evolution in a solidifying Al-0.4Sc dilute alloy melt revealed in operando by synchrotron X-ray total scattering and modelling. Scr. Mater. 2022, 211, 114484. [Google Scholar] [CrossRef]
- Choudhuri, D.; Majumdar, B.S.; Wilkinson, H. Investigation of in-liquid ordering mediated transformations in Al-Sc via ab initio molecular dynamics and unsupervised learning. Phys. Rev. Mater. 2022, 6, 103406. [Google Scholar] [CrossRef]
- Li, X.G.; Chen, C.; Zheng, H.; Zuo, Y.; Ong, S.P. Complex strengthening mechanisms in the NbMoTaW multi-principal element alloy. npj Comput. Mater. 2020, 6, 70. [Google Scholar] [CrossRef]
- Zhao, L.; Zong, H.; Ding, X.; Lookman, T. Anomalous dislocation core structure in shock compressed bcc high-entropy alloys. Acta Mater. 2021, 209, 116801. [Google Scholar] [CrossRef]
- Yin, S.; Zuo, Y.; Abu-Odeh, A.; Zheng, H.; Li, X.G.; Ding, J.; Ong, S.P.; Asta, M.; Ritchie, R.O. Atomistic simulations of dislocation mobility in refractory high-entropy alloys and the effect of chemical short-range order. Nat. Commun. 2021, 12, 4873. [Google Scholar] [CrossRef]
- Zheng, H.; Fey, L.T.W.; Li, X.G.; Hu, Y.J.; Qi, L.; Chen, C.; Xu, S.; Beyerlein, I.J.; Ong, S.P. Multi-scale Investigation of Chemical Short-Range Order and Dislocation Glide in the MoNbTi and TaNbTi Refractory Multi-Principal Element Alloys. npj Comput. Mater. 2023, 9, 89. [Google Scholar] [CrossRef]
- An, X.H.; Wu, S.D.; Wang, Z.G.; Zhang, Z.F. Significance of stacking fault energy in bulk nanostructured materials: Insights from Cu and its binary alloys as model systems. Prog. Mater. Sci. 2019, 101, 1–45. [Google Scholar] [CrossRef]
- Ziehl, T.J.; Morris, D.; Zhang, P. Detection and impact of short-range order in medium/high-entropy alloys. iScience 2023, 26, 106209. [Google Scholar] [CrossRef]
- Birbilis, N.; Choudhary, S.; Scully, J.R.; Taheri, M.L. A perspective on corrosion of multi-principal element alloys. npj Mater. Degrad. 2021, 5, 14. [Google Scholar] [CrossRef]
- Wang, S.D.; Liu, X.J.; Lei, Z.F.; Lin, D.Y.; Bian, F.G.; Yang, C.M.; Jiao, M.Y.; Du, Q.; Wang, H.; Wu, Y.; et al. Chemical short-range ordering and its strengthening effect in refractory high-entropy alloys. Phys. Rev. B 2021, 103, 104107. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Li, Q.J.; Sheng, H.; Ma, E. Strengthening in multi-principal element alloys with local-chemical-order roughened dislocation pathways. Nat. Commun. 2019, 10, 3563. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.H.; Wang, Y.J.; Dai, L.H. Novel atomic-scale mechanism of incipient plasticity in a chemically complex CrCoNi medium-entropy alloy associated with inhomogeneity in local chemical environment. Acta Mater. 2020, 194, 283–294. [Google Scholar] [CrossRef]
- Xu, S.; Jian, W.R.; Beyerlein, I.J. Ideal simple shear strengths of two HfNbTaTi-based quinary refractory multi-principal element alloys. APL Mater. 2022, 10, 111107. [Google Scholar] [CrossRef]
- Xu, S.; Chavoshi, S.Z.; Su, Y. On calculations of basic structural parameters in multi-principal element alloys using small atomistic models. Comput. Mater. Sci 2022, 202, 110942. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef]
- Xu, S.; Hwang, E.; Jian, W.R.; Su, Y.; Beyerlein, I.J. Atomistic calculations of the generalized stacking fault energies in two refractory multi-principal element alloys. Intermetallics 2020, 124, 106844. [Google Scholar] [CrossRef]
- Rao, S.I.; Akdim, B.; Antillon, E.; Woodward, C.; Parthasarathy, T.A.; Senkov, O.N. Modeling solution hardening in BCC refractory complex concentrated alloys: NbTiZr, Nb1.5TiZr0.5 and Nb0.5TiZr1.5. Acta Mater. 2019, 168, 222–236. [Google Scholar] [CrossRef]
- Maresca, F.; Curtin, W.A. Theory of screw dislocation strengthening in random BCC alloys from dilute to “High-Entropy” alloys. Acta Mater. 2020, 182, 144–162. [Google Scholar] [CrossRef]
- Maresca, F.; Curtin, W.A. Mechanistic origin of high strength in refractory BCC high entropy alloys up to 1900K. Acta Mater. 2020, 182, 235–249. [Google Scholar] [CrossRef]
- Yan, S.; Qi, Z.; Chen, Y.; Cao, Y.; Zhang, J.; Zheng, G.; Chen, F.; Bian, T.; Chen, G. Interlamellar boundaries govern cracking. Acta Mater. 2021, 215, 117091. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, Y.; Qi, Z.; Chen, G. Increasing high-temperature fatigue resistance of polysynthetic twinned TiAl single crystal by plastic strain delocalization. J. Mater. Sci. Technol. 2021, 93, 53–59. [Google Scholar] [CrossRef]
- Ikehata, H.; Nagasako, N.; Furuta, T.; Fukumoto, A.; Miwa, K.; Saito, T. First-principles calculations for development of low elastic modulus Ti alloys. Phys. Rev. B 2004, 70, 174113. [Google Scholar] [CrossRef]
- Jian, W.R.; Xie, Z.; Xu, S.; Su, Y.; Yao, X.; Beyerlein, I.J. Effects of lattice distortion and chemical short-range order on the mechanisms of deformation in medium entropy alloy CoCrNi. Acta Mater. 2020, 199, 352–369. [Google Scholar] [CrossRef]
- Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Comput. Phys. Commu. 2015, 197, 212–219. [Google Scholar] [CrossRef]
- Cowley, J.M. An Approximate Theory of Order in Alloys. Phys. Rev. 1950, 77, 669–675. [Google Scholar] [CrossRef]
- De Fontaine, D. The number of independent pair-correlation functions in multicomponent systems. J. Appl. Crystallogr. 1971, 4, 15–19. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Modell. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Moravcik, I.; Cizek, J.; Kovacova, Z.; Nejezchlebova, J.; Kitzmantel, M.; Neubauer, E.; Kubena, I.; Hornik, V.; Dlouhy, I. Mechanical and microstructural characterization of powder metallurgy CoCrNi medium entropy alloy. Mater. Sci. Eng. A 2017, 701, 370–380. [Google Scholar] [CrossRef]
- Ye, Y.F.; Zhang, Y.H.; He, Q.F.; Zhuang, Y.; Wang, S.; Shi, S.Q.; Hu, A.; Fan, J.; Yang, Y. Atomic-scale distorted lattice in chemically disordered equimolar complex alloys. Acta Mater. 2018, 150, 182–194. [Google Scholar] [CrossRef]
- Ge, H.; Song, H.; Shen, J.; Tian, F. Effect of alloying on the thermal-elastic properties of 3d high-entropy alloys. Mater. Chem. Phys. 2018, 210, 320–326. [Google Scholar] [CrossRef]
- Dirras, G.; Lilensten, L.; Djemia, P.; Laurent-Brocq, M.; Tingaud, D.; Couzinié, J.P.; Perrière, L.; Chauveau, T.; Guillot, I. Elastic and plastic properties of as-cast equimolar TiHfZrTaNb high-entropy alloy. Mater. Sci. Eng. A 2016, 654, 30–38. [Google Scholar] [CrossRef]
- Fazakas, E.; Zadorozhnyy, V.; Varga, L.K.; Inoue, A.; Louzguine-Luzgin, D.V.; Tian, F.; Vitos, L. Experimental and theoretical study of Ti20Zr20Hf20Nb20X20 (X=V or Cr) refractory high-entropy alloys. Int. J. Refrac. Met. Hard Mater. 2014, 47, 131–138. [Google Scholar] [CrossRef]
- Wang, X.; Xu, S.; Jian, W.R.; Li, X.G.; Su, Y.; Beyerlein, I.J. Generalized stacking fault energies and Peierls stresses in refractory body-centered cubic metals from machine learning-based interatomic potentials. Comput. Mater. Sci. 2021, 192, 110364. [Google Scholar] [CrossRef]
- Jian, W.R.; Xu, S.; Beyerlein, I.J. On the significance of model design in atomistic calculations of the Peierls stress in Nb. Comput. Mater. Sci. 2021, 188, 110150. [Google Scholar] [CrossRef]
- Ray, J.R.; Rahman, A. Statistical ensembles and molecular dynamics studies of anisotropic solids. J. Chem. Phys. 1984, 80, 4423–4428. [Google Scholar] [CrossRef]
- Huang, J.; Fang, W.; Xue, C.; Peng, T.; Yu, H.; Li, J.; Sun, L.; He, X.; Liu, B.; Yang, Y.; et al. Exploring the relationship between lattice distortion and phase stability in a multi-principal element alloy system based on machine learning method. Comput. Mater. Sci. 2023, 221, 112089. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, H.; Li, W.; Ding, X.; Wang, Y.; Vitos, L. Generalized Stacking Fault Energy of Al-Doped CrMnFeCoNi High-Entropy Alloy. Nanomaterials 2019, 10, 59. [Google Scholar] [CrossRef]
- Geslin, P.A.; Rodney, D. Microelasticity model of random alloys. Part I: Mean square displacements and stresses. J. Mech. Phys. Solids 2021, 153, 104479. [Google Scholar] [CrossRef]
- Zhang, W.; Chabok, A.; Kooi, B.J.; Pei, Y. Additive manufactured high entropy alloys: A review of the microstructure and properties. Mater. Des. 2022, 220, 110875. [Google Scholar] [CrossRef]
- Feng, R.; Kim, G.; Yu, D.; Chen, Y.; Chen, W.; Liaw, P.K.; An, K. Elastic behavior of binary and ternary refractory multi-principal-element alloys. Mater. Des. 2022, 219, 110820. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, S.; Ding, J.; Chong, Y.; Jia, T.; Ophus, C.; Asta, M.; Ritchie, R.O.; Minor, A.M. Short-range order and its impact on the CrCoNi medium-entropy alloy. Nature 2020, 581, 283–287. [Google Scholar] [CrossRef]
- Romero, R.A.; Xu, S.; Jian, W.R.; Beyerlein, I.J.; Ramana, C.V. Atomistic simulations of the local slip resistances in four refractory multi-principal element alloys. Int. J. Plast. 2022, 149, 103157. [Google Scholar] [CrossRef]
- Huang, H.; Li, X.; Dong, Z.; Li, W.; Huang, S.; Meng, D.; Lai, X.; Liu, T.; Zhu, S.; Vitos, L. Critical stress for twinning nucleation in CrCoNi-based medium and high entropy alloys. Acta Mater. 2018, 149, 388–396. [Google Scholar] [CrossRef]
- Ding, J.; Yu, Q.; Asta, M.; Ritchie, R.O. Tunable stacking fault energies by tailoring local chemical order in CrCoNi medium-entropy alloys. Proc. Natl. Acad. Sci. USA 2018, 115, 8919–8924. [Google Scholar] [CrossRef]
- Wang, H.; Pan, R.; Tang, A.; She, J.; Mi, X.; Wu, L.; Tan, J. Effects of Nb concentration and temperature on generalized stacking fault energy of Zr–Nb alloys by molecular dynamics simulations. Mater. Res. Express 2021, 8, 016540. [Google Scholar] [CrossRef]
- Pei, Z.; Zhang, S.; Lei, Y.; Zhang, F.; Chen, M. Decoupling between Shockley partials and stacking faults strengthens multiprincipal element alloys. Proc. Natl. Acad. Sci. USA 2021, 118, e2114167118. [Google Scholar] [CrossRef]
- Zhao, B.; Huang, P.; Zhang, L.; Li, S.; Zhang, Z.; Yu, Q. Temperature Effect on Stacking Fault Energy and Deformation Mechanisms in Titanium and Titanium-aluminium Alloy. Sci. Rep. 2020, 10, 3086. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Xu, S.; Su, Y.; Smith, L.T.W.; Beyerlein, I.J. Frank-Read source operation in six body-centered cubic refractory metals. J. Mech. Phys. Solids 2020, 141, 104017. [Google Scholar] [CrossRef]
- Su, Y.; Ardeljan, M.; Knezevic, M.; Jain, M.; Pathak, S.; Beyerlein, I.J. Elastic constants of pure body-centered cubic Mg in nanolaminates. Comput. Mater. Sci 2020, 174, 109501. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).