
Ovarian Cancer: Treatment and Resistance to Pharmacotherapy


Abstract
:1. Introduction
2. Histological Subtypes of OC
Tumor Heterogeneity
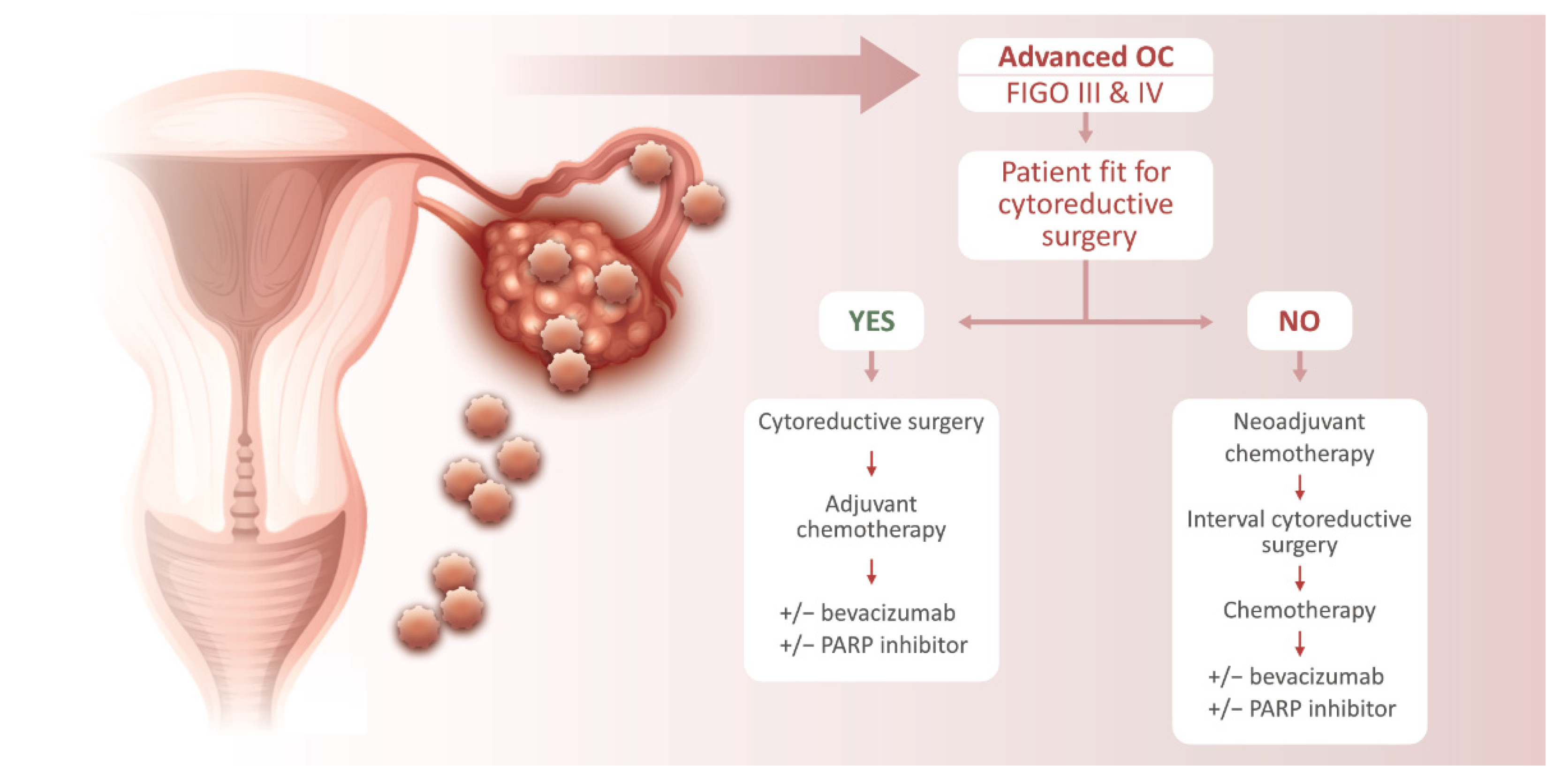
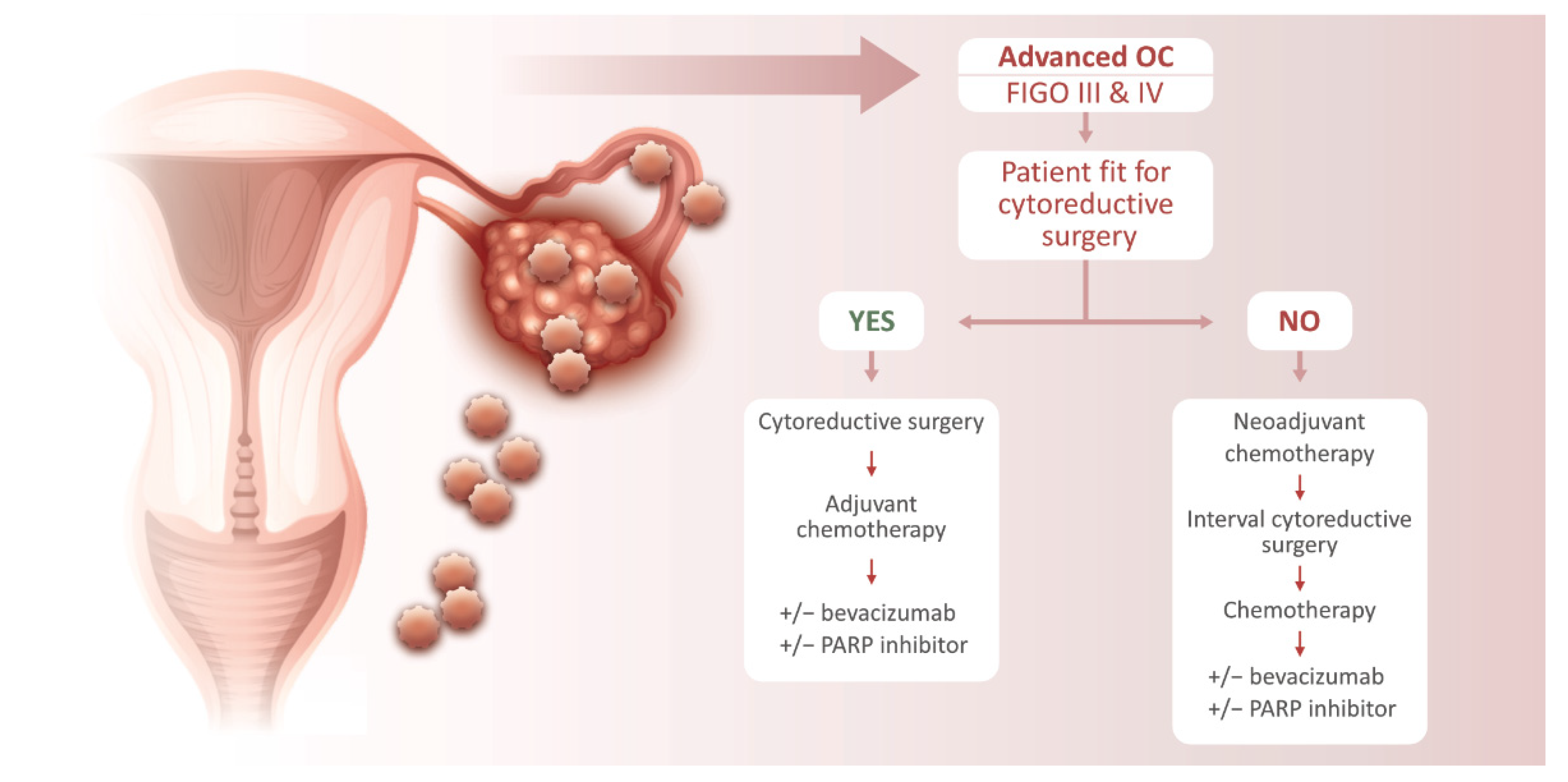
3. Ovarian Cancer Treatment
4. Resistance to Treatment
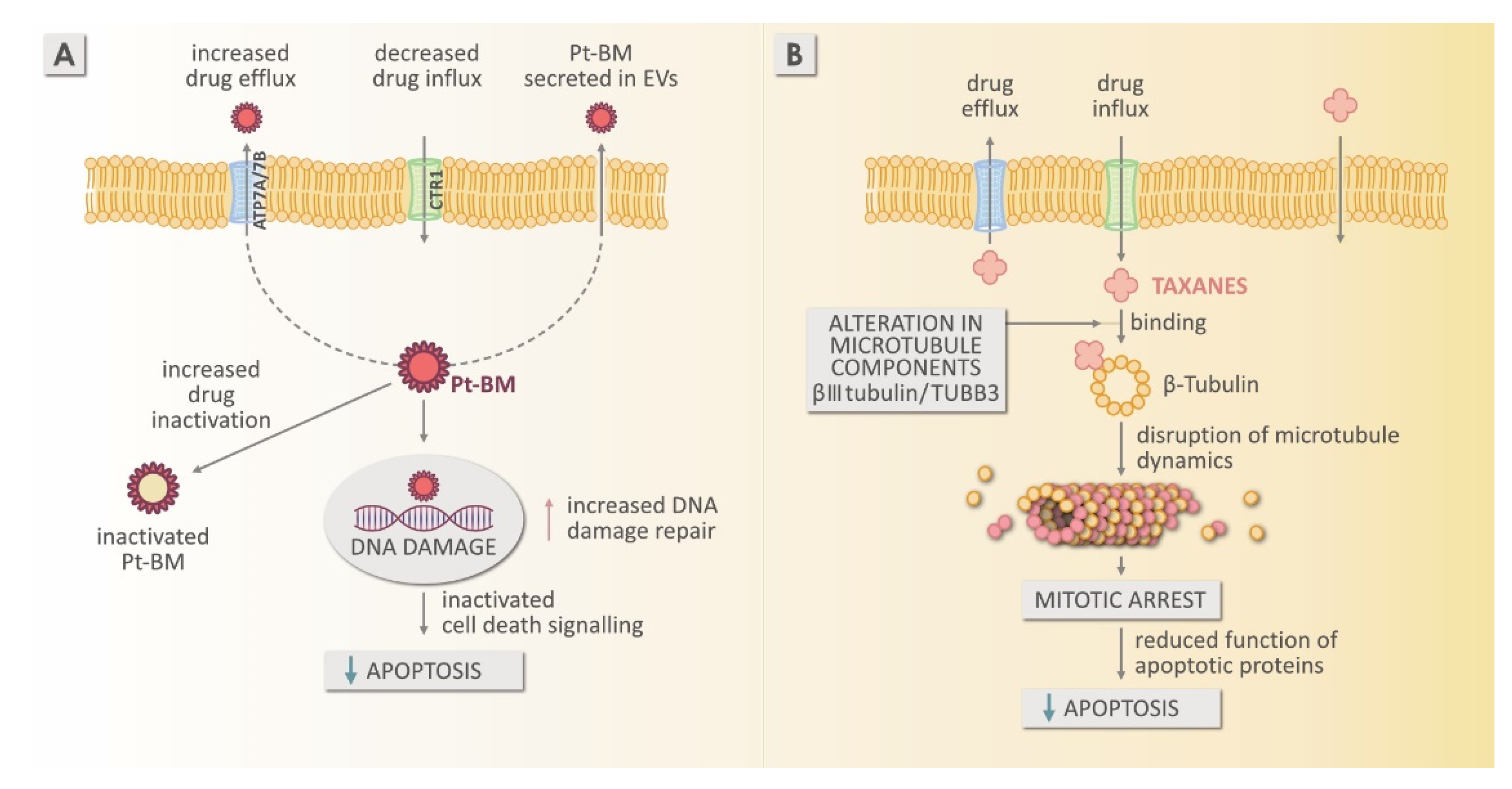
4.1. Resistance to Platinum-Based Chemotherapy
Mechanism of Resistance to Platinum-Based Medicines
4.2. Mechanism of Resistance to Taxanes
4.3. Mechanism of Resistance to PARP Inhibitors
4.4. Mechanism of Resistance to Bevacizumab
5. The Role of the Tumor Microenvironment
6. Resistance Study Models
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidents and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sant, M.; Chirlaque Lopez, R.; Agresti, R.; Sanchez Perez, M.J.; Holleczek, B.; Bielska-Lasota, M.; Dimitrova, N.; Innos, K.; Katalinic, A.; Langseth, H.; et al. EUROCARE-5 Working Group, Survival of women with cancers of breast and genital organs in Europe 1999–2007: Results of the EUROCARE-5 study. Eur. J. Cancer 2015, 51, 2191–2205. [Google Scholar] [CrossRef]
- Kobal, B. Operativno zdravljenje v ginekološki onkologiji. In Ginekologija in Perinatologija, 1st ed.; Takac, I., Gersak, K., Borko, E., Rakar, S., Sabec, N., Valh Lopert, A., Kobal, G., Rems, V., Eds.; Medicinska Fakulteta: Maribor, Slovenia, 2016; pp. 320–331. [Google Scholar]
- Zobec Logar, H.B.; Smrkolj, Š.; Merlo, S.; Bebar, S.; Kobal, B.; Škof, E.; Cerar, O.; Cvjetićanin, B.; Vakselj, A.; Šešek, M.; et al. Smernice za Obravnavo Bolnic z Rakom Jajčnikov, Jajcevodov in s Primarnim Peritonealnim Seroznim Rakom, 2nd ed.; Združenje za Radioterapijo in Onkologijo SZD, Združenje za Ginekološko Onkologijo, Kolposkopijo in Cervikalno Patologijo SZD, Onkološki Inštitut: Ljubljana, Slovenia, 2016. [Google Scholar]
- Zadnik, V.; Primic Zakelj, M.; Lokar, K.; Jarm, K.; Ivanus, U.; Zagar, T. Cancer burden in Slovenia with the time trends analysis. Radiol. Oncol. 2017, 51, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukanović, D.; Herzog, M.; Kobal, B.; Černe, K. The contribution of copper efflux transporters ATP7A and ATP7B to chemoresistance and personalized medicine in ovarian cancer. Biomed. Pharm. 2020, 129, 110401. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar]
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberts, A.; Johnson, J.; Lewis, M.; Raff, K.; Roberts, P.W. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002; pp. 1091–1144. [Google Scholar]
- Colombo, N.; Sessa, C.; du Bois, A.; Ladermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO Ovarian Cancer Consensus Conference Working Group, ESMO–ESGO consensus conference recommendations on ovarian cancer: Pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef] [Green Version]
- Cornelison, R.; Llaneza, D.C.; Landen, C.N. Emerging therapeutics to overcome chemoresistance in epithelial ovarian cancer: A mini-review. Int. J. Mol. Sci. 2017, 18, 2171. [Google Scholar] [CrossRef] [Green Version]
- Berek, J.S.; Kehoe, S.T.; Kumar, L.; Friedlander, M. Cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obs. 2018, 143 (Suppl. S2), 59–78. [Google Scholar] [CrossRef]
- Kroeger, P.T., Jr.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obs. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef]
- Prat, J. Pathology of cancers of the female genital tract. Int. J. Gynaecol. Obs. 2015, 131 (Suppl. S2), S132–S145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular characterization of epithelial ovarian cancer: Implications for diagnosis and treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodnik, L. Ovarian cancer: Resistance to treatment. Med. Razgl. 2021, 60, 55–64. [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Yueng, C.L.A.; Wong, S.T.C.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A review in the theme: Cell and molecular processes in cancer metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, L.; Karimnia, N.; Stephens, A.; Bilandzic, M. Therapeutic targeting of collective invasion in ovarian cancer. Int. J. Mol. Sci. 2019, 20, 1466. [Google Scholar] [CrossRef] [Green Version]
- Černe, K.; Kobal, B. Ascites in advanced ovarian cancer. In Ovarian Cancer; Devaja, O., Papadopoulos, A.J., Eds.; IntechOpen: London, UK, 2017; pp. 1–20. [Google Scholar]
- Paracchini, L.; Mannarino, L.; Craparotta, I.; Romualdi, C.; Fruscio, R.; Grassi, T.; Fotia, V.; Caratti, G.; Perego, P.; Calura, E.; et al. Regional and temporal heterogeneity of epithelial ovarian cancer tumor biopsies: Implications for therapeutic strategies. Oncotarget 2016, 12, 2404–2417. [Google Scholar] [CrossRef] [Green Version]
- McGuire, W.P., 3rd; Markman, M. Primary ovarian cancer chemotherapy: Current standards of care. Br. J. Cancer 2003, 89 (Suppl. S3), S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Lawrie, T.A.; Rabbie, R.; Thoma, C.; Morrison, J.; Cochrane Gynaecological, Neuro-oncology and Orphan Cancer Group. Pegylated liposomal doxorubicin for first-line treatment of epithelial ovarian cancer. Cochrane Database Syst. Rev. 2013, 10, CD010482. [Google Scholar]
- Pignata, S.; Scambia, G.; Katsaros, D.; Gallo, C.; Pujade-Lauraine, E.; De Placido, S.; Bologna, A.; Weber, B.; Raspagliesi, F.; Panici, P.B.; et al. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 396–405. [Google Scholar] [CrossRef]
- Falandry, C.; Rousseau, F.; Mouret-Reynier, M.A.; Tinquaut, F.; Lorusso, D.; Herrstedt, J.; Savoye, A.M.; Stefani, L.; Bourbouloux, E.; Sverdlin, R.; et al. Efficacy and safety of first-line single-agent carboplatin vs carboplatin plus paclitaxel for vulnerable older adult women with ovarian cancer: A GINECO/GCIG randomized clinical trial. JAMA Oncol. 2021, 7, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kobal, B.; Cvjetičanin, B. Kirurško zdravljenje epitelnega raka jajčnikov (ERJ), ocena resektabilnosti. In Rak Jajčnikov: Zbornik Predavanj; Sola o Ginekološkem Raku: Ljubljana, Slovenia, 2015; pp. 47–53. [Google Scholar]
- Jiang, X.; Li, W.; Li, X.; Bai, H.; Zhang, Z. Current status and future prospects of PARP inhibitor clinical trials in ovarian cancer. Cancer Manag. Res. 2019, 11, 4371–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/glossary/summary-product-characteristics (accessed on 18 February 2022).
- Harrap, K.R. Preclinical studies identifying carboplatin as a viable cisplatin alternative. Cancer Treat. Rev. 1985, 12, 21–33. [Google Scholar] [CrossRef]
- Chatelut, E. Pharmacology of platinum compounds: Differences between the three molecules and factors of interpatient variability. Bull. Cancer 2011, 98, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhang, P.Y. Recent perspectives of epithelial ovarian carcinoma. Oncol. Lett. 2016, 12, 3055–3058. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.X.; Chan, D.W.; Ngan, H.Y.S. Mechanisms of chemoresistance in human ovarian cancer at a glance. Gynecol. Obs. 2012, 2, e104. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Eno, M.L.; Im, D.D.; Rossenshein, N.B. Chemotherapy time interval and development of platinum and taxane resistance in ovarian, fallopian, and peritoneal carcinomas. Arch. Gynecol. Obs. 2010, 281, 325–328. [Google Scholar] [CrossRef]
- Rauh-Hain, J.A.; Nitschmann, C.C.; Worley, M.J., Jr.; Bradford, L.S.; Berkowitz, R.S.; Schorge, J.O.; Campos, S.M.; del Carmen, M.G.; Horowitz, N.S. Platinum resistance after neoadjuvant chemotherapy compared to primary surgery in patients with advanced epithelial ovarian carcinoma. Gynecol. Oncol. 2013, 129, 63–68. [Google Scholar] [CrossRef]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T.; Luesley, D.; Perren, T.; Bannoo, S.; Mascarenhas, M.; et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Škof, E.; Cerar, O. Epitelijski rak jajčnikov, rak jajcevodov, primarni peritonealni serozni karcinom (PPSC)—Priporočila za sistemsko zdravljenje. Onkologija 2015, 19, 17–21. [Google Scholar]
- Cole, C.B.; Bates, S.; Fojo, A. Overview of mechanisms of chemoresistance in ovarian cancer. In Overcoming Ovarian Cancer Chemoresistance; Samimi, G., Annunziate, C., Eds.; Academic Press: London, UK, 2020; pp. 26–38. [Google Scholar]
- Kardos, J.; Héja, L.; Simon, A.; Jablonkai, I.; Kovacs, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Palm, M.E.; Weise, C.F.; Lundin, C.; Wingsle, G.; Nygren, Y.; Björn, E.; Naredi, P.; Wolf-Watz, M.; Wittung-Stafshede, P. Cisplatin binds human copper chaperone Atox1 and promotes unfolding in vitro. Proc. Natl. Acad. Sci. USA 2011, 108, 6951–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadini-Buoninsegni, A.F.; Bartolommei, G.; Moncelli, M.R.; Inesi, G.; Galliani, A.; Sinisi, M.; Losacco, M.; Natile, G.; Arnesano, F. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew. Chem. 2014, 53, 1297–1301. [Google Scholar] [CrossRef] [Green Version]
- Safaei, R.; Maktabi, M.H.; Blair, B.G.; Larson, C.A.; Howel, S.B. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J. Inorg. Biochem. 2009, 103, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Katano, K.; Safaei, R.; Samimi, G.; Holzer, A.; Tomioka, M.; Goodman, M.; Howell, S.B. Confocal microscopic analysis of the interaction between cisplatin and the copper Transporter ATP7B in human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 4578–4588. [Google Scholar] [CrossRef] [Green Version]
- Arnesano, F.; Natile, G. Interference between copper transport systems and platinum drugs. Semin. Cancer Biol. 2021, 76, 173–188. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Okuda, T.; Holzer, A.; Howell, S.B. The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae. Mol. Pharmacol. 2002, 62, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- eIncrease in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin. Cancer Res. 2003, 9, 5853–5859.
- Katano, K.; Safaei, R.; Samimi, G.; Holzer, A.; Rochdi, M.; Howell, S.B. The copper export pump ATP7B modulates the cellular pharmacology of carboplatin in ovarian carcinoma cells. Mol. Pharmacol. 2003, 64, 466–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar] [PubMed]
- Hua, H.; Günther, V.; Georgiev, O.; Schaffner, W. Distorted copper homeostasis with decreased sensitivity to cisplatin upon chaperone Atox1 deletion in Drosophila. BioMetals 2011, 24, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of taxane resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]
- Škof, E. Experience with olaparib in the treatment of recurrent ovarian epithelial cancer with mutations in the BRCA 1 and BRCA 2 genes. Oncology 2021, 25, 12–16. [Google Scholar]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Lee, E.K.; Matulonis, U.A. PARP Inhibitor resistance mechanisms and implications for post-progression combination therapies. Cancers 2020, 12, 2054. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol 2021, 18, 773–791. [Google Scholar] [CrossRef]
- Montemagno, C.; Pagès, G. Resistance to anti-angiogenic therapies: A mechanism depending on the time of exposure to the drugs. Front. Cell Dev. Biol. 2020, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Wieser, V.; Marth, C. Resistance to chemotherapy and anti-angiogenic therapy in ovarian cancer. Memo 2019, 12, 144–148. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Shen, F.; Hu, W.; Coleman, R.L.; Sood, A.K. New ways to successfully target tumor vasculature in ovarian cancer. Curr. Opin. Obs. Gynecol. 2015, 27, 5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat. 2012, 15, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthebane, D.A.; Rovve, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Cesi, G.; Walbrecq, G.; Margue, C.; Kreis, S. Transferring intercellular signals and traits between cancer cells: Extracellular vesicles as “homing pigeons”. Celli Commun. Signal. 2016, 14, 13. [Google Scholar] [CrossRef] [Green Version]
- Tkach, M.; Théery, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Giallombardo, M.; Taverna, S.; Alessandro, R.; Hong, D.; Rolfo, C. Exosome-mediated drug resistance in cancer: The near future is here. Ther. Adv. Med. Oncol. 2016, 8, 320–322. [Google Scholar] [CrossRef] [Green Version]
- Trzpis, M.; McLaughlin, P.M.; de Leij, L.M.F.H.; Harmsen, M.C. Epithelial cell adhesion molecule: More than a carcinoma marker and adhesion molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Schmelzer, E.; Reid, L.M. EpCAM expression in normal, non-pathological tissues. Front. Biosci. 2008, 13, 3096–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heredia-Soto, V.; López-Guerrero, J.A.; Redondo, A.; Mendiola, M. The hallmarks of ovarian cancer: Focus on angiogenesis and micro-environment and new models for their characterisation. EJC Suppl. 2020, 15, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2015, 10, e0122284. [Google Scholar] [CrossRef]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High grade serous ovarian cancer: Basic sciences, clinical and therapeutic standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Medicines | Monotherapy | Combination | Prior Response to | Special Consideration |
|---|---|---|---|---|
| Pt-Based Therapy | ||||
| (Complete or Partial) | ||||
| Primary/Front-Line Therapy | ||||
| bevacizumab | – | 1. carboplatin/paclitaxel | – | 1. FIGO stages IIIB, IIIC, IV. |
| Maintenance Therapy Following Completion of First-Line Pt-Based CT | ||||
| olaparib | yes | – | yes | 1. BRCA1/2-mutation. |
| 2. High-grade EOC, FIGO stages III, IV. | ||||
| olaparib | – | 1. bevacizumab | yes | 1. HRD positive status. |
| 2. High-grade EOC, FIGO stages III, IV. | ||||
| 3. Following first-line Pt-based CT in combination with bevacizumab. | ||||
| niraparib | yes | – | yes | 1. High-grade EOC, FIGO stages III, IV. |
| Therapy of Relapsed Cancer | ||||
| bevacizumab | – | 1. carboplatin/gemcitabin | yes | 1. First recurrence. |
| 2. carboplatin/Paclitaxel | 2. No prior anti-VEGF therapy. | |||
| bevacizumab | – | 1. paclitaxel | no | 1. No more than 2 prior CT regimens. |
| 2. topotecan | 2. No prior anti-VEGF therapy. | |||
| 3. doxorubicin PL | ||||
| rucaparib | yes | – | – | 1. BRCA1/2-mutation. |
| 2. High-grade EOC. | ||||
| 3. Progression after two or more prior Pt-based CT. | ||||
| 3. Un-tolerant to further Pt-based CT. | ||||
| Maintenance Treatment of Relapsed Cancer | ||||
| olaparib | yes | – | yes | 1. BRCA1/2-mutation. |
| 2. High-grade EOC. | ||||
| niraparib | yes | – | yes | 1. High-grade serous EOC. |
| rucaparib | yes | – | yes | 1. High-grade EOC. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukanović, D.; Kobal, B.; Černe, K. Ovarian Cancer: Treatment and Resistance to Pharmacotherapy. Reprod. Med. 2022, 3, 127-140. https://doi.org/10.3390/reprodmed3020011
Lukanović D, Kobal B, Černe K. Ovarian Cancer: Treatment and Resistance to Pharmacotherapy. Reproductive Medicine. 2022; 3(2):127-140. https://doi.org/10.3390/reprodmed3020011
Chicago/Turabian StyleLukanović, David, Borut Kobal, and Katarina Černe. 2022. "Ovarian Cancer: Treatment and Resistance to Pharmacotherapy" Reproductive Medicine 3, no. 2: 127-140. https://doi.org/10.3390/reprodmed3020011
APA StyleLukanović, D., Kobal, B., & Černe, K. (2022). Ovarian Cancer: Treatment and Resistance to Pharmacotherapy. Reproductive Medicine, 3(2), 127-140. https://doi.org/10.3390/reprodmed3020011